

Abstract
The exposure of phosphatidylserine (PS) molecules from the inner to the outer leaflet of the plasma membrane has been recognized as a well-defined molecular epitope of cells undergoing apoptosis. Examination and monitoring of PS exposure is an extensively used molecular marker in non-invasive apoptosis imaging under a variety of clinical conditions, including the assessment of therapeutic anti-cancer agents and myocardial infarction. Herein, we report the identification of a PS-recognizing peptide which was identified by the screening of an M13 phage display peptide library onto PS-coated ELISA plates. Repeated biopanning for a total of four rounds revealed a predominant enrichment of the phage clone displaying peptide sequence, CLSYYPSYC (46%). The identified phage clone evidenced enhanced binding to a number of apoptotic cells over non-apoptotic cells, and this binding was inhibited by both annexin V and synthesized peptide displayed on the phage. The binding of the fluorescein-labelled CLSYYPSYC peptide to apoptotic versus normal cells was assessed by both FACS analysis and fluorescence microscopy. Optical imaging after the systemic administration of fluorescein-labelled CLSYYPSYC peptide to tumour-bearing nude mice (H460 cells xenograft model) treated with a single dose of an anticancer drug (camp-tothecin) indicated peptide homing to the tumour. The histological examination of tumour tissues showed intense staining of the tumour vasculature and apoptotic tumour cells. With these results, the CLSYYPSYC peptide is recognized as a novel PS-recognizing moiety which may possibly be developed into a molecular probe for the imaging of apoptosis in vivo. This application would clearly be relevant to assessments of the efficacy of anticancer therapy in tumours.
Keywords: phosphatidylserine, apoptosis, phage display, annexin V, peptide
Introduction
Phosphatidylserine (PS) is an anionic phospholipid which is confined to the cytoplasmic side of the plasma membrane [1]. The translocation of PS from the inner to the outer leaflet of plasma membrane occurs in apoptotic cells, and is the most commonly assessed molecular marker for the detection and measurement of apoptotic cells under both in vitro and in vivo conditions [2]. The activated platelets, endothelial cells in the tumour vascula-ture and apoptotic tumour cells have also been shown to expose PS molecules on their cell surfaces [3–5]. The precise measurement and monitoring of apoptosis is a matter of substantial value in assessing the therapeutic response of tumours to radiation, chemo- and photodynamic-therapies [6]; monitoring the course of disease in myocardial infarction [7] and in the early diagnosis of neurodegenerative diseases [8]. Additionally, constitutive exposure in the tumour vasculature makes PS an attractive target molecule for the selective delivery of therapeutic agents to tumours [9]. Kuriyama et al. (2006) demonstrated the use of the PS-interacting peptide as a carrier molecule for the genes [10]. Thus far, annexin V is the most extensively employed means of measuring apoptosis under both in vitro and in vivo conditions [11], and this has been coupled with a variety of molecular probes (magnetic, radioactive or optical) for in vivo apoptosis imaging. However, the requirement of a micromolar range of calcium for the optimal binding of annexin V to PS, as well as the activation of transmembrane scramblase activity at this concentration of calcium, resulting in non-specific exposure of PS, are some of the limitations associated with the use of annexin V in molecular apoptosis imaging [12].
The aforementioned studies attempted to seek alternatives to annexin V, and demonstrated that fluorescent dyes [13], the zinc-dipicolylamine co-ordination complex [14], dye-labelled cationic liposome [15], peptide-labelled nanoparticles [16], short peptides [17] and larger proteins including lactadherine and the C2A domain of synaptotagmin I [18, 19] can detect the PS molecules on the surfaces of apoptotic cells. Peptides specific to molecules do have advantages over proteins and antibodies for a number of reasons, including superior stability, non-immunogenicity, low cost, easy labelling and rapid clearance and tissue penetration [20]. Thus, the discovery of a peptide endowed with PS specificity could be extremely useful, and may result in the development of a diagnostic means for the non-invasive imaging of apoptosis under a variety of clinical conditions, and as a carrier molecule for targeted therapy. Importantly, the construction of nanoparticles coated with multiple numbers of peptides will enable the multiva-lent interaction of peptides with its target molecule, thus enhancing the affinity of the peptide to a considerable extent.
The principal objective of the present study was to discover novel PS-recognizing peptidic ligands that can be utilized for a variety of applications, including the molecular imaging of apoptosis or targeting moieties in PS-exposed cells. We screened a phage display peptide library via the conventional method of repeated biopanning of the phage library to PS-coated plates prior to examining the enrichment of the consensus peptide sequences. The ability of the identified phage clone and its corresponding peptide to recognize and bind to the PS molecules on the apoptotic cell surface were demonstrated herein. The targeting of fluores-cein-labelled peptides to apoptotic tumour cells following systemic administration into tumour-bearing mice, and its possible use in the in vivo detection of apoptosis by using optical imaging system were evaluated herein.
Materials and methods
Materials
L-α-phosphatidylserine (PS) and L-α-phosphatidylcholine (PC) were purchased from Avanti Polar Lipids, Inc. USA. The his-tagged annexin V protein was purified from Escherichia coli transformed with cDNA encoding for annexin V in a pET29b expression vector system (Novagen, Madison, WI).
Cell culture
H460 and H157 human lung cancer cells and U937 leukaemia cells were maintained in RMPI 1640 medium containing 10% foetal bovine serum supplemented with antibiotics.
Screening of PS binding phages
The M13 phage library evidencing random cyclic peptides of 7-mer fused to pIII (New England Biolabs, Ipswich, MA, USA) was used in this study. PC or PS dissolved in ethanol (3 μg/ml, 100 μl) was added to 96-well Immulon 1B microtitre plates (Thermo, Milford, MA, USA) and allowed to air-dry for 6 hrs. Non-specific binding sites in the wells were blocked with 10 mg/ml of bovine serum albumin (BSA) in Tris buffered saline (TBS) (Tris-HCl 50 mM, pH 7.4, NaCl 150 mM) for 1 hr at room temperature (RT). The coating of PC or PS into the plates was assessed via an annexin V binding test as described below in the description of phage ELISA. The 2 × 1011 plaque-forming units (pfu) of the original phage library (10 μl) in 100 μl of TBS containing 10 mg/ml of BSA were added to PC-coated wells for 1 hr at RT with gentle shaking (subtraction step). Then, the PC-subtracted phage library was transferred to PS-coated wells and incubated for an additional 1 hr at RT with gentle shaking. The wells were extensively washed to remove the unbound and feebly bound phages via at least 10 washings in TBS-T (TBS containing 0.05% Tween-20). The phages bound to the PS-coated wells were eluted via 10 min. of incubation with 100 μl of 0.2 M glycine-HCl (pH 2.2) containing 1 mg/ml BSA at RT. The elutes were immediately neutralized with 1M Tris-HCl (pH 9.1) and amplified in 20 ml of ER2738 bacteria in accordance with the manufacturer's instructions. Phages from the culture supernatant were precipitated by polyethylene glycol/NaCl and the titres were determined. LB plates containing isopropyl-beta-D-thiogalactopyronoside (IPTG) and X-gal were used and the blue colonies were counted after overnight incubation at 37°C. For subsequent rounds of selection, the same inputs of 2 × 1011 pfu from previous selection rounds were maintained. To ensure the enrichment of PS-specific phages, the PC-coated wells were also included in each selection round; phage titres from the PC- and PS-coated wells were compared. A total of four selection rounds were conducted prior to the isolation of single-stranded DNA from individual phage clones. DNA from at least 30 individual phage clones were sequenced using an automated DNA sequencer (Genotech, Rep. of Korea) with the primer -96 pIII provided by the manufacturer (New England Biolabs). The deduced amino acid sequences were aligned using the CLUSTALW program to determine the consensus peptide sequence. An advanced BLAST search (EMBL/GenBank/DDBJ) was conducted to detect proteins with significant homology to the peptide sequences.
Characterization of selected phage clones
The binding specificity of the selected phage clones to PS was evaluated via phage-binding tests. In brief, PC- or PS-coated ELISA plates were prepared as described above. After 1 hr of blocking with 10 mg/ml of BSA in TBS at RT, the selected phage clone and amplified M13 phage library (control) (1 × 109 pfu) in 100 μl TBS was added to PC- or PS-coated wells, then incubated for 1 hr at RT with gentle shaking. The plate wells were extensively washed in TBS-T (10 times) prior to the elution of the bound phage with 100 μl of 0.2 M glycine-HCl (pH 2.2) containing 1 mg/ml BSA for 10 min. at RT, then immediately neutralized with 1M Tris-HCl (pH 9.1). The phage titres were determined in each of the elutes.
Phage ELISA
Additionally, the binding specificity of the selected phage clone to PS was assessed via phage ELISA. In brief, Immulon 1B microtitre plates were coated with PC or PS as mentioned above, then blocked with TBS containing 10 mg/ml of BSA for 1 hr at RT. The selected phage clone (1 × 109 pfu) in 100 μl of TBS-T was added to PC- or PS-coated wells and incubated for 1 hr at RT with gentle shaking. The amplified phage library was also included as a control. After extensive washing with TBS-T six times, the bound phages were detected via 1 hr of incubation with HRP-conjugated anti-M13 antibody (New England Biolabs) (dilution, 1:4000 in TBS-T) at RT, followed by colour development with Horseradish peroxidase (HRP) substrate solution (TMB, Pierce). Colour development was halted via the addition of 2N H2SO4 and then measured at 450 nm using a microplate reader (Bio-Rad, model 550).
Phage ELISA was also conducted in microplates coated with PS lipo-somes. For the preparation of PC or PC/PS liposome (50:50 Mol.%), phospholipids in chloroform were dried under a high-speed vacuum, hydrated in TBS, then sonicated for 5–10 min. on ice. The Immulon 1B microtitre plates were coated with 100 μl of liposome (20 μg/ml) at 4°C overnight. After blocking the plate with TBS containing 10 mg/ml BSA for 1 hr at RT, all the procedures were conducted exactly as mentioned above. The specificity of the selected phage clone was assessed via an inhibition study of phage binding to PS-coated plates in the presence of annexin V. The phages were incubated in the presence of annexin V protein (10 nM), and phages bound to the plates were detected as described above.
Phospholipids coating the wells were assessed via annexin V binding tests by ELISA. For annexin V binding, his-tagged recombinant annexin V protein (20 μg/ml) in TBS containing 10 mg/ml of BSA was added to ELISA plates coated with PC or PS, then incubated for 1 hr at RT. After three washings with TBS-T, the bound annexin V protein was determined via additional incubation with HRP-conjugated mouse anti-His IgG antibody (Santa Cruz Biotechnology) and 3,3’,5,5-tetramethylbenzidine (TBM) substrate, as described above.
Phage clone binding to apoptotic cells
The binding of the selected phage clone to various apoptotic cells was assessed by a phage plaque assay. Apoptosis was induced in H460, H157 and U937 cells via treatment with etoposide (50 μm) (Sigma). U937 cells were treated for 4 hrs, whereas H460 and H157 cells were treated for 18 hrs. In order to ensure the exposure of PS molecules on the apoptotic cell surfaces, fluorescence activated cell sorting (FACS) analyses were conducted after annexin V (BD Biosciences) staining, in accordance with the manufacturer's instructions. For the phage-binding assay, apoptotic cells were washed with Phosphate buffered saline (PBS) and pre-incubated for 30 min. in Dubelco's modified Eagle's medium (DMEM) medium containing 10 mg/ml BSA at RT. The cells were then incubated with 1 × 109 pfu of either an amplified phage library or the selected phage clone at 4°C for 1 hr with gentle shaking. The unbound phages were extensively washed away with DMEM medium containing 10 mg/ml of BSA and 0.05% Tween-20. The bound phages were eluted via 10 min. of incubation with 1 ml of 0.2 M glycine-HCl (pH 2.2) containing 1 mg/ml BSA at RT. The elutes were immediately neutralized with 1M Tris-HCl (pH 9.1). Phage titres in the elutes were determined after serial dilution and plague forming assay.
The PS specificity of bound phages to apoptotic cells was evaluated by competition with annexin V. For this, the apoptotic cells were pre-incubated for 30 min. with annexin V at final concentration of 10 μm prior to incubation with phages. The binding of the phages to the apoptotic cells was also performed in the presence of synthesized peptide (50 μm) displayed on the phage clone. The bound phages were eluted as described above, and the phage titres were determined.
Peptide synthesis
Peptides were synthesized via the standard Fmoc method, conjugated with fluorescein at the N-terminus, and purified via HPLC (Peptron Co.).
FACS analysis
The binding of peptides to apoptotic cells were evaluated via FACS analysis. The induction of apoptosis and PS exposure on the apoptotic cell surfaces were assessed via FACS analysis after annexin V staining, as described above. Peptide or control peptide at a final concentration of 2.5 μm in Hepes buffer (pH 7.4, Hepes 10 mM; NaCl2 138 mM and with/without 2 mM CaCl2) was incubated with normal or apoptotic cells (1 × 105 cells/ml) at a final volume of 0.5 ml for 15 min. at RT. The cells were washed with binding buffer and FACS analysis was immediately conducted (FACSScan, Becton Dickinson, San Jose, CA, USA).
Microscopy
For fluorescence microscopy, H460 cells cultured on eight-chamber slides (Nalgen Nunc Int.) were induced to undergo apoptosis, as described above. The cells were washed in PBS and incubated for 30 min. with 10 μm fluorescein-labelled peptides in Hepes buffer, and then with annexin V Alexa fluor 594 (Molecular Probes) for 15 min. at RT. The cells were washed extensively prior to 5 min. of fixation with 4% paraformaldehyde (PFA). The cells were counterstained with the nuclear stain, 4’,6-diamidi-no-2-phenylindole (DAPI), prior to mounting (Molecular Probes, Oregon) and the pictures were taken with a fluorescence microscope (Zeiss, Oberkochen, Germany).
For confocal microscopy, cells were stained with peptide and annexinV Alexa fluor 568 (Molecular Probes) as described above and confocal images were taken using laser confocal microscopy (LEICA DM IRB).
In vivo homing of CLSYYPSYC peptide into H460 cells via tumour xenograft and optical imaging
All animal experiments were conducted in accordance with the guidelines established by the Kyungpook National University. To prepare the tumour xenografts, 6-week-old BALB/c male nude mice (Japan, SLC, Inc.) were injected subcutaneously in the right shoulder with 1 × 107 H460 cells suspended in RPMI medium containing 10% FBS. The tumours were then grown to a size of 0.5–1 cm within a 3 week period.
Tumour-bearing mice either treated or untreated with a single dose of campthothecin (Sigma) (10 mg/kg) 24 hrs prior to peptide injection received intravenous injections of fluorescein-labelled CLSYYPSYC or control peptides (50 μm each) through the tail vein under isoflurane anaesthesia. Peptide homing to tumours was detected using an optical imaging system (ART Advanced Research Technologies Inc., Montreal, Canada) using a 470 nM/Green fluorescence protein (GFP) filter at different time intervals after peptide injection. For each mouse used, baseline fluorescence was assessed prior to peptide injection. The images obtained were processed using eXplore Optix optiView Software and normalized. Ex vivo imaging of tumour tissues was conducted after removing the tumours from mice 2 hrs after peptide injection, and the images were processed as described above.
For histological examination, the mice were deeply anaesthetized and opened. After perfusion with PBS and fixation with 4% PFA via heart puncture, the tumour tissues and organs were removed. Cryosections were prepared and fluorescence microscopy and immunohistochemical studies were performed. The tumour vessels were stained with antibody to mouse CD31 (BD Pharmigen). Apoptosis in the tumour tissues were assessed by in vitro terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL) staining in accordance with the manufacturer's instructions (Chemicon Int., Temecula, CA, USA).
Results
Screening of M13 phage library and validation of selected phage clones
The M13 phage library evidencing CX7C random peptides was screened to enrich phages that bind selectively to PS over PC. The simple schematic diagram of selection strategy is shown in Fig. 1A. Phospholipids coating the ELISA plates were assessed via the annexin V binding test prior to the initiation of biopanning rounds (Fig. 1B). To exclude the phages that bind to PC, subtraction steps were included in each round of selection. The phage enrichment to PS was observed in the second round (approximately 114-fold) and did not increase further in subsequent rounds (Fig. 1C). The four total rounds of biopanning were conducted prior to the isolation of the individual phage clones for DNA sequencing.
1.
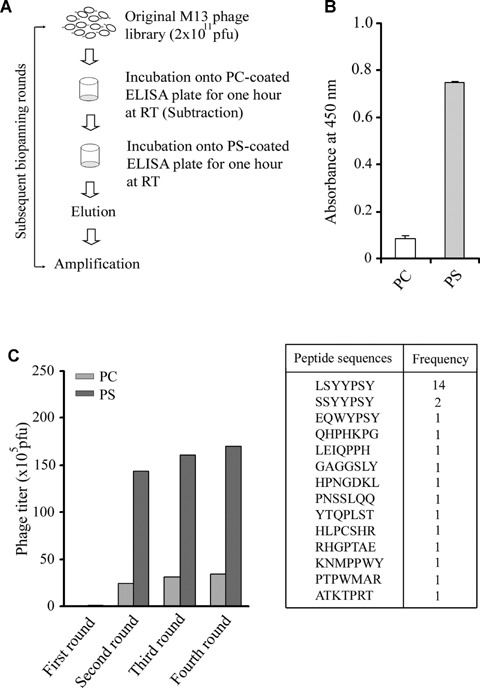
Screening of M13 phage display peptide library. (A) Schematic diagram of biopanning strategy. The M13 phage library (2 × 1011 pfu) was incubated with phosphatidylcholine (PC)-coated well before transferring to phos-phatidylserine (PS)-coated well. After incubating for 1 hr at room temperature (RT), unbound phages were removed by extensive washing. The bound phages were eluted and amplified. The total four rounds of biopanning were carried out. The phage titre of 2 × 1011 pfu was maintained in each round of biopanning. (B) Coating of phospholipids to ELISA plate were assessed by ELISA test. (C) Both PC and PS were included in each round of biopanning to ensure that enrichment during biopanning is occurring for PS versus PC. In each round, the phages binding to PC and PS were eluted and titre was determined. The peptide sequences displayed on phage clones were determined.
Thirty individual phage clones were picked from the third and fourth rounds of screening, and DNA sequencing was conducted (Genotech, Korea). The obtained DNA sequences were translated into the corresponding amino acids. The 14 phage clones among 30 evidenced the same peptide sequence (Fig. 1C), and peptide alignment using the Clustal W program revealed one predominant motif, YYPSY. The database search was conducted in order to determine the human proteins possessing significant homology with the peptide sequences. Some of the proteins possessing amino acid sequences homologous to the peptide sequences are shown in Table 1. The phage-displayed peptides may mimic one or more of the proteins that bind to PS.
1.
Example of human proteins containing identical amino acid sequences to peptide
| Peptide sequence | Homologous amino acids | Name of proteins | Accession number | |||||
|---|---|---|---|---|---|---|---|---|
| CLSYYPSYC | 297LSYYRSY303 | Vacuolar protein sorting-associated protein | Q8N1B4 | |||||
| 63LSYSPSY69 | E-selectin precursor | P16581 | ||||||
| 293LSYYP297 | Tenascin-N precursor | Q9UQP3 | ||||||
| 715YYPSY719 | Protein transport protein Sec24A | Q95486 | ||||||
| 291YYPSY295 | Transmembrane protein 66 precursor | Q5R491 | ||||||
| 149YYPSY153 | Transcription factor E2-α | P15923 | ||||||
Peptide was analysed using the NCBI BLAST search against the SWISSPROT database, using the option for short nearly exact matches to identify human proteins with homologous amino acid sequences. Amino acid with bold letter indicates difference in amino acid sequence between peptide and protein. The numbers indicate the positions of amino acids in the proteins.
The phage clone displaying the consensus peptide sequence, CLSYYPSY, was selected and validated for selective binding to PS over PC. The binding of the selected phage was assessed via phage plague assay (Fig. 2A) and phage-ELISA assay (Fig. 2B). For the control phage, the M13 phage library was amplified one time and used as a control phage. Empty ELISA plate-wells were also included in order to ensure that phage selection occurred for PS, and not for the ELISA plate surface (data not shown). We determined that the selected phage clone was bound with equal activity to the PS liposome over the PC liposome coated onto 96-well ELISA (Fig. 2C) plates, thereby indicating the possibility of phage binding to the PS molecule exposed on the apoptotic cell surfaces. Additionally, the binding of the selected phage clone, CLSYYPSYC, to the PS-coated plate was inhibited by annexin V (Fig. 2D).
2.
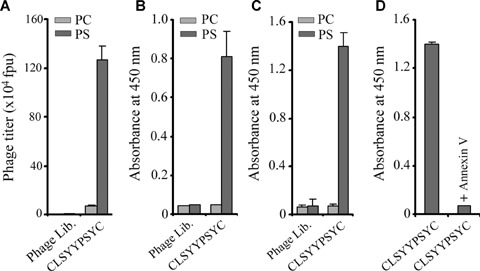
Selection of phage clones. Phage clone showing predominant consensus sequence, CLSYYPSYC was selected for further study. (A) Phage CLSYYPSYC and amplified phage library (1 × 109 pfu) were incubated in ELISA plate coated with PC or PS for 1 hr at RT. The bound phages were eluted and phage titre determined. (B) Higher binding of selected phage clone to PS in ELISA. (C) Higher binding of selected phage clone to PS liposome in ELISA. (D) Binding of selected phage clone to PS was inhibited in the presence of annexin V. Data is representative of three independent experiments.
Binding of phage to apoptotic cells
Apoptosis was induced in a variety of cell types, including H460, H157 and U937 by etoposide treatment. The selective binding of the phage clone to apoptotic cells over normal cells was assessed via phage plague assay (Fig. 3A, B and C). PS exposure in the apoptotic cells was confirmed via FACS analysis using annexin V staining. Some of the cancer cell lines, B16F10 and MCF-7, which evidenced minimal PS exposure after etoposide treatment, also exhibited less significant binding of the phage to the treated cells versus the untreated cells (data not shown). The specificity of interaction to PS molecules on the surfaces of apoptotic cells was verified via the inhibition of phage binding by annexin V (Fig. 3D) in H460 cells. However, we were unable to demonstrate the PS specificity of phage binding to apoptotic cells using the PS liposome in the inhibition study. The M13 phage library itself evidenced increased binding to apoptotic cells in the presence of the PS liposome (data not shown). Furthermore, the specificity of phage binding was also verified via the inhibition of phage binding to apoptotic H460 cells in the presence of synthesized peptides, but not by the irrelevant peptide (Fig. 3D). Additionally, the binding of the phage clone to PS was unaffected by 4 mM ethylenediaminetetraacetic acid (EDTA), thereby indicating calcium independency in binding to PS molecules (data not shown).
3.
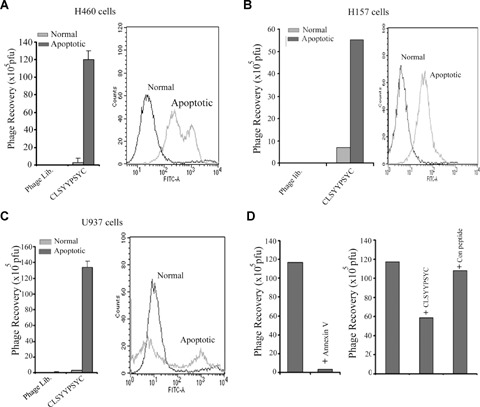
Binding of CLSYYPSYC phage to apoptotic cells. Apoptosis of cells was induced as described in material and methods. The PS exposure on cell membrane of apoptotic cells is examined by fluorescence activated cell sorting (FACS) analysis (right column). The amplified phage library or phage clone CLSYYPSYC (1 × 109 pfu) was incubated with apoptotic cells or normal cells at 4°C for 1 hr. After extensive washing, bound phages were eluted and phage titre determined (A, H460 cells; B, H157 cells; C, U937 cells). (D) Specificity of phage CLSYYPSYC binding to apoptotic H460 cells was examined by inhibition study by annexin V (10 μm) and CLSYYPSYC peptide or control peptide (50 μm). Cells were pre-incubated with annexin V or peptides before incubating with phage clone, and bound phage was determined by plague assay as described in ‘Materials and methods’. The results are expressed as mean±S.D. and are representative of 2–3 independent experiments.
Peptide binding to apoptotic cells
The ability of the synthesized peptide to distinguish between apop-totic and non-apoptotic cells was assessed via both FACS analysis and fluorescence microscopy. Fluorescence-labelled CLSYYPSYC peptide binding was observed in nearly 40% of the H460 cells treated with etoposide, but not to the untreated cells on the FACS analysis (Fig. 4A). Annexin V staining indicated that more than 80% of treated cells are apoptotic. The FACS data in Fig. 3A could represent this result. This result suggests that annexin V with four well-defined PS-binding sites may bind strongly to apoptotic cells than the peptide. Similar results were obtained when the etoposide-treated A549 and H157 cells were utilized (data not shown). The unrelated peptide used in the experiment (control peptide) evidenced no binding to the treated cells.
4.
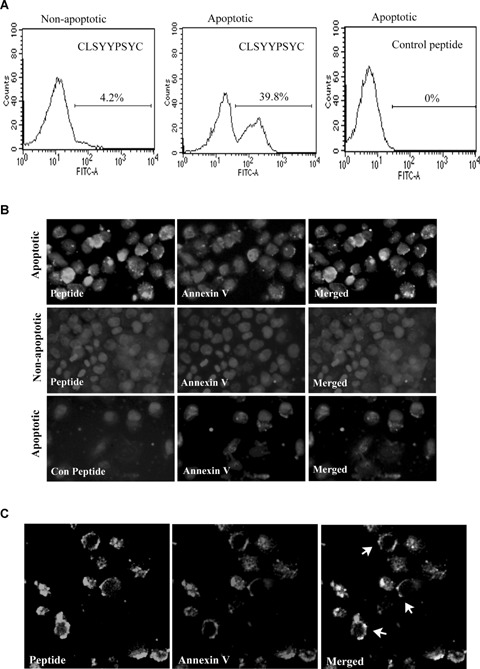
Binding of FITC-labelled CLSYYPSYC peptide to apoptotic H460 cells. (A) Apoptotic H460 cells were detached from the culture plate after trypsin treatment and incubated with FITC-labelled CLSYYPSYC peptide or control peptide (2.5 μm) in Hepes buffer for 15 min. at RT and analysed by FACS analysis. (B) For immunofluorescence microscopy, apoptotic cells in chamber slide were incubated for 30 min. in Hepes buffer containing peptide or control peptide (10 μm each) and annexin V. Cells were washed and fixed with 4% paraformaldehyde (PFA) before staining with DAPI (images were taken at magnification of 400x). (C) For confocal microscopy, apoptotic H460 cells was stained with peptide and Alexa 568-labelled annexin V as described above and confocal images were taken using laser confocal microscopy (LEICA DM IRB) (magnification, x400).
Fluorescence microscopy indicated that the binding of the peptide to the etoposide-treated cells was significantly higher than to the untreated cells (Fig. 4B). The cellular distribution of peptide to apoptotic cells revealed that discrete regions of the apoptotic cell membrane were stained by peptide. Many cells exhibited colocalization of signals when costained with peptide and annexin V-Alexa 594 (Fig. 4B). The confocal microscopy performed on apoptotic H460 cells after staining with fluorescein-labelled CLSYYPSYC and annexinV Alexa fluor 568 further confirmed the colocalization of annexin V and peptide (Fig. 4C). The binding/recognition of PS molecules by peptide occurred regardless of the presence or absence of calcium ions (data not shown).
Peptide homing to H460 cell tumour xenografts and optical imaging
We assessed the ability of the CLSYYPSYC peptide to detect cell apoptosis in vivo, and its possible application in the molecular imaging of apoptosis using an H460 cell tumour xenograft model in nude mice. After a single dose of camptothecin treatment, the mice were injected systematically with fluorescein-labelled peptide through the tail vein. Peptide homing to tumour was evaluated via an optical imaging system at different time intervals after peptide injection. The representative images from each group (n= 3) is shown in Fig. 5A. The treatment of tumour-bearing mice with a single dose of anticancer drug induced significantly enhanced peptide homing, as the induction of tumour cell apoptosis is the common mode of the antitumour activity of camptothecin. Some signals were observed in the untreated group and the basal apoptosis of the tumour may have contributed this. Although the Fluorescein isothjocynate (FITC) signal is absorbed to a significant degree by surrounding tissues, the signal generated by homing peptides in tumour tissues was detectable within 1–2 hrs of peptide injection, and even at 18 hrs post-injection. In addition, peptide homing to tumour tissues was assessed by ex vivo imaging (Fig. 5B). The systemic administration of control peptide to tumour-bearing mice treated with camtothecin did not evidence a fluorescence signal of peptide homing to tumour in either in vivo or ex vivo imaging.
5.
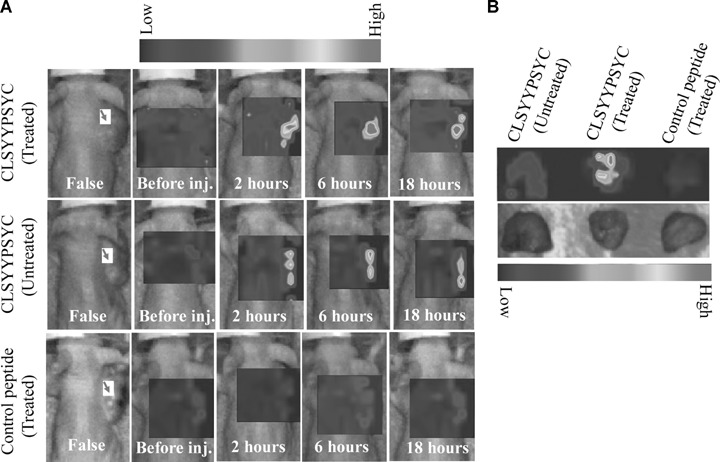
In vivo homing of peptide to tumour. (A) Fluorescein-labelled CLSYYPSYC peptide or control peptide was systematically injected through tail vein of tumour-bearing nude mice treated or untreated with camptothecin 24 hrs before the peptide injection. The homing of peptides was examined by using the optical imaging system as described in ‘Materials and methods’. The red arrows indicate the tumours. (B) For ex vivo imaging, tumours from mice were removed 2 hrs after post injection of the peptides and image were taken similar as in vivo imaging
For histological examination, the cryosections were prepared from the tumour tissues. Fluorescein microscopic observation indicated the homing of the peptide to tumour vasculature and tumour cells. The fluorescein signal of CLSYYPSYC peptide homing was more prominent in the tissue sections prepared from mice treated with anti-cancer drugs (Fig. 6A). Presence of apoptotic/necrotic areas inside the tumour tissue caused extensive binding of peptide giving the appearance of regularly distributed bright spots. No fluorescein signals were observed in tumour tissues from mice injected with control peptides. In addition, the binding of CLSYYPSYC peptide to control organs such as liver and lung was minimal, while being observed in the glomeruli of kidney that represents the excretion route for the peptide (Fig. 6A). The tumour vasculature was stained intensely by the peptide, as demonstrated by the fact that fluorescein signals in tumour vasculature were highly colocalized with the Alexa 594 used for murine CD31 staining (Fig. 6A). Histological examinations with consecutive tissue slides showed that the fluorescein signals of peptides correlate with haematoxylin-eosin (data not shown) and TUNEL staining showing an increased area of in vivo apoptosis (Fig. 6B).
6.
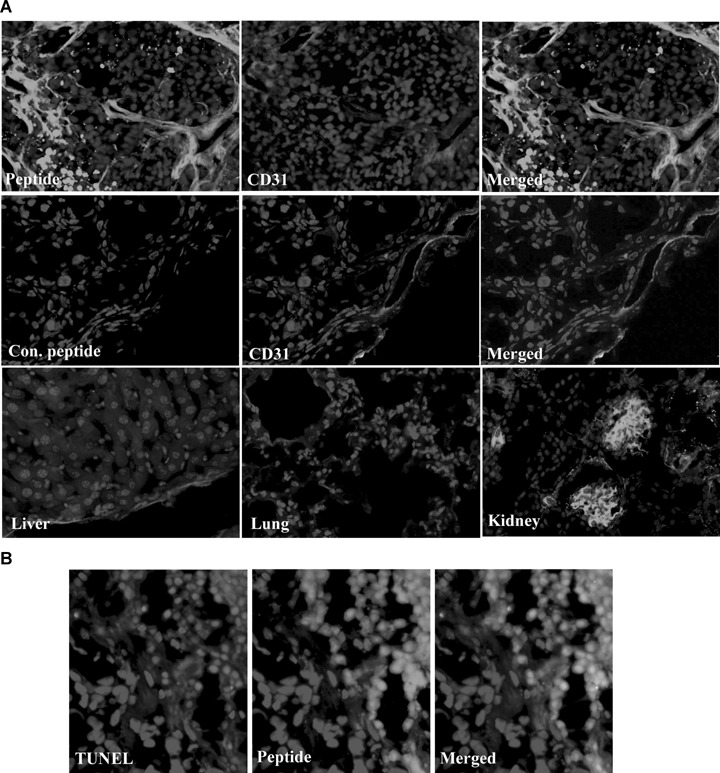
Histological examination. (A) Cryosections were prepared from the tumour tissues and peptide signals were observed using fluorescence microscopy. For endothelial cells staining, mice anti-CD31 antibody was used followed by Alexa 568-labelled secondary antibody. (B) Tumour cell apoptosis was observed by TUNEL staining as described in ‘Materials and methods’.
Discussion
In the present study, we have described the identification of a novel PS-interacting peptide via a phage display technique, and recognized the discovered peptide as a PS-recognizing moiety which could be utilized for the in vivo molecular imaging of apoptosis. Repeated biopanning of the phage library to PS and the subtraction of PC-recognizing phages in each round of selection allowed us to isolate the phage clone displaying the consensus peptide sequence, CLSYYPSYC. PS molecules are translocated from the inner to the outer membranes of apoptotic cells. The NH3+ head groups in the PS molecules are located below the CO2− and PO4− in the cell membrane surface and the anionic nature of the PS molecules is largely attributed to its CO2” and PO4” groups [21]. The experimental strategy we undertook in this study could result in the isolation of phage clones that may or may not recognize PS molecules existing in the apoptotic cell membranes as phage screening was conducted on PS-coated plates. In an effort to exclude this possibility, we first assessed the binding of the selected phage clone to the PS versus the PC liposome, and then to a number of apoptotic cells versus normal cells. The PS specificity of binding was confirmed via competition with annexin V.
The molecular basis of interaction between the identified peptide, CLSYYPSYC and the PS molecule has yet to be clearly elucidated. In the previous study, the isolation of PS-interacting peptides rich in basic amino acid residues was characterized using a phage display technique [22]. It is interesting to note that basic amino acid residues are not present in the CLSYYPSYC peptide, which could impart an overall positive charge to the peptide. Thus, electrostatic interaction may play a minimal role in the interaction between the CLSYYPSYC peptide and anionic PS molecules. Furthermore, we synthesized linear and cyclic forms of CLSYYPSYC peptide. The cyclic form showed more specific binding to apoptotic cells versus non-apoptotic cells. Thus, cyclic form of peptide was used throughout this study. In the previous study, it was shown that the affinity of the displayed peptide in the phage might be significantly higher than that of the synthesized peptide, largely due to the favourable presentation of the peptide in the macromolecular structures of the phage particles [17]. We believe that the construction of suitable nanoparticles harbouring multiple numbers of CLSYYPSYC peptides could increase the affinity of the peptide to a considerable degree.
The NCBI database search revealed a number of proteins that evidenced homology to CLSYYPSYC, and further study into whether this peptide sequence could represent the true PS-interacting motif or part of the motif in proteins such as sec24A, κ-E2-binding factor, transmembrane protein 66 precursors, tenascin-N-precursor and transcription factor E2-α will be an interesting area for further studies. It is possible that these proteins may represent novel PS-interacting proteins in biological systems.
We also assessed the performance of the CLSYYPSYC peptide in binding to apoptotic cells in vivo using a H460 cell tumour xenograft model. The early assessment of the efficacy of anti-cancer agents is highly desirable and represents an unmet need in clinical oncology studies [23]. Molecular apoptosis imaging is highly useful in addressing this need as the induction of tumour cell-apoptosis is the primary mechanism of action of the majority of anticancer drugs [24]. We first demonstrated the homing of the fluorescein-labelled CLSYYPSYC peptide to tumours using an optical imaging system. The tumour vasculature and apoptotic tumour cells were the most intensely stained cells in the histological examination. The previous study showed that PS exposure is induced by anticancer drug treatment in the tumour vasculature and tumour cells [4, 5]. Although FITC signals are highly absorbed by tissues, and are only rarely utilized for in vivo imaging, we were able to observe a distinct peptide homing signal, using FITC-labelled CLSYYPSYC peptide to the tumour, which was enhanced further by anticancer drug treatment to mice. We believe that the labelling of the CLSYYPSYC peptide with suitable imaging probes including radioisotopes and MR imaging contrasts may further improve the sensitivity of the peptide to a considerable degree, and may be useful for clinical purposes. Additionally, in situ amplification mechanism may further enhance the binding and homing of CLSYYPSYC peptide to tumour as tumour apoptosis and PS exposure progress during tumour therapy. Furthermore, small peptides that can target the tumour vasculature may be an important tool for development into therapeutic agents for tumour therapy, largely due to its non-immunogenic and highly penetrative nature in tumour tissues [25]. Tumour vasculature is a suitable target for cancer therapy due to its non-malignant nature and thus, low probability to evidence drug-resistance [26]. Thus, the tumour vasculature is more accessible to anticancer drugs and evidences a more intrinsic amplification mechanism [26]. Thus, the examination of CLSYYPSYC peptide as carrier molecule for anticancer drugs to tumour vessels may prove an interesting area for future studies.
Recently, two studies described the identification of PS-recognizing peptides using phage display technique [27, 28]. However, PS-binding affinity, in vivo bio-distribution, stability etc. has yet to be described for these peptides. Importantly, each candidate peptide should be validated after their synthesis as peptides present in phages and their synthesized forms are distinctly different.
PS serve as an ‘eat-me signal’ for the removal of apoptotic cells by phagocytes in vivo and pre-incubation of apoptotic cells with annexin V inhibits their recognition and removal by phagocytes. Immunogenecity of apoptotic cells can be increased if their PS is masked by annexin V before injecting into the mice [29]. In this context, study of CLSYYPSYC peptide as adjuvant in apoptotic cells will be interesting as it could be useful in increasing the efficacy of apoptotic cell-based vaccines.
In conclusion, we identified a novel PS-interacting peptide using a phage display technique, and this peptide may be developed into a molecular imaging probe for monitoring and measuring apoptosis in vivo and as a carrier molecule for the targeting of the tumour vasculature.
Acknowledgments
This study was supported by the grant No. RTI04-01-01 from the Regional Technology Innovation Program of the MOCIE, Advanced Medical Technology Cluster for Diagnosis and Prediction at Kyungpook National University from MOCIE, MOST GRL program and Brain Korea 21 programs.
References
- 1.Yamaji-Hasegawa A, Tsujimoto M. Asymmetric distribution of phospholipids in biomembranes. Biol Pharm Bull. 2006;29:1547–53. doi: 10.1248/bpb.29.1547. [DOI] [PubMed] [Google Scholar]
- 2.Zwaal RF, Comfurius P, Bevers EM. Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci. 2005;62:971–88. doi: 10.1007/s00018-005-4527-3. [DOI] [PubMed] [Google Scholar]
- 3.Dasgupta SK, Guchhait P, Thiagarajan P. Lactoadherin binding and phosphatidylserine expression on cell surface-comparison with annexin A5. Transl Res. 2006;148:19–25. doi: 10.1016/j.lab.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Ran S, He J, Huang X, Soares M, Scothorn D, Thorpe PE. Antitumor effects of a monoclonal antibody that binds anion-ic phospholipids on the surface of tumor blood vessels in mice. Clin Cancer Res. 2005;11:1551–62. doi: 10.1158/1078-0432.CCR-04-1645. [DOI] [PubMed] [Google Scholar]
- 5.Ran S, Downes A, Thorpe PE. Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 2000;62:6132–40. [PubMed] [Google Scholar]
- 6.Corsten MF, Hofstra L, Narula J, Reutelingsperger CP. Counting heads in the war against cancer: defining the role of annexin A5 imaging in cancer. Cancer Res. 2006;66:1255–60. doi: 10.1158/0008-5472.CAN-05-3000. [DOI] [PubMed] [Google Scholar]
- 7.Zhao M, Zhu X, Ji S, Zhou J, Ozker KS, Fang W, Molthen RC, Hellman RS. 99mTc -labeled C2A domain of synaptotagmin I as a target-specific molecular probe for noninvansive imaging of acute myocardial infarction. J Nucl Med. 2006;47:1367–74. [PubMed] [Google Scholar]
- 8.Lampl Y, Lorberboym M, Blankenberg FG, Sadeh M, Gilad R. Annexin V SPECT imaging of phosphatidylserine expression in patients with dementia. Neurobiology. 2006;66:1253–64. doi: 10.1212/01.wnl.0000208436.75615.8c. [DOI] [PubMed] [Google Scholar]
- 9.Ran S, Downes A, Thorpe PE. Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 2002;62:6132–40. [PubMed] [Google Scholar]
- 10.Kuriyama S, Taguchi Y, Nishimura K, Mizuguchi K, Kobayashi K, Katayama Y, Yanagibashi K, Niidome T. Peptide vector for gene delivery with high affinity for phosphatidylserine. J Pept Sci. 2006;12:626–32. doi: 10.1002/psc.768. [DOI] [PubMed] [Google Scholar]
- 11.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis, flow cytometry detection of phosphatidylserine expression on early apoptotic cells using fluorescein labeled annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 12.DiVittorio KM, Johnson JR, Johansson E, Reynolds AJ, Jolliffe KA, Smith BD. Synthetic peptides with selective affinity for apoptotic cells. Org Biomol Chem. 2006;4:1966–76. doi: 10.1039/b514748d. [DOI] [PubMed] [Google Scholar]
- 13.Laakko T, King L, Fraker P. Versatility of merocyanine 540 for the flow cytometric detection of apoptosis in human and murine cells. J Immunol Methods. 2002;261:129–39. doi: 10.1016/s0022-1759(01)00562-2. [DOI] [PubMed] [Google Scholar]
- 14.Hanshaw RG, Lakshmi C, Lambert TN, Johnson JR, Smith BD. Fluorescent detection of apoptotic cells by using zinc coordination complexes with a selective affinity for membrane surfaces enriched with phosphatidylserine. Chem Bio Chem. 2005;6:2214–20. doi: 10.1002/cbic.200500149. [DOI] [PubMed] [Google Scholar]
- 15.Bose S, Tuunainen I, Parry M, Medina OP, Mancini G, Kinnunen PK. Binding of cationic liposomes to apoptotic cells. Analy Biochem. 2004;331:385–94. doi: 10.1016/j.ab.2004.05.033. [DOI] [PubMed] [Google Scholar]
- 16.Quinti L, Weissleder R, Tung C-H. A fluorescent nanosensor for apoptotic cells. Nano Let. 2006;6:488–90. doi: 10.1021/nl0524694. [DOI] [PubMed] [Google Scholar]
- 17.Laumonier C, Segers J, Laurent S, Michel A, Coppee F, Belayew A, Elst LV, Muller R. A new peptidic vector for molecular imaging of apoptosis, identified by phage display technology. J Biomol Screening. 2006;11:537–45. doi: 10.1177/1087057106288220. [DOI] [PubMed] [Google Scholar]
- 18.Shi J, Shi Y, Waehrens LN, Rasmussen JT, Heegard CW, Gilbert GE. Lactadherin detects early phosphatidylserine exposure on immobilized leukemia cells undergoing programmed cell death. Cytometry Part A. 2007;69A:1193–201. doi: 10.1002/cyto.a.20345. [DOI] [PubMed] [Google Scholar]
- 19.Jung HI, Kettunen MI, Davletov B, Brindle KM. Detection of apoptosis using the C2A domain of synaptotagmin I. Bioconjug Chem. 2004;15:983–7. doi: 10.1021/bc049899q. [DOI] [PubMed] [Google Scholar]
- 20.Signore A, Annovazzi A, Chianelli M, Corsetti F, Van De Wiele C, Watherhouse RN. Peptide radiopharmaceuticals for diagnosis and therapy. Eur J Nucl Med. 2001;28:1555–65. doi: 10.1007/s002590100583. [DOI] [PubMed] [Google Scholar]
- 21.Kimura T. Human opioid peptide metenkephalin binds to anionic phosphatidylserine in high preference to zwitterionic phosphatidylchoine: natural-abundance 13C NMR study on the binding state in large unilamellar vesicles. Biochemistry. 2006;45:15601–9. doi: 10.1021/bi061641v. [DOI] [PubMed] [Google Scholar]
- 22.Nakai Y, Nomura Y, Sato T, Shiratsuchi A, Nakanishi Y. Isolation of a Drosophila gene coding for a protein containing a novel phosphatidylserine-binding motif. J Biochem. 2005;137:593–9. doi: 10.1093/jb/mvi072. [DOI] [PubMed] [Google Scholar]
- 23.Corsten MF, Hofstra L, Narula J, Reutelingsperger CPM. Counting heads in the war against cancer: defining the role of annexin V imaging in cancer treatment and surveillance. Cancer Res. 2006;66:1255–60. doi: 10.1158/0008-5472.CAN-05-3000. [DOI] [PubMed] [Google Scholar]
- 24.Neves AA, Brindle KM. Assessing responses to cancer therapy using molecular imaging. Biochim Biophys Acta. 2006;1766:242–61. doi: 10.1016/j.bbcan.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 25.Blankenberg FG, Katsikis PD, Tait JF, Davis RE, Raumovski L, Ohtsuki K, Kopiwoda S, Abrams MJ, Daekes M, Robbins RC, Maecker HT, Strauss HW. In vivo detection and imaging of phosphatidylserine expression during programmed cell death. Proc Natl Acad Sci USA. 1998;95:6349–54. doi: 10.1073/pnas.95.11.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arap W, Pasqualini R, Ruoslahati Cancer treatment by targeted drugs delivery to tumor vasculature in a mouse model. Science. 1998;279:377–80. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 27.Shao R, Xiong C, Wen X, Gelovani JG, Li C. Targeting phosphatidylserine on apoptotic cells with phages and peptides selected from a bacteriophage display library. Mol Imaging. 2007;6:417–26. [PubMed] [Google Scholar]
- 28.Segers J, Laumonier C, Burtea C, Laurent S, Elst LV, Muller RN. From phage display to magnetophage, a new tool for magnetic resonance molecular imaging. Bioconjugate Chem. 2007;18:1251–8. doi: 10.1021/bc060377f. [DOI] [PubMed] [Google Scholar]
- 29.Stach CM, Turnay X, Voll RE, Kern PM, Kolowos W, Beyer TD, Kalden JR, Herrmann M. Treatment with annexin V increases immunogenicity of apoptotic human T-cells in balb/c mice. Cell Death Differ. 2000;7:911–5. doi: 10.1038/sj.cdd.4400715. [DOI] [PubMed] [Google Scholar]