

Abstract
Objective: Hyperhomocysteinemia induces endothelial dysfunction and promotes atherosclerotic vascular disease. Infiltrates of activated macrophages and lymphocytes are observed in human and experimental atherosclerotic lesions, their emigration being guided by endothelial-leukocyte adhesion molecules and chemoattractants. The CXC-chemokine CXCL16 functions as an adhesion molecule by interacting with its receptor (CXCR6) and also as a scavenger for oxidized low density lipoprotein (oxLDL). We investigated the modulation of CXCL16 on cultured endothelial cells (EC) and the recruitment of CXCR6+ lymphocytes in response to homocysteine (Hcy), in vitro and in vivo. Methods and Results: Hcy-stimulated EC show a significant increase in CXCL16 mRNA and protein expression. Incubation of EC with d,l-Hcy and l-Hcy significantly increased CXCR6+ lymphocyte adhesion to EC while l-Cysteine (l-Cys) had no effect. Furthermore, EC stimulation with Hcy increased uptake of DiI-oxLDL. An anti-CXCL16 monoclonal antibody, antioxidants (Tiron) and PPAR-γ agonists (Pioglitazone) considerably reduced CXCR6+ lymphocyte adhesion and uptake of DiI-oxLDL. Upon injection in the peritoneal cavities of mice, l-Hcy and not l-Cys, increased the number of CXCR6+ lymphocytes, which was reduced by coinjection with Pioglitazone or anti-human CXCL16 antibody. Conclusions: Hyperhomocysteinemia up-regulates CXCL16 leading to increased recruitment of CXCR6+ lymphocytes and scavenging of modified lipids via a potential involvement of a PPAR-γ-dependent mechanism. CXCL16 may therefore contribute to the formation and progression of atherosclerotic lesions under conditions of hyperhomocysteinemia.
Keywords: homocysteine, atherosclerosis, scavenger, chemokines, PPAR-γ
Introduction
Inflammation plays a central role in the development and progression of atherosclerosis.
An early stage in the development of this disease involves circulating leukocytes that adhere to the activated endothelium and migrate into the subendothelial space. Infiltrates of activated macrophages and T lymphocytes are observed in human and experimental atherosclerotic lesions [1].
In the subendothelial space, macrophages take up ox-low-density lipoprotein (LDL) through their scavenger receptors and develop into foam cells, a hallmark of atherosclerotic disease. In addition, activated endothelial cells (EC) express scavenger receptors, take up modified lipids and get charged with lipid drops [2]. Chemokines, such as Monocyte chemoattractant protein 1 (MCP-1) and Regulated on Activation, Normal T Expressed and Secreted Protein (RANTES) are important for monocyte/macrophage and T cell recruitment to the lesion [3, 4]. CXCL16, a recently discovered CXC chemokine [5, 6], has been found to be expressed in human atherosclerotic lesions [7, 8]. It is identical to the scavenger receptor SR-PSOX, which mediates uptake of oxidized LDL [9]. As a chemokine, CXCL16 is special because it is expressed in a membrane-bound form inducing cell-cell adhesion by ligating CXCR6 chemokine receptor on lymphocyte subsets [10] and also shed to soluble form [11], guiding the migration of activated Th1 and Tc1 cells to the sites of inflammation [5].
Hyperhomocysteinemia has been considered an independent risk factor for atherosclerosis but the underlying mechanism remains elusive. Elevated concentrations of Hcy seem to be harmful for EC [12], whereas pathophysiologically relevant concentrations as found in patients with mild hyperhomocysteinemia seem to induce functional endothelial changes summarized as endothelial dysfunction [13]. Recently, it has been shown that exposure of cultured EC to Hcy leads to endothelial activation with increased expression of chemokines, like MCP-1 and IL-8 [14] and adhesion molecules [15, 16].
It has been reported that Hcy increases the production of reactive oxygen species (ROS) from T lymphocytes and their proliferation rate in ApoE-deficient mice maintained under high-methionine diet, insinuating that hyperhomocysteinemia may be involved in the pathogenesis of atherosclerosis by enhancing T cell response [17]. Moreover, Hcy enhances ROS production in EC and activates NF-κB which triggers increased Intercellular Adhesion Molecule 1 (ICAM-1) expression and adhesion of monocytes [18].
Due to its anti-inflammatory, and antioxidant actions, the perox-isome proliferator-activated receptor-7 (PPAR-γ) family of nuclear hormone receptors has been intensively investigated in models of vascular inflammation. Activation of PPAR-γ counteracts the induction of pro-inflammatory chemokines, such as MCP-1 in cytokine-treated human vascular EC [19]. Recently, it has been observed that PPAR-γ agonists decrease hyperhomocysteinemia and cardiac dysfunction [20]. In addition, PPAR-γ which is expressed in vascular EC [21] ameliorates the Hcy-induced EC activation [22].
In this study we investigated whether relevant pathophysiolog-ical levels of Hcy modulate the CXCL16 expression on EC and recruitment of CXCR6+ lymphocytes in vitro and in vivo via a PPAR–γ-dependent mechanism.
Materials and methods
Chemicals
d,l-Hcy, l-Hcy thiolactone hydrochloride and l-Cys were from Sigma Chemical Co. and freshly prepared first as 100 mM stock solutions in Dulbecco's Modified Eagle's Medium (DMEM) medium supplemented with 10% FBS. l-Hcy was synthesized from its thiolactone by hydrolysis in 0.1 M NaOH [23]. Tumour necrosis factor (TNF)-α, interferon (IFN)–γ and Tiron were from Sigma Chemical Co. Pioglitazone was from Takeda Chemical Industries® (Osaka, Japan) and GW9662 from Tocris (Bristol, UK). Monoclonal anti-human PE conjugated CXCR6 antibody, monoclonal goat anti-human CXCL16 antibody, Fluorescein Isothiocyanate (FITC) conjugated secondary antibody anti goat IgG, PE-conjugated anti goat IgG antibody and goat IgG isotype control were from R&D Systems. The Omniscript kit for reverse transcription was obtained from Qiagen (Hilden Germany). GoTaq DNA polymerase and the PCR reaction buffer were purchased from Promega (Mannheim, Germany). DiI-oxLDL was purchased from Intracel (Frederick, USA). Cell culture media, Foetal Bovine Serum (FBS), antibiotics, and accutase were from PAA Laboratories; TRIZOL was from Invitrogen and Ficoll from GE Healthcare.
Cell lines
Human embryonic kidney epithelial cell (HEK293) line was transfected with the human CXCR6 receptor [24]. EA.hy926 cell line [25] and primary human umbilical vein EC (HUVEC) were used as experimental models of endothelial monolayer. EA.hy926 cells, derived by fusing HUVEC with the permanent human cell line A549/8 as described [25] are widely used to study EC specific gene expression and endothelial cell-leukocyte interactions, as they express EC markers, adhesion molecules [26] and secrete chemokines [27]. HUVEC were isolated from umbilical cord by digestion with collagenase and cultured in EC growth medium (PromoCell) containing 10% heat-inactivated FBS, and antibiotics. Passages 2 and 3 were used for the experiments. EA.hy926 and HEK293 cells were maintained in DMEM containing 4.500 mg\l D-glucose, 10% heat-inactivated FBS and antibiotics.
Sorting human CXCR6+ lymphocytes
CXCR6+ human peripheral lymphocytes were isolated from buffy coats of five healthy human volunteers by Ficoll separation followed by CXCR6 staining with the monoclonal anti-human CXCR6-PE antibody (2.5μxg/∼1 × 106 cells), sorting using the FACS Aria (Becton Dickinson and propagated with human recombinant Interleukin-2 (25 U/ml) as reported previously [28].
Fluorescent labelling of cells
CXCR6-HEK293 cells or human primary lymphocytes were washed in serum-free Hank's balanced salt solution (HBSS) and 1 × 107 cells/ml were incubated with 5 μM BCECF/AM (Molecular Probes) in serum free HBSS (prepared as 1 mg/ml stock in anhydrous Dimethylsulfoxid (DMSO) and stored at (20°C) for 30 min. at 37°C and 5% CO2. Cells were then washed with HBSS and re-suspended in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS at a density of 5 × 105 cells/ml.
Static adhesion assay
Adhesion of CXCR6-HEK293, respectively human CXCR6+ lymphocytes to EC was studied under static conditions as described previously [18]. Briefly, EC grown to confluence on 24-well plate were incubated for 24 hrs with varying test substances. After washing, they were co-incubated with 2′, 7′-bis-(2-carboxyethyl)-5-(6')-carboxyfluorescein acerox-ymethyl ester (BCECF/AM) labelled CXCR6-HEK293 cells or human CXCR6+ lymphocytes for 30 min. at 37°C on a rotating platform. After removing the non-adherent cells by washing, the relative fluorescence was determined using a fluorimeter TECAN Spectra Fluor Plus (excitation 485 nm and emission 535 nm).
Determination of CXCL16 mRNA by RT-PCR
Total RNA from EC stimulated 24 hrs with Hcy was isolated by Trizol method. After reverse transcription using the Omniscript kit, the cDNA was amplified using the specific pair of primers for human CXCL16 with the following sequences: 5′-ACTCGTCCCAATGAAACCAC-3′ and 5′-ATGAA-GATGATGGCCAGGAG-3′.
Determination of CXCL16 surface expression by flow cytometry
After 24 hrs incubation with the test chemicals including Hcy, EC were washed with PBS and labelled with the goat anti-human CXCL16, followed by a secondary labelling with the FITC-labelled antibody against goat IgG. The fluorescence was quantified using the FACS Calibur (Becton Dickinson) and CellQuest software. The median of the specific fluorescence intensity was used as a marker for expression of the respective epi-tope. Nonspecific fluorescence was detected by using isotype-matched non-binding antibody and subtracted.
Uptake of DiI-labelled oxidized human LDL
EC were stimulated for 24 hrs with Hcy and/or other chemicals and after washing, the cells were incubated at 37°C for 4 hrs with 10 μg/ml DiI-oxLDL. Then, the DiI-oxLDL media was gently removed from cells, which were washed 3–4 times with probe-free media. Cells were then detached from the plate by treatment with Accutase for 5 min. at 37°C and fixed with 2% formaldehyde in PBS for 20 min. at RT. The fluorescence was quantified using the FACS Calibur and the 514 nm excitation respectively 550 nm emission filters.
in vivo model of peritoneal inflammation
One millilitre of Hcy and/or other chemicals dissolved in PBS was injected in the peritoneum of each wild-type C57-Black6 mouse used for the experiment (three independent experiments, three mice per condition). Phosphate Buffer Saline (PBS) and 4% thyoglycolate were used as control. Peritoneal cells were isolated by peritoneal lavage from the peritoneal cavity 4 days after injection. 2 × 106 cells were incubated for 1 hr at Room Temperature (RT) in FACS buffer containing rat anti-mouse CXCR6 antibody. PE-conjugated secondary antibody was then added, and the mixture was incubated at RT for 45 min. The cells were then washed and fixed in 2% formaldehyde before undergoing flow cytometry analysis.
In parallel, cell free peritoneal lavage was investigated for the presence of soluble CXCL16 using a validated and sensitive sandwich ELISA system for murine CXCL16 from R&D systems.
Animal experiments and study protocols were approved by local authorities, complying with German animal protection laws.
Statistical analysis
Data are reported as means ± SEM and analysed by factorial anova and post-hoc comparisons. Differences in dose-responses were tested with two-way repeated measures anova with post-hoc analysis performed with Fisher's PLSD and Bonferonni/Dunn procedures. Statistical significance was defined as a P-value < 0.05.
Results
CXCL16 expression on homocysteine-activated endothelial cells
To investigate whether pathophysiological relevant concentrations of Hcy would influence the endothelial expression of CXCL16, RT-PCR analysis of Hcy-stimulated EC using CXCL16 specific primers was performed. As shown in Figure 1, 24 hrs treatment with d,l-Hcy considerably up-regulated the CXCL 16 mRNA expression in cultured EA.hy926 cells to a similar extent co-stimulation with the two cytokines TNF-α and IFN-γ (both 20 ng/ml), that are known to synergize in CXCL16 expression [11]. By contrast, in l-Cys-treated ECs the CXCL16 expression was similar to that in unstimulated cells.
1.
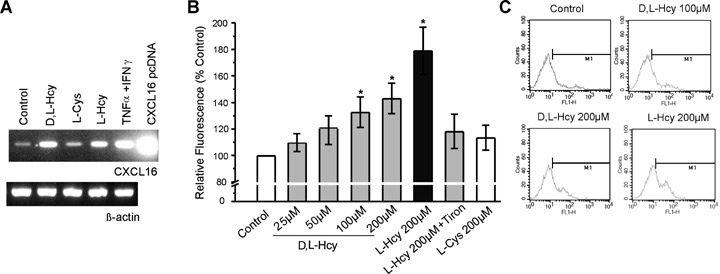
CXCL16 expression in homocysteine-treated endothelial cells. (A) EA.hy926 cells were incubated for 24 hrs with d,l-Hcy, l-Hcy, l-Cys, and investigated for the expression level of CXCL16 and β-actin mRNA by RT-PCR. Untreated cells and cytokine-stimulated cells (tumour necrosis factor [TNF]-α+ interferon [IFN]-γ) were used as control. The CXCL16 pcDNA plasmid (100 pg/ml) was used as positive control for the PCR. (B) 24 hrs incubation with d,l-Hcy and l-Hcy but not l-Cys significantly and dose-dependently increased the expression level of CXCL16 on the surface of EA.hy926 cells as determined by the change in relative fluorescence intensity (n = 4 experiments, *P < 0.05 versus control). In the presence of Tiron, Hcy's stimulatory effect was diminished ($P < 0.05 versusl-Hcy). In (C) representative FACS histograms for CXCL16 expression on Hcy-stimulated EC are shown.
Next, CXCL16 surface expression was determined by flow cytometry (Fig. 1B and C). d,l-Hcy dose-dependently increased endothelial surface expression of CXCL16. The effect reached significance by incubation with 100 and 200 μM d,l- Hcy and l- Hcy. This effect was specific for L-stereoisomer of Hcy but not for l-Cys. These observations suggest that Hcy but not other related thiols induce the de novo synthesis and surface expression of CXCL16. Notably, Hcy-induced CXCL16 expression was significantly diminished in the presence of the antioxidant Tiron (Figure 1B). The functional consequences of these effects will be addressed in detail in the following experiments.
Dose-dependent and stereospecific increase in adherent human primary lymphocytes to homocysteine-stimulated endothelial cells
Membrane bound CXCL16 serves as an adhesion molecule by binding the specific receptor CXCR6 which is known to be expressed on some sets of lymphocytes [10]. CXCR6-expressing HEK293 cells were investigated for adhesion to EA.hy926 mono-layer (Fig. 2A). Pre-incubation of EC with 200 μM d,l-Hcy or l-Hcy for 24 hrs significantly increased the number of adhering CXCR6-HEK293 cells (Fig. 2B and C) compared to untreated EC.
2.
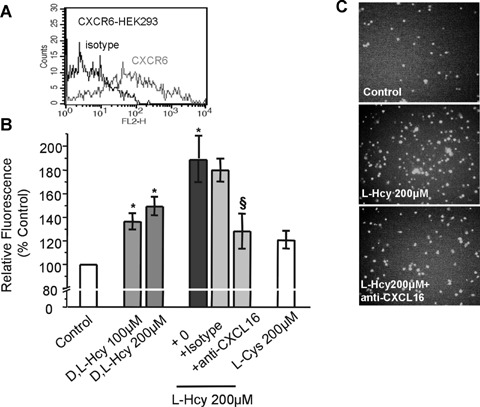
Adhesion of CXCR6-HEK293 cells to homocysteine-stimulated endothelial cells. (A) HEK293 cells were transfected to stably express CXCR6 as shown by flow cytometry. (B) As determined by the fluorescence signal of adherent CXCR6-HEK293 cells, Hcy but not l-Cys significantly increased the adhesiveness of EC (n = 5 experiments; *P < 0.05 versus control). Pre-treatment of Hcy-stimu-lated EC with a neutralizing antibody against CXCL16 diminished Hcy's stimulatory effect ($P < 0.05 versusl-Hcy only). In (C) representative microscopy pictures showing adhesion of CXCR6-HEK293 cells to unstim-ulated and Hcy-stimulated EC are presented.
Next, human peripheral lymphocytes were sorted for CXCR6 expression (Fig. 3A) and expanded for 3 days by culturing with IL-2. Lymphocytes uniformly expressing CXCR6 (Fig. 3B) were then assessed for adhesion to Hcy-activated EA.hy926 cells. As shown in Figure 3C, l-Hcy induced more than twofold up-regulation of CXCR6+ lymphocyte adhesion. TNF-α (20 ng/ml) was used as positive control. By contrast, l-Cys, had no significant effect on adhesion of either CXCR6-HEK293 cells or CXCR6+ lymphocytes.
3.
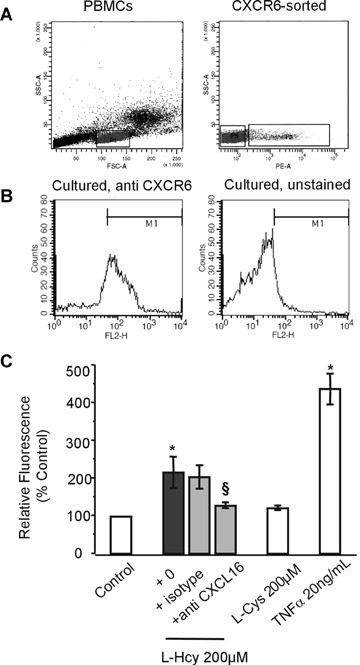
Adhesion of CXCR6+ lymphocytes to homocysteine-stimulated endothelial cells. (A) Peripheral blood CXCR6+ lymphocytes were sorted and after 3 days culture with IL-2, CXCR6+ lymphocytes (B) were assayed for adhesion to Hcy-stimulated EA.hy926 cells (C). Hcy, but not l-Cys increased the adhesion of CXCR6-sorted lymphocytes (n = 5 experiments; *P < 0.05 versus control). Tumour necrosis factor (TNF)-a was used as positive control. The neutralizing antibody against CXCL16 significantly decreased Hcy-induced lymphocyte adhesion ($P < 0.05 vs. l-Hcy).
To confirm the functional relevance of increased CXCL16 expression on Hcy-incubated EC, EA.hy926 cells were incubated with 200 μM l-Hcy for 24 hrs, followed by incubation with a blocking antibody against human CXCL16 (10 μg/ml) or an iso-type control, and the adhesion assays were performed. Blocking CXCL16 significantly reduced Hcy-induced CXCR6-HEK293 cell adhesion (Fig. 2B and C) and, more importantly, also human CXCR6+ lymphocyte adhesion to EC (Fig. 3C). These findings demonstrate a significant involvement of CXCL16 in increased adhesiveness of Hcy-activated EC.
CXCL16-mediated uptake of oxidized LDL by homocysteine-stimulated endothelial cells
The membrane bound CXCL16-variant functions not only as adhesion molecule by binding the specific receptor CXCR6, but also as scavenger for modified lipids [10]. To explore whether the scavenger activity of cxcl16 might be enhanced by Hcy, the uptake of DiI-oxLDL by ec was studied using facs analysis. d,l- and l-Hcy-incubated EA.hy926 cells showed a significant increase in DiI staining (Fig. 4), whereas l-Cys had no significant effect.
4.

Hcy's stimulatory effect on uptake of DiI-labelled oxLDL by endothelial cells. (A) EA.hy926 cells were assayed for binding of DiI-oxLDL by flow cytometry. (B) Binding of DiI-oxLDL to EC was significantly increased by 24 hrs stimulation of EC with Hcy but not by treatment with l-Cys (n = 4 experiments; *P <0.05 versus untreated control cells). Pre-incubation of Hcy-activated ECs with neutralizing antibody against CXCL16 significantly dimished the uptake of oxLDL ($P <0.05 versusl-Hcy only). (C) Representative photomicrographs indicate increased uptake of oxLDL by Hcy-treated EC.
To confirm the involvement of CXCL16 in the uptake of oxLDL by Hcy-incubated EC, EA.hy926 cells were incubated with 200 μM l-Hcy for 24 hrs, followed by incubation with a blocking antibody against human CXCL16 (10 μg/ml) or an isotype control, and the DiI-oxLDL binding assays were performed. Blocking CXCL16 significantly reduced uptake of oxLDL by Hcy-treated EC (Fig. 4). These data indicate that the scavenger receptor activity of CXCL16 contributes to increased oxLDL uptake by Hcy-activated EC.
Effect of antioxidants and PPAR-γ modulators on homocysteine-induced CXCL16 expression and function
Since the antioxidant Tiron significantly reduced Hcy-induced CXCL16 expression we further investigated whether oxidative stress modulators would affect CXCL16 up-regulation and function. Co-incubation of EC with l-Hcy together with the antioxidant Tiron significantly diminished Hcy's stimulatory effect on oxLDL binding and adhesion of CXCR6-HEK293 cells or CXCR6+ lymphocytes (Fig. 5A, B and C). Similarly, the PPAR-γ agonist Pioglitazone significantly reduced oxLDL binding and cell adhesion. Co-incubation with GW9662, an antagonist of PPAR-γ, slightly increased Hcy-induced oxLDL uptake and CXCR6+ lymphocyte adhesion to EC (Fig. 5A and B).
5.

Effect of reactive oxygen species (ROS) scavenger (Tiron), agonists (Pioglitazone) and antagonists (GW9662) of peroxisome proliferator-activated receptor-7 (PPAR-γ) on oxLDL binding and adhesion of CXCR6+ lymphocytes to homocysteine-stimulated endothelial cells. 24 hrs incubation of EA.hy926 cells with Hey together with Tiron or Pioglitazone but not GW9662 (5μM) significantly diminished oxLDL binding (A), adhesion of CXCR6-HEK293 cells (B) and CXCR6-sorted primary human lymphocytes (C). (n = five experiments, *P < 0.05 versus control; P < 0.05 versusl-Hcy 200 μM).
Expression and function of CXCL16 in hcy-incubated primary endothelial cells
In order to reproduce the major findings using primary EC, HUVECs were stimulated 24 hrs with Hcy, with/without other chemicals, and assessed for FACS analysis, adhesion assay and oxLDL uptake. The results presented in Table 1 show that Hcy significantly increased the CXCL16 surface expression on HUVEC and triggered adhesion of CXCR6+ lymphocytes and binding of oxLDL in a CXCL16-dependent manner. These events could be significantly diminished by Tiron and PPAR-γ agonist.
1.
Expression and function of CXCL16 in hcy-incubated primary endothelial cells
| Condition | CXCL16 surface expression (% Control) | Adhesion of CXCR6+ lymphocytes (% Control) | DiI-oxLDL uptake (% Control) |
|---|---|---|---|
| Control | 100 | 100 | 100 |
| l-Hcy 200 μmol/L | 182,27 ± 15,73* | 191,80 ± 18,73* | 180,27 ± 8,71* |
| l-Hcy 200 μmol/L+Tiron | 120,29 ± 12,17 | 128,29 ± 6,17 | 115,29 ± 2,70 |
| l-Hcy 200 μmol/L+isotype | - | 190,10 ± 8,73 | - |
| l-Hcy 200 μmol/L +antiCXCL16 | - | 120,10 ± 8,52 | 125,06 ± 2,57 |
| l-Hcy 200 μmol/L+Pioglitazone | - | 138,29 ± 4,17 | 122,03 ± 7,90 |
| l-Hcy 200 μmol/L+GW9662 | - | 195,06 ± 8,81 | 195,063 ± 18,81 |
| l-Cys 200 μmol/L | 113,41 ± 5,15 | 118,41 ± 6,18 | 110,41 ± 5,21 |
n = 4 experiments; *P< 0.05 versus Control.
Peritoneal recruitment of CXCR6+ lymphocytes after homocysteine injection
To study the potential involvement of CXCL16 in the proinflamma-tory activity of Hcy in vivo, Hcy or l-Cys were injected in the murine peritoneum and after 4 days cells were harvested from the peritoneal cavity by lavage. Flow cytometric analysis of these cells for expression of CXCR6 revealed that injection of Hcy but not l-Cys induced an accumulation of CXCR6+ lymphocytes (Fig. 6). Co-injection together with Pioglitazone or anti-CXCL16 antibody reversed the stimulatory effect of Hcy on the recruitment of CXCR6+ lymphocytes in the peritoneum which shows that the inflammatory effect of Hcy in this model involves the activity of CXCL16 and is down-regulated by activators of PPAR-γ. An ELISA sandwich system used to measure the soluble murine CXCL16 in the liquid collected after the peritoneal lavage showed an increase in CXCL16 after 200 μM l-Hcy injection to 131.78% of control. Co-injection with an antibody against CXCL16 or Pioglitazone diminished the detectable amount of CXCL16 to 96.69% or 90.17% of control, respectively. These data demonstrate that the recruitment of CXCR6+ cells is associated with the up-regulation of CXCL16.
6.

Recruitment of CXCR6+ lymphocytes following homocysteine injection in an in vivo model of peritoneal inflammation. l-Hcy increased the number of CXCR6+ lymphocytes as shown as% gated cells but l-Cys not. Co-injection of Hcy together with Pioglitazone or anti-CXCL16 antibody reversed Hcy'stimulatory effect on the recruitment of CXCR6+ lymphocytes in the peritoneum (n = 3 experiments, *P < 0.05 versus control; $P < 0.05 versusl-Hcy 200 μM).
Discussion
Hyperhomocysteinemia has been recognized as a risk factor for atherosclerosis, but the pathobiological mechanisms by which it promotes vascular disease are still not completely understood. An association between Hcy, the transmembrane chemokine CXCL16 and recruitment of CXCR6+ lymphocytes has not been described in the literature. Our data show for the first time, that pathophysiolog-ical relevant concentration of Hcy up-regulates the CXCL16 expression on EC at mRNA and protein levels. The adhesion experiments of the present study provide conclusive evidence that Hcy induces more than twofold increase in adhesion of CXCR6+ lymphocytes to EC. Moreover, Hcy markedly up-regulates the scavenging activity of EC for oxLDL. Both effects are attributable to considerably enhanced CXCL16 expression, as they could be effectively blocked by an anti-human CXCL16 antibody. Therefore, the up-regulation and function of CXCL16 may provide a mechanistic link between hyperhomocysteinemia and its influences on the development and progression of vascular inflammatorydiseases.
Emigration and accumulation of T lymphocytes in the lesions already occurs during the earlier stages of atherosclerosis, perhaps even preceding that of monocytes [29]. As seen in ApoE-deficient mice maintained under high methionine diet, hyperhomocysteinemia may be involved in the pathogenesis of atherosclerosis by enhancing the T cell response [17]. Previous findings have indicated that Hcy induces the expression of MCP-1 and IL-8 in human aortic EC [14, 30] and in human monocytes [31]. These chemokines are key attractants for neutrophils and monocytes [4]. Our data provide evidence that Hcy also induces CXCL16 which functions as a potent attractant and adhesionmolecule for CXCR6+ T lymphocytes [5, 6]. The involvement of CXCL16 in atherosclerosis has first been suggested by the observation of its expression in atherosclerotic plaques [7, 8]. Moreover, T cells co-localize with CXCL16-expressing EC in plaques [8] and T cell infiltration into the lesions has been found to be reduced in ApoE-deficient mice with targeted disruption of the CXCR6 gene [32]. In fact, our in vitro data demonstrate that CXCL16 significantly contributes to adhesion of CXCR6+ lymphocyte to Hcy-activated EC as demonstrated by inhibition experiments with a neutralizing antibody to CXCL16.
Lipid accumulation in the growing lesions and foam cell formation is a result of excessive lipid uptake by macrophages but also by EC [2]. Several recent studies have indicated that scavenging of oxLDL is also mediated by CXCL16 [8, 9]. Our findings show that Hcy enhances binding and uptake of DiI-oxLDL by EC via CXCL16 in a dose-dependent and stereospecific way. The observation is supported by the efficiency of anti-human CXCL16 antibody to block the uptake of DiI-oxLDL by Hcy-treated EC. The residual scavenging activity of EC which was not blocked by neutralizing CXCL16 could show a potential synergistic collaboration between CXCL16 and other scavenger receptors, such as endothelial receptor for oxidized LDL (LOX-1) [33] that has been found to be up-regulated by homocysteine through oxidative stress at the gene expression level in cultured aortic EC [34].
Recently, CXCL16 gene deficiency in LDL receptor deficient mice was associated with accelerated atherosclerosis, despite a reduced capacity of macrophages to accumulate oxLDL. This could imply that the effecs of CXCL16 as a lipid scavenger in macrophages or EC are atheroprotective, while its function in T cell recruitment promotes lesion formation which would explain the lesion reduction observed in CXCR6-deficient mice [32, 35].
Oxidative stress has been implicated in hyperhomocysteine-mia-induced endothelial dysfunction. PPARs known as proantiox-idants are expressed in vascular EC [21, 36, 37] and a negative correlation between high levels of Hcy and PPAR expression has been already demonstrated [38]. In a previous study we have shown that incubation of EA.hy926 cells and HUVECs with l-, but not withD-Hcy, increases ROS accumulation, activation of nf-kb and expression of ICAM-1 resulting in increased monocyte adhesion. Co-administration of Tiron reversed all these events and reduced NF-κB activity to control level [18]. Similar findings were reported by an independent study showing that incubation of EC with 100 and 200 μM Hcy induced NF-κB nuclear translocation due to superoxide generation [39]. Our present study demonstrates that oxidative stress signals in Hcy-activated EC also affect the expression and function of CXCL16. Both the antioxidant Tiron and the PPAR agonist Pioglitazone are able to counteract Hcy's stimulatory effect on CXCR6+ lymphocyte adhesion to EC and uptake of ox-LDL by EC. These observations are in line with a previous study reporting that treatment with PPAR agonists, such as Rosiglitazone and Pioglitazone also down-regulate the expression of CXCL16 by human macrophages [40].
Taken together, these data suggest that Hcy, through production of ROS and by potential involvement of a PPAR-γ-dependent mechanism, induces up-regulation of CXCL16 and increases adhesion of lymphocytes to EC.
Whereas several studies describe the effects of Hcy on cultured cells, limited information exists on the effects of Hcy in vivo. Our in vivo experiments show that intraperitoneal injection of Hcy induces the recruitment of CXCR6+ inflammatory cells to the peritoneal cavity. This effect is at least in part mediated by CXCL16 as it is blocked by co-injection of Hcy with the CXCL16 neutralizing antibody. ELISA measurement of soluble CXCL16 in the peritoneal lavage of l-Hcy-treated mice confirmed that the enhanced recruitment of CXCR6+ cells is associated with the up-regulation of CXCL16 and both effects were consistently suppressed by co-administration of Pioglitazone. Although these experiments only represent a surrogate model for recruitment during atherosclerotic disease, they nevertheless underscore the pro-inflammatory effect induced by Hcy, which is at least partly mediated by the CXCL16/CXCR6 axis. Therefore, treatment with activators of PPAR-γ may be beneficial to counteract CXCL16 up-regulation in EC by Hcy and thereby limit CXCR6+ lymphocyte recruitment and inflammation within atherosclerotic lesions. These studies can be extended in the future to investigate the potential correlation between diet-induced hyperhomocysteinemia and presence of
CXCL16 and CXCR6 in the atherosclerotic lesions in ApoE-defi-cient mice. Future studies may thus bewarranted to check possible correlation between hyperhomocysteinemia and plasma levels of soluble CXCL16 in human subjects with different cardiovascular diseases and receiving different forms of treatment.
Acknowledgments
This work was supported in part by the German Research Foundation (FOR809, TP5) and the IZKF Biomat RWTH Aachen. We thank Tanja Kogel for expert technical assistance.
References
- 1.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 2.Ross R, Masuda J, Raines EW. Cellular interactions, growth factors, and smooth muscle proliferation in atherogenesis. Ann N Y Acad Sci. 1990;598:102–12. doi: 10.1111/j.1749-6632.1990.tb42282.x. [DOI] [PubMed] [Google Scholar]
- 3.Liehn EA, Zernecke A, Postea O, Weber C. Chemokines: Inflammatory mediators of atherosclerosis. Arch Physiol Biochem. 2006;112:229–38. doi: 10.1080/13813450601093583. [DOI] [PubMed] [Google Scholar]
- 4.Weber C, Schober A, Zernecke A. Chemokines: key regulators of mononu-clear cell recruitment in atherosclerotic vascular disease. Arterioscler Thromb Vasc Biol. 2004;24:1997–2008. doi: 10.1161/01.ATV.0000142812.03840.6f. [DOI] [PubMed] [Google Scholar]
- 5.Matloubian M, David A, Engel S, Ryan JE, Cyster JG. A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat Immunol. 2000;1:298–304. doi: 10.1038/79738. [DOI] [PubMed] [Google Scholar]
- 6.Wilbanks A, Zondlo SC, Murphy K, Mak S, Soler D, Langdon P, Andrew DP, Wu L, Briskin M. Expression cloning of the STRL33/BONZO/TYMSTR ligand reveals elements of CC, CXC, and CX3C chemokines. J Immunol. 2001;166:5145–54. doi: 10.4049/jimmunol.166.8.5145. [DOI] [PubMed] [Google Scholar]
- 7.Minami M, Kume N, Shimaoka T, Kataoka H, Hayashida K, Akiyama Y, Nagata I, Ando K, Nobuyoshi M, Hanyuu M, Komeda M, Yonehara S, Kita T. Expression of SR-PSOX, a novel cell-surface scavenger receptor for phos-phatidylserine and oxidized LDL in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2001;21:1796–800. doi: 10.1161/hq1001.096652. [DOI] [PubMed] [Google Scholar]
- 8.Wuttge DM, Zhou X, Sheikine Y, Wagsater D, Stemme V, Hedin U, Stemme S, Hansson GK, Sirsjo A. CXCL16/SR-PSOX is an interferon-gamma-regulated chemokine and scavenger receptor expressed in atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2004;24:750–5. doi: 10.1161/01.ATV.0000124102.11472.36. [DOI] [PubMed] [Google Scholar]
- 9.Shimaoka T, Kume N, Minami M, Hayashida K, Kataoka H, Kita T, Yonehara S. Molecular cloning of a novel scavenger receptor for oxidized low density lipoprotein, SR-PSOX, on macrophages. J Biol Chem. 2000;275:40663–6. doi: 10.1074/jbc.C000761200. [DOI] [PubMed] [Google Scholar]
- 10.Shimaoka T, Nakayama T, Fukumoto N, Kume N, Takahashi S, Yamaguchi J, Minami M, Hayashida K, Kita T, Ohsumi J, Yoshie O, Yonehara S. Cell surface-anchored SR-PSOX/CXC chemokine ligand 16 mediates firm adhesion of CXC chemokine receptor 6-expressing cells. J Leukoc Biol. 2004;75:267–74. doi: 10.1189/jlb.1003465. [DOI] [PubMed] [Google Scholar]
- 11.Abel S, Hundhausen C, Mentlein R, Schulte A, Berkhout TA, Broadway N, Hartmann D, Sedlacek R, Dietrich S, Muetze B, Schuster B, Kallen KJ, Saftig P, Rose-John S, Ludwig A. The trans-membrane CXC-chemokine ligand 16 is induced by IFN-gamma and TNF-alpha and shed by the activity of the disintegrin-like metalloproteinase ADAM10. J Immunol. 2004;172:6362–72. doi: 10.4049/jimmunol.172.10.6362. [DOI] [PubMed] [Google Scholar]
- 12.Harker LA, Ross R, Slichter SJ, Scott CR. Homocystine-induced arteriosclerosis. The role of endothelial cell injury and platelet response in its genesis. J Clin Invest. 1976;58:731–41. doi: 10.1172/JCI108520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weiss N, Heydrick SJ, Postea O, Keller C, Keaney JF, Jr, Loscalzo J. Influence of hyperhomocysteinemia on the cellular redox state–impact on homocysteine-induced endothelial dysfunction. Clin Chem Lab Med. 2003;41:1455–61. doi: 10.1515/CCLM.2003.223. [DOI] [PubMed] [Google Scholar]
- 14.Poddar R, Sivasubramanian N, DiBello PM, Robinson K, Jacobsen DW. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: implications for vascular disease. Circulation. 2001;103:2717–23. doi: 10.1161/01.cir.103.22.2717. [DOI] [PubMed] [Google Scholar]
- 15.Koga T, Claycombe K, Meydani M. Homocysteine increases monocyte and T-cell adhesion to human aortic endothelial cells. Atherosclerosis. 2002;161:365–74. doi: 10.1016/s0021-9150(01)00670-0. [DOI] [PubMed] [Google Scholar]
- 16.Wang G, Woo CW, Sung FL, Siow YL,OK. Increased monocyte adhesion to aortic endothelium in rats with hyperhomocys-teinemia: role of chemokine and adhesion molecules. Arterioscler Thromb Vasc Biol. 2002;22:1777–83. doi: 10.1161/01.atv.0000035404.18281.37. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Q, Zeng X, Guo J, Wang X. Oxidant stress mechanism of homocysteine potentiating Con A-induced proliferation in murine splenic T lymphocytes. Cardiovasc Res. 2002;53:1035–42. doi: 10.1016/s0008-6363(01)00541-7. [DOI] [PubMed] [Google Scholar]
- 18.Postea O, Krotz F, Henger A, Keller C, Weiss N. Stereospecific and redox-sensi-tive increase in monocyte adhesion to endothelial cells by homocysteine. Arterioscler Thromb Vasc Biol. 2006;26:508–13. doi: 10.1161/01.ATV.0000201039.21705.dc. [DOI] [PubMed] [Google Scholar]
- 19.Murao K, Imachi H, Momoi A, Sayo Y, Hosokawa H, Sato M, Ishida T, Takahara J. Thiazolidinedione inhibits the production of monocyte chemoattractant protein-1 in cytokine-treated human vascular endothelial cells. FEBS Lett. 1999;454:27–30. doi: 10.1016/s0014-5793(99)00765-6. [DOI] [PubMed] [Google Scholar]
- 20.Rocic P, Rezk B, Lucchesi PA. PPAR-gamma agonists decrease hyperhomcys-teinemia and cardiac dysfunction: new hope for ailing diabetic hearts? Am J Physiol Heart Circ Physiol. 2006;291:H26–8. doi: 10.1152/ajpheart.00277.2006. [DOI] [PubMed] [Google Scholar]
- 21.Marx N, Bourcier T, Sukhova GK, Libby P, Plutzky J. PPARgamma activation in human endothelial cells increases plas-minogen activator inhibitor type-1 expression: PPARgamma as a potential mediator in vascular disease. Arterioscler Thromb Vasc Biol. 1999;19:546–51. doi: 10.1161/01.atv.19.3.546. [DOI] [PubMed] [Google Scholar]
- 22.Hunt MJ, Tyagi SC. Peroxisome prolifera-tors compete and ameliorate Hcy-mediated endocardial endothelial cell activation. Am J Physiol Cell Physiol. 2002;283:C1073–9. doi: 10.1152/ajpcell.00152.2002. [DOI] [PubMed] [Google Scholar]
- 23.Jakubowski H. Biosynthesis and reactions of homocysteine thiolactone. In: Carmel R, Jacobson DW, editors. Homocysteine in health and disease. Cambrigde: Cambridge University Press; 2001. [Google Scholar]
- 24.Scholz F, Schulte A, Adamski F, Hundhausen C, Mittag J, Schwarz A, Kruse ML, Proksch E, Ludwig A. Constitutive expression and regulated release of the transmembrane chemokine CXCL16 in human and murine skin. J Invest Dermatol. 2007;127:1444–55. doi: 10.1038/sj.jid.5700751. [DOI] [PubMed] [Google Scholar]
- 25.Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci USA. 1983;80:3734–7. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mutin M, Dignat-George F, Sampol J. Immunologic phenotype of cultured endothelial cells: quantitative analysis of cell surface molecules. Tissue Antigens. 1997;50:449–58. doi: 10.1111/j.1399-0039.1997.tb02899.x. [DOI] [PubMed] [Google Scholar]
- 27.Dwivedi A, Anggard EE, Carrier MJ. Oxidized LDL-mediated monocyte adhesion to endothelial cells does not involve NFkappaB. Biochem Biophys Res Commun. 2001;284:239–44. doi: 10.1006/bbrc.2001.4955. [DOI] [PubMed] [Google Scholar]
- 28.Ryan QC, Goonewardene IM, Murasko DM. Maintenance of normal T lymphocyte function after transfection with SV40 large T. Cell Immunol. 1993;149:65–81. doi: 10.1006/cimm.1993.1136. [DOI] [PubMed] [Google Scholar]
- 29.Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 30.Zhang HS, Cao EH, Qin JF. Intracellular redox status modulates monocyte chemoattractant protein-1 expression stimulated by homocysteine in endothelial cells. J Cardiovasc Pharmacol. 2003;42:258–65. doi: 10.1097/00005344-200308000-00016. [DOI] [PubMed] [Google Scholar]
- 31.Zeng X, Dai J, Remick DG, Wang X. Homocysteine mediated expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human monocytes. Circ Res. 2003;93:311–20. doi: 10.1161/01.RES.0000087642.01082.E4. [DOI] [PubMed] [Google Scholar]
- 32.Galkina E, Harry BL, Ludwig A, Liehn EA, Sanders JM, Bruce A, Weber C, Ley K. CXCR6 promotes atherosclerosis by supporting T cell homing, interferon gamma production and macrophage accumulation in the aortic wall. Circulation. 2007;116:1801–11. doi: 10.1161/CIRCULATIONAHA.106.678474. [DOI] [PubMed] [Google Scholar]
- 33.Chen XP, Zhang TT, Du GH. Lectin-like oxidized low-density lipoprotein receptor-1, a new promising target for the therapy of atherosclerosis? Cardiovasc Drug Rev. 2007;25:146–61. doi: 10.1111/j.1527-3466.2007.00009.x. [DOI] [PubMed] [Google Scholar]
- 34.Nagase M, Ando K, Nagase T, Kaname S, Sawamura T, Fujita T. Redox-sensitive regulation of lox-1 gene expression in vascular endothelium. Biochem Biophys Res Commun. 2001;281:720–5. doi: 10.1006/bbrc.2001.4374. [DOI] [PubMed] [Google Scholar]
- 35.Aslanian AM, Charo IF. Targeted disruption of the scavenger receptor and chemokine CXCL16 accelerates atherosclerosis. Circulation. 2006;114:583–90. doi: 10.1161/CIRCULATIONAHA.105.540583. [DOI] [PubMed] [Google Scholar]
- 36.Delerive P, Martin-Nizard F, Chinetti G, Trottein F, Fruchart JC, Najib J, Duriez P, Staels B. Peroxisome proliferator-acti-vated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ Res. 1999;85:394–402. doi: 10.1161/01.res.85.5.394. [DOI] [PubMed] [Google Scholar]
- 37.Jackson SM, Parhami F, Xi XP, Berliner JA, Hsueh WA, Law RE, Demer LL. Peroxisome proliferator-activated receptor activators target human endothelial cells to inhibit leukocyte-endothelial cell interaction. Arterioscler Thromb Vasc Biol. 1999;19:2094–104. doi: 10.1161/01.atv.19.9.2094. [DOI] [PubMed] [Google Scholar]
- 38.Brude IR, Finstad HS, Seljeflot I, Drevon CA, Solvoll K, Sandstad B, Hjermann I, Arnesen H, Nenseter MS. Plasma homocys-teine concentration related to diet, endothe-lial function and mononuclear cell gene expression among male hyperlipidaemic smokers. Eur J Clin Invest. 1999;29:100–8. doi: 10.1046/j.1365-2362.1999.00419.x. [DOI] [PubMed] [Google Scholar]
- 39.Au-Yeung KK, Woo CW, Sung FL, Yip JC, Siow YL,OK. Hyperhomocysteinemia activates nuclear factor-kappaB in endothelial cells via oxidative stress. Circ Res. 2004;94:28–36. doi: 10.1161/01.RES.0000108264.67601.2C. [DOI] [PubMed] [Google Scholar]
- 40.Lehrke M, Millington SC, Lefterova M, Cumaranatunge RG, Szapary P, Wilensky R, Rader DJ, Lazar MA, Reilly MP. CXCL16 is a marker of inflammation, atherosclerosis, and acute coronary syndromes in humans. J Am Coll Cardiol. 2007;49:442–9. doi: 10.1016/j.jacc.2006.09.034. [DOI] [PubMed] [Google Scholar]