

Abstract
We previously showed that interleukin-1β (IL-1β) down-regulation of type II TGFβ receptor (TβRII) involves NFκB pathway and requires de novo synthesis of a yet unknown protein. Here, we demonstrate that this effect is mediated through Sp1 site located at position -25 of human TβRII promoter. Inhibition of transcription factors binding (decoy oligonucleotides or mithramycin) abolished IL-1β effect. EMSA and ChIP revealed that this treatment induced Sp3 binding to cis-sequence whereby IL-1β exerts its transcriptional effects whereas it decreased that of Sp1. Moreover, although the cytokine did not modulate Sp1 expression, it increased that of Sp3 via NFκB pathway. Experiments of gain and loss of function clearly showed that Sp3 inhibited TβRII expression whereas its silencing abolished IL-1β effect. In addition, both Sp1 and Sp3 were found to interact with NFκB, which therefore may indirectly interact with TβRII pro moter. Altogether, these data suggest that IL-1β decreases TβRII expression by inducing Sp3 via NFκB and its binding on core promote at the expense of Sp1, which could explain the loss of cell responsiveness in certain conditions. These findings bring new insights in the knowledge of the interference between two antagonistic transduction pathways involved in multiple physiopathological processes.
Keywords: interleukin-1, TGFß receptors, Sp transcription factors, NFκB
Introduction
Transforming growth factor β (TGFβ) belongs to a superfamily of polypeptides that governs a number of cellular processes, including cell proliferation, differentiation, immunosuppression and synthesis of extra-cellular matrix proteins.
TGFβ exerts its action by binding to specific cell surface receptors. Three major types of receptors, named receptor type I, II and III, have been identified. The type II TGFβ receptor (TβRII) is a constitutively active cell surface serine/threonine kinase receptor [1]. TβRII has affinity for TGF-β1 and TGF-β3 isoforms, but requires the accessory type III TGFβ receptor (TβRIII, betaglycan) to bind TGFβ2 [2, 3]. Upon ligand binding, TβRII forms a complex with the type I TGFβ receptor (TβRI) and phosphorylates the juxtamembrane GS domain of TβRI to activate its serine/threonine kinase [4, 5]. In turn, transcription factors Smad2 and Smad3 are phosphorylated by TβRI [6]. Activated Smad2/3 then bind Smad4 and translocate to the nucleus to enable interaction with other transcription factors and regulate the transcription of TGFβ responsive genes.
Because TβRII is essential to TGFβ signalling, alteration of its expression is often associated with a loss of TGFβ sensitivity and correlated to numerous pathology, such tumour progression or osteoarthritis process. The reduction of TβRII expression can be due to a decrease of TβRII gene transcription. The promoter of the TβRII gene has been cloned and characterized. Sequence analysis revealed that neither TATA box nor initiator element upstream the six transcription start sites of the human TβRII gene. Several target sequences regulating gene transcription have been identified in the TβRII promoter. Preliminary analysis has revealed four major regulatory elements [7]. They are divided into two positive regulatory elements (PRE1 and PRE2, located at -219 to -172 and +1 to +50, respectively), a negative regulatory element (NRE, located at -100 to -67) and a core promoter (located between -47 and -1). Like other promoters that lack TATA boxes, Sp1 is required for transcription of TβRII gene. Four regulatory sites at position -25, -59, -102 and -147 were identified to bind Sp family factors. Moreover, the promoter of the TβRII gene contains binding sites for AP1 (located in the PRE1) and an Ets binding site that is located in the PRE2 of the TpRII promoter.
Several studies have related that IL-1β exerts numerous opposite effects, and it is able to act on TGFβ system. For instance, this pro-inflammatory cytokine inhibits TGFβ2 and TGFβ3 expression, whereas it stimulates TGFβ1. In addition, a cross-talk between both IL-1β and TGFβ pathways was recently described [8]. Moreover, we have recently shown that IL-1β counteracts TGFfi signalling through TpRII down-regulation via NFκB pathway, and this effect needs de novo synthesis of a yet unknown protein. The aim of the present study was to determine the mechanism of IL-1β-induced repression of TpRII gene expression. We demonstrated, here, for the first time that the Sp1 site located at −25 is required for IL-1β-induced down-regulation of TβRII. Furthermore, we found that, whereas NFκB is indispensable for IL-1β effects, this transcriptional factor acts indirectly on TβRII down-regulation by induction of Sp3.
Material and methods
Cell culture and treatments
Human articular chondrocytes (HACs) were prepared from femoral head as previously described [9]. Cells were seeded at 4 x 104 cells/cm2 in 6-well plates or 100-mm dishes and cultured in Dulbeccos modified Eagles medium (DMEM) supplemented with 10% heat-inactivated foetal calf serum (FCS), 100 IU/ml penicillin, 100 μg/ml streptomycin and 0.25 μg/ml fungizone, in a 5% CO2 atmosphere. Cells were used as primary cultures except for nucleofection assay where they were passaged once. Cells were treated with human recombinant IL-1β (Sigma-Aldrich Co., St Quentin Fallavier, France) in DMEM supplemented with 2% FCS. In some experiments, mithramycin (from Sigma, dissolved in water) was added 30 min. before treatment for 24 hrs with IL-1β.
Expression vectors and reporter genes
The TβRII-luciferase reporter vectors (pTbRII1670, pTbRII219, pTbRII100 and pTbRII47) corresponding to the region −1670/+36, −219/+36, −100/+36 and −47/+36, respectively driven luciferase reporter gene, were kindly provided by S. J. Kim (National Cancer Institute, Bethesda, MD, USA). Additional plasmids have been generated. The pTbRII15 containing the sequence −15/+36 of the human TβRII promoter-driven luciferase cDNA and the vectors corresponding to the constructs −219/+36 and −47/+36 mutated at the Sp1 site (GG → CC at position −22), named pTbRII219mut and TbRII47mut, respectively were obtained by PCR using the construct pTbRII47 or pTbRII219 as a matrix and the following primers:
−15/+36 Forward: 5′AATTGGTACCAGGTCCTGCCCAGCT 3′
Reverse (GL primer 2): 5′ CTTTATGTTTTTGGCGTCTTCCA 3′
pGL2Sacmut Forward: 5′ GCTAACATAACCCGGGAGGTACCGGGCGCTT 3′
TbRIImutSp1Reverse: 5′ CAACAGCTGGGCAGGACCTCTCTGGGCC 3′
Amplicons were checked by sequencing. Amplicons and the promoter-less luciferase reporter plasmid, basic pGL2 (Promega, Madison, WI, USA) were digested by KpnI and HindIII before ligation.
p65, Sp1 and Sp3 expression vectors (pSG5-p65, pEVR2-Sp1 and pCMV-Sp3) were obtained from Dr Jalinot (Laboratoire de Biologie Moleculaire et Cellulaire, ENS, Lyon, France) and Dr Suske (Institut fur Molekularbiologie and tumorforschung, Marburg, Germany), respectively.
Transfection experiments and luciferase assay
Chondrocytes were transiently transfected by nucleofection. A reporter plasmid containing the appropriate TβRII promoter constructs (4 μg) was cotransfected with pSV40-βGal expression vector (1 μg). This latter encodes for β-galactosidase, which serves as an internal control to normalize transfection efficiency. It was previously verified that the transcriptional activity of this promoter was not affected by IL-1β treatment (data not shown). In other experiments, 4 μg of reporter plasmids were cotransfected with 2 μg of p65 or Sp3 expression vectors (pSG-p65 and pCMVSp3).
After 6 hrs of transfection, the culture medium was replaced with DMEM containing 10% FCS and the chondrocytes cultures were incubated with or without IL-1β (2 ng/ml) for 24 hrs. At the end of experiments, cells were washed once with PBS and harvested in lysis buffer. Luciferase activity was assayed on total cell extracts (Luciferase Assay kit, Promega, Charbonnières, France) in a luminometer (Berthold Lumat LB 9501; Berthold, Thoiry, France) and β–Galactosidase activity was assayed by a colourimetric assay. Luciferase activities were normalized to transfection efficiency, and transcriptional activities were expressed as relative luciferase units (RLU) (means ± S.D. of triplicates).
RNA extraction and real-time RT-PCR
Total RNA were extracted using Trizol (Invitrogen). Following extraction, 1 μg of DNase-I-treated RNA was reverse transcribed into cDNA in the presence of random hexamers (Applied Biosystems, Courtaboeuf, France) and Moloney murine leukaemia virus reverse transcriptase (Invitrogen Cergy Pontoise, France). The reaction was carried out at 37°C for 1 hr followed by a further 10-min. step at 95°C. Amplification of the generated cDNA were performed by real-time PCR in Applied Biosystems SDS7000 apparatus. The relative mRNA level was calculated with the 2-δδCT method.
Gene-silencing experiments
RNA interference targeting p65 was performed as previously described using the plasmid shRNAp65 (short hairpin RNA p65) [9]. For other siRNA experiments, 1.5 μg of double-strand siRNA against Sp1 or Sp3 (SantaCruz Biotechnology, by TeBU-bio SA, Le Perray en Yuelines, France), were nucle-ofected in HACs. Negative control siRNA (SantaCruz Biotechnology) known to not lead to specific degradation of any cellular message was used in silencing experiments. After 6 hrs of transfection, the culture medium was replaced with DMEM containing 10% FCS and cells were incubated with IL-1β (2 ng/ml) for 24 hrs.
Synthesis and transfection of decoy oligonucleotides
Decoy oligonucleotides sequences used are
TβRIIwt decoy: Forward 5′- GAGGAGAGGGAGAAGGCTCTCGGGCGGA-GAGA -3′ and Reverse, 5′- TCTCTCCGCCCGAGAGCCTTCTCCCTCTCCTC -3′
TβRIImut decoy: Forward 5′- GAGGAGAGGGAGAAGGCTCTCGGGCCCA-GAGA -3′ and Reverse 5′- TCTCTGGGCCCGAGAGCCTTCTCCCTCTCCTC -3′,
Sp1 decoy Forward 5′- ATTCGATCGGGGCGGGGCGAGC -3′ and Reverse 5′- CGACGCCCCGCCCCGATCGAAT -3′ (consensus sequences are underlined).
NFκB decoy Forward 5′- AGTTGAGGGGACTTTCCCAGGC -3′ and Reverse 5′- GCCTGGGAAAGTCCCCTCAACT -3′.
They were annealed overnight and transfected into cells using the Oligofectamine reagent according to the manufacturers instructions (Invitrogen). Decoy oligonucleotides were previously shown to bind Sp1 and NFκB, respectively (data not shown).
Nuclear extracts and EMSA
Nuclear extracts were prepared as previously described [10]. For EMSA, five fentomoles of [γ-32P]ATP-labelled probes (sequence from −47/−15 of the human TβRII promoter) and 6 μg of nuclear extracts were incubated at room temperature for 15 min. in binding buffer (20 mM Tris-HCl pH 7.5, 1 mM DTT, 100 mM NaCl, 5% bovine serum albumin, 0.1% Nonidet P-40, 10% glycerol, 25 μM ZnCl2) and 2 μg of poly(dI-dC).(dI-dC). For competition, 100-fold molar excess of unlabelled oligonucleotides was used. Alternatively, antibodies (Santa Cruz Biotechnology) were added and incubated for additional 30 min. Samples were resolved on a 7% non-denatu-rating polyacrylamide gel and exposed for autoradiography.
Chromatin immunoprecipitation assay
The chromatin immunoprecipitation (ChIP) experiments were performed using the ChIP-IT Enzymatic kit from Active Motif (Rixensart, Belgium) according to manufacturers instructions. For each experimental condition 107 cells were cross-linked by 1% formaldehyde for 10 min. Isolated nuclei were then subjected to enzymatic digestion for 15 min. DNA fragments obtained ranged between 200 and 1000 bp. Chromatin fractions were pre-cleared with protein G beads followed by immunoprecipitation with 4 μg of rabbit polyclonal Sp1, Sp3 or p65 antibodies or IgG as a control. Cross-linking was reversed and the proteins were removed by proteinase K treatment. Column purified DNA was subjected to conventional PCR (40 cycles) using Taq Polymerase (Invitrogen) to amplify Sp3 promoter (Forward: TGATCCTGTCAGCATCGGCGGT; Reverse: TTGTGTGATATGTGATGTGCTGG) or SYBR-Green real-time PCR amplification to amplify an 81-bp segment of the TβRII core promoter using the following primers: Forward: TGGCAGCTACGAGAGAGCTAGG; Reverse: AACAGCTGGGCAGGACCTCT. The specificity of the amplifications was confirmed both by dissociation curve and by generation of a single PCR product at the predicted size.
Immunoprecipitation
Fifty micrograms of nuclear extracts were pre-cleared with protein A beads (Sigma) followed by immunoprecipitation with 5 μg polyclonal p65 antibody overnight at 4°C. Then, they were precipitated with protein A beads, washed five times and boiled in SDS sample buffer containingβ-mercap-toethanol. The supernatants were submitted to Western blot for Sp1 and Sp3 detection.
Western blot
Total protein, extracted with RIPA, were separated by SDS-PAGE and detected by Western blot analysis using TβRII antibody as previously described [9]. For Sp1 and Sp3 detection, immunoprecipitated proteins or nuclear extracts (20 μg) were subjected to fractionation in a 10% SDS-PAGE minigel, transferred to PVDF membranes (Amersham Biosciences, Orsay, France) by electroblotting, and reacted with rabbit Sp1 or Sp3 polyclonal antibodies. Subsequently, membranes were incubated with anti-rabbit secondary peroxidase-conjugated antibody. The signals were revealed with SuperSignal West Pico Chemiluminescent Substrate (Pierce Perbio Science, Brébières, France) and exposed to X-ray film. The membranes were also reacted with anti-human β-actin or lamin A antibodies (1/500 dilution) to verify equal loading.
Statistical analysis
All experiments were repeated with different donors at least three times with similar results, and representative experiments are shown in the figures. Data are presented as means of triplicates ± S.D. Statistical significance was determined by Students t-test. Differences were considered to be statistically significant at P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***).
Results
The Sp1 binding site in position -25 is essential to the IL-1β-induced inhibition of TβRII expression
In a previous study, we demonstrated that IL-1β decreased the response of chondrocytes to TGFβ and that this effect was due to differences in TβRII transcriptional activity. Transient transfection of deleted TβRII promoter, followed by IL-1β treatment, revealed that IL-1β inhibits the transcriptional activity of all deletion constructs, except the shortest construct (Fig. 1A). This suggests that IL-1β mediates its effect through a short sequence of human TβRII core promoter, located between −47 and −15 bp upstream of the transcription start site.
1.
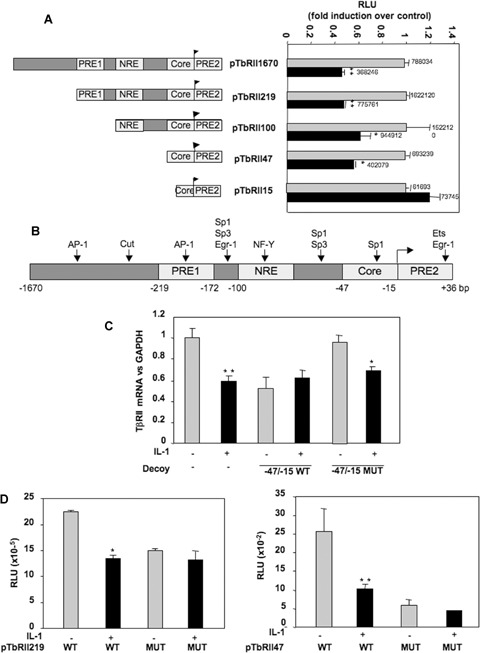
IL-1β inhibits transcriptional activity of the human TβRII promoter via–47/–15 region. A: HACs were cultured for 5–6 days in 10% FCS-containing DMEM and transiently transfected by nucleofection with TβRII promoter constructs as described in Methods. Then, they were treated with IL-1β for 24 hrs, and luciferase activities were determined and expressed as RLU fold induction over control. Means of triplicate absolute values are given on each histogram. B: Schematic representation of the human TβRII promoter. Putative binding sites are represented: transcription initiation site; PRE1/2: Positive regulatory element 1/2; NRE: Negative regulatory element. C: HACs were transfected with double-strand oligonucleotides corresponding to the wild-type (WT) or mutated (Mut) −47/−15 sequence of the human TβRII promoter. Then, they were incubated in DMEM + 2% FCS in the presence or not of IL-1β (1 ng/ml) for 24 hrs. Thereafter, TβRII mRNA levels were analysed by real-time RT-PCR. The modulation of TβRII mRNA expression was expressed as per cent of controls, after normalization to GAPDH signal. D: HACs were transiently transfected by nucleofection with wild-type or mutated p219TbRII or p47TbRII vectors as described in Methods. Then, they were treated with IL-1β for 24 hrs, and luciferase activities were determined and expressed as RLU.
Analysis of this 31-bp region showed a putative binding site for Sp1 family factors (Fig. 1B). To confirm the role of the region −47/−15 and to define whether the Sp1 site plays a role in regulation of TβRII transcription induced by IL-1β, decoy experiments were carried out with wild-type or mutated double-strand oligonucleotides corresponding to the −47/−15 region of the TβRII promoter. TβRII mRNA level was determined after a 24-hrs incubation period in the presence or the absence of IL-1β treatment. WT decoy oligonucleotide prevented IL-1β-induced inhibition of TβRII expression, whereas the Sp1-mutated oligonucleotide was unable to abolish the negative effect of IL-1β (Fig. 1C). In addition, constructs containing the −219/+36 or −47/+36 regions mutated at position −25 were used in transient transfection and their transcriptional activity was compared with that of the wild-type corresponding constructs. As shown in Fig. 1D, the mutation is sufficient to inhibit TβRII transcription and to block IL-1β-induced repression of TβRII.
These data strongly suggest that the Sp1 element at −25 exerts a key role in the basal expression of the TβRII and is essential to the IL-1β-induced repression of TβRII gene transcription.
Sp1 binding is necessary to IL-1β effect on TβRII expression
To further verify the role played by Sp1 family factors in down-regulation of TβRII by IL-1β, TβRII expression was analysed in the presence of a decoy oligonucleotide for Sp1 or by using mithramycine, an inhibitor of DNA binding of Sp1 family members. The data presented in Fig. 2 demonstrated that IL-1β effect on TβRII expression was dependent on transcription factors binding to Sp1 site on DNA, since competition or inhibition of −25 site binding prevented IL-1β-induced repression of TβRII expression.
2.
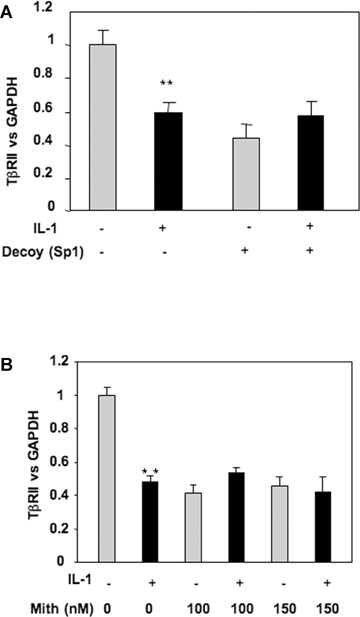
IL-1β represses TβRII expression via Sp1 site at position −25. A: HAC, transfected with an Sp1 consensus sequence (Sp1 decoy), were incubated in DMEM + 2% FCS in the presence or not of IL-1β (1 ng/ml) for 24 hrs. Thereafter, TβRII mRNA versus GAPDH level was determined by RT-PCR. B: Subconfluent cultures of chondrocytes were treated with IL-1β (1 ng/ml) for 48 hrs in the presence or absence of mithramycin (100 or 150 nM). TβRII expression was analysed at mRNA level by real-time RT-PCR (B).
Analysis of binding activity on the −47/−15 sequence of the human TβRII promoter
Since the Sp1 site at position −25 was found to participate in the regulation of the TβRII promoter under IL-1β treatment, Sp1 family proteins should bind to this sequence. To define the DNA-bind-ing activity of nuclear proteins to the −47/−15 sequence, EMSAs were performed using the −47/−15 sequence as a probe. The wild-type double-strand 5' labelled oligonucleotides were generated and incubated with nuclear extracts of primary HACs treated or not with IL-1β (1 ng/ml) for 1 or 3 hrs. One delayed protein–DNA complex was observed and named a (Fig. 3A). This complex was enhanced by IL-1β, and the binding is specific, since complex formation was diminished by addition of molar excess of unlabelled probe, but not by the addition of an oligonucleotide in which the Sp1 site was mutated. To further characterize the transcription factors interacting with the Sp1 site at −25 position under IL-1β treatment, additional EMSA competition experiments were performed using competitors representing some consensus DNA-binding sites from different transcriptional factors. Thus, the binding activity of the complex a is decreased when consensus sequence for Sp1 was used, whereas competition with consensus sequence for NFκB or AP-1 did not (Fig. 3A).
3.
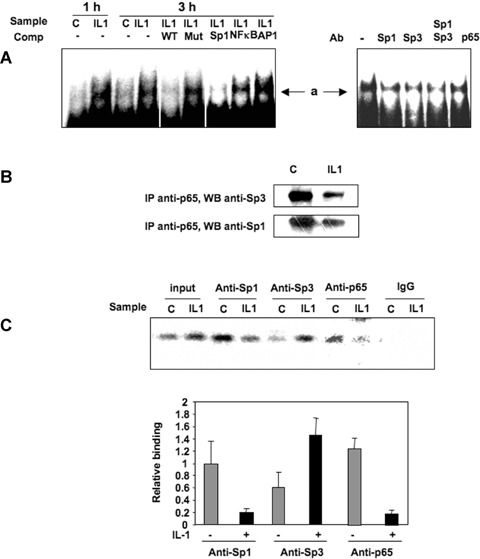
IL-1β enhances Sp1/Sp3 binding to TβRII promoter (position −25). A: Electrophoretic mobility shift assay (EMSA) reactions were performed using the 5′ end-labelled −47/−15 probe, incubated with 6 μg of nuclear extract from chondrocytes treated in the absence or in the presence of IL-1β for 1 or 3 hrs. EMSA reactions were also performed with 3-hrs IL-1β sample in the presence of 100-fold molar excess of the wild-type, mutated −47/−15 unlabelled probe or Sp1, NFκB and AP1 consensus sequences. Untreated samples were also pre-incubated with Sp1, Sp3 and p65 antibodies (Ab) before incubation with the probe. B: Nuclear extracts were immunoprecipitated with anti-p65 antibody and immunoblotted with anti-Sp1 or anti-Sp3 antibodies. C: After 24 hrs of incubation with IL-1β, cells were fixed with formaldehyde, lysed and underwent digestion. in vivo cross-linked chromatin was then precipitated independently using Sp1, Sp3, p65 antibodies or IgG. The recovered immunoprecipitated DNA was then used for PCR with specific primers for the TβRII promoter segment of interest. The amplicons were analysed by electrophoresis on a 3% agarose gel. Sp1, Sp3 and p65 relative binding of three independent experiments are shown in histograms.
Moreover, we tested the effect of mithramycin on the binding activity of the −47/−15 sequence (data not shown). This inhibitor, which prevents the effect of IL-1β on TβRII expression, induces the disappearance of complex a, suggesting an involvement of Sp1 family transcription factors in the complex and a key role in TβRII regulation.
Binding of Sp1 and Sp3 to the TβRII core promoter is modulated by IL-1β treatment
To get further insights into the nature of nuclear proteins binding to the core promoter, antibody interference assays were performed with specific antibodies. Nuclear extracts from untreated HACs were pre-incubated with Sp1, Sp3 and p65 antibodies prior to the addition of the labelled probe. The binding activity of complex a to the probe was decreased in the presence of the Sp1 and Sp3 antibodies, indicating that this complex contains both Sp1 and Sp3 factors. Surprisingly, p65 antibody was also able to reduce the binding of complex a. Since competition with NFκB consensus oligonucleotides was not able to modify this binding, we suggest that p65 could participate to the complex through protein–protein interaction. Immunoprecipitation of p65 (from nuclear extracts) followed by Western blot with Sp1 or Sp3 antibodies, showed that both factors were able to interact with p65 in the nucleus (Fig. 3B) and that IL-1β seemed to decrease these interactions. Similar results were obtained when Sp3 (or Sp1) were immunoprecipitated before immunoblotting with p65 antibody (data not shown). This suggests that NFκB may indirectly bind to TβRII core promoter via a physical interaction with Sp1 or Sp3.
To determine whether Sp1, Sp3 and p65 were recruited to the endogenous TβRII promoter in vivo, ChIP was performed and followed by real-time PCR. As illustrated in Fig. 3C, Sp1, Sp3 and p65 were involved, and interestingly, IL-1β decreased Sp1 and p65 binding to the TβRII core promoter region, whereas Sp3 interaction was markedly increased.
In conclusion, these experiments suggest that IL-1β-induced inhibition of TβRII transcription implies a multimeric complex involving, at least, Sp1 and Sp3, which bind to the Sp1 site at position −25, and where NFκB could be recruited through protein–protein interaction.
IL-1β induces Sp3 expression but does not modulate Sp1 level
Changes Sp1/Sp3 binding activity could result from modification of an existing form of the protein rendering it more active or from increased expression. To define the effect of IL-1β on Sp1 and Sp3 expression, reverse transcriptase-PCR using total RNA extracted from primary HACs, treated with or without IL-1β, was performed. As shown in Fig. 4A, the Sp1 mRNA was not modulated by IL-1β, whereas Sp3 mRNA was increased. To determine whether there was a correlation with the respective amounts of Sp1 and Sp3 proteins present in the nucleus, Western blot experiments were performed using nuclear protein extracts. The amount of Sp1 was not changed by IL-1β treatment. On the contrary, the two isoforms of Sp3 (60 and 100 kD) were found to be increased (Fig. 4B). Moreover, to define whether Sp3 could be the de novo protein required for IL-1β effect on TβRII [9], we reproduced the experiment in the presence of cycloheximide. Our result (Fig. 4C) showed that the stimulation of Sp3 expression is partially reduced suggesting a mechanism both dependent and independent of protein synthesis.
4.
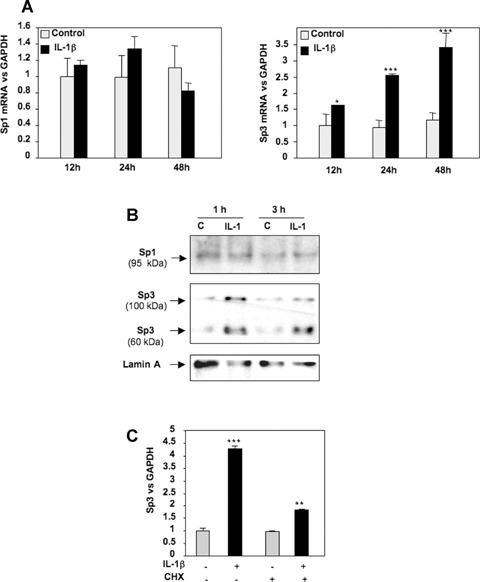
IL-1β up-regulates Sp3 mRNA and protein expression. A, B: HAC were cultured for 5–6 days and incubated in DMEM + 2% FCS for 24 hrs, before addition of IL-1β (1 ng/ml) or vehicle (control) for indicated times. mRNA (A) and nuclear protein (B) levels of Sp1 and Sp3 were analysed by real-time RT-PCR and Western blot, respectively. C: Subconfluent HAC were incubated for 24 hrs with DMEM + 2% FCS and treated with cycloheximide for 30 min. before IL-1β addition (1 ng/ml). Thereafter, Sp3 mRNA level was assayed by real-time RT-PCR.
Sp1 and Sp3 differentially modulate TβRII expression
To determine whether the modulation of the Sp1/Sp3 ratio governs the repression of TβRII expression, gain and loss of function experiments were performed. Forced expression of Sp1 and Sp3, checked by Western blot, were followed by analysis of mRNA steady-state level. Ectopic expression of Sp1 does not change TβRII expression but counteracts IL-1β-induced repression. Moreover, Sp3 over-expression mimes IL-1β effects (Fig. 5A). Silencing of Sp1 or Sp3, proven by both Western blot and RT-PCR, led to inhibition of TβRII expression (Sp1 siRNA) or abolition of IL-1β-induced repression of TβRII (Sp3 siRNA) (Fig. 5B). In addition, transfections of wild-type or mutated TβRII promoter constructs demonstrated that Sp3 represses TβRII promoter activity via−47/−15 region as shown in Fig. 6A.
5.
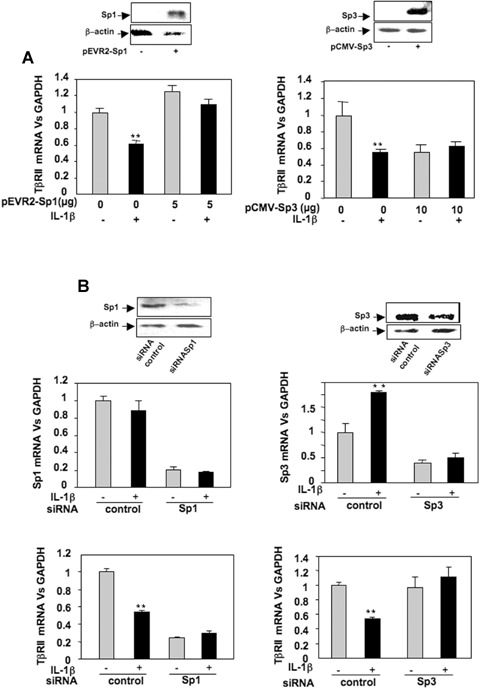
Sp1 and Sp3 exert opposite effect on TβRII expression. A: HAC were trans-fected overnight with pEVR2-Sp1 or pCMV-Sp3 expression vectors (or with insertless plasmid as controls). Thereafter, media were replaced with DMEM + 2% FCS for 24 hrs in the absence or in the presence of IL-1β (1 ng/ml). Sp1 and Sp3 over-expression were checked by Western blot, and TβRII mRNA levels were analysed and expressed as relative expression versus GAPDH. B: HAC were nucleo-fected with Sp1 or Sp3 siRNA oligonu-cleotides. Thereafter, the medium was replaced with DMEM + 10% FCS for 24 hrs in the presence or in the absence of IL-1β (1 ng/ml). Then, Sp1 and Sp3 protein levels were defined by Western blot. Total RNA was also extracted, and real-time RT-PCR analysis was performed. Histograms represent the relative Sp1, Sp3 or TβRII mRNA levels versus GAPDH.
6.
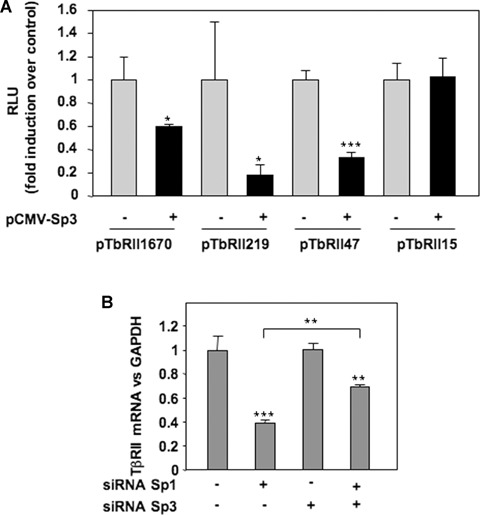
Sp3 inhibits TβRII transcription through the Sp1 site at −25 of human TβRII promoter. A: HAC were nucleofected with 2 μg of pCMV-Sp3 (or insertless pCMV vector), and TβRII promoter constructs. After 24 hrs, luciferase activities (RLU) were determined and expressed as per cent of controls. B: Human chondro-cytes were nucleofected with siRNA targeted Sp1 and/or Sp3, and TβRII mRNA levels were analysed by real-time RT-PCR after 24 hrs of incubation.
Taken together, these data suggest that IL-1β reduces TβRII transcription through the induction of Sp3 expression and its binding on Sp1 site at −25 in TβRII core promoter. To investigate whether Sp3 acts as a direct inhibitor of TβRII transcription and/or as a repressor of an activator (Sp1), we investigated TβRII mRNA expression after the silencing of both Sp1 and Sp3 (Fig. 6B). As expected, Sp1 silencing reduced TβRII mRNA level, while that of Sp3 did not. Interestingly, when both were simultaneously silenced, the decrease of TβRII expression was significantly weaker, indicating that Sp3 acts not only as a competitor of the activator Sp1 but also as an intrinsic repressor.
NFiκB/p65 mediates IL-1β effect on TβRII transcription via core promoter and by inducing Sp3 expression
We previously demonstrated that IL-1β down-regulates TβRII through NFκB/p65 pathway. However, EMSA experiments (Fig. 3) showed that this transcriptional factor weakly bind to the sequence of TβRII promoter which mediates IL-1β effect. We attempted to understand its mechanism of action. Transient cotransfection of luciferase-driven TβRII promoter constructs with p65 expression vector indicated that p65 decreases TβRII transcription through the same region (−47/−15 bp) of the human promoter that Sp3 did at the −25 Sp1 site (Fig. 7). This result confirms that p65 is a mediator of IL-1β effect on TβRII expression. Furthermore, given that p65 is not able to strongly bind to the core promoter of TβRII, we suggest that this factor could act indirectly through the modulation of Sp3 level. This hypothesis was confirmed since ectopic p65 over-expression led to a great induction of Sp3 expression (Fig. 8A). In contrast, silencing of p65 by siRNA induced Sp3 mRNA decrease (Fig. 8B). In the same way, blockade of NFκB by PDTC or decoy oligonucleotide prevents induction of Sp3 by IL-1β (Fig. 8C and D). Finally, using ChIP assay, we showed that p65 is able to bind in vivo to Sp3 promoter (Fig. 8E).
7.
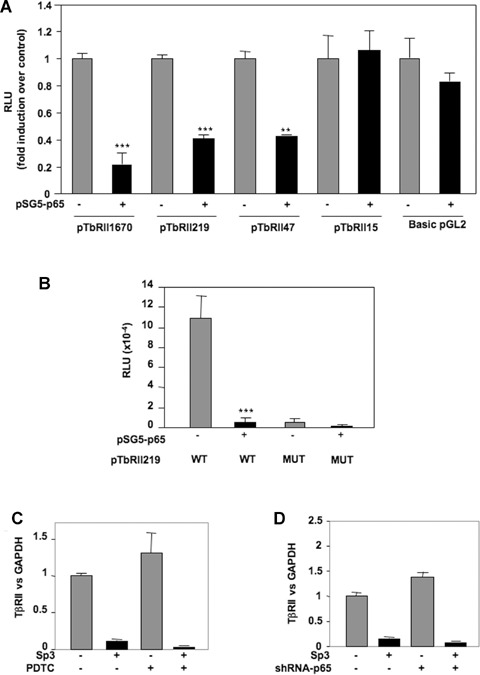
NFκB p65 subunit inhibits TβRII transcription through the Sp1 site at −25 of human TβRII promoter. HAC were nucleofected with 2 μg of pSG5-p65 (or insertless pSG5 vector), and wild-type (A) or mutated (B) TβRII promoter constructs. After 24 hrs, luciferase activities were determined and expressed as RLU.
8.
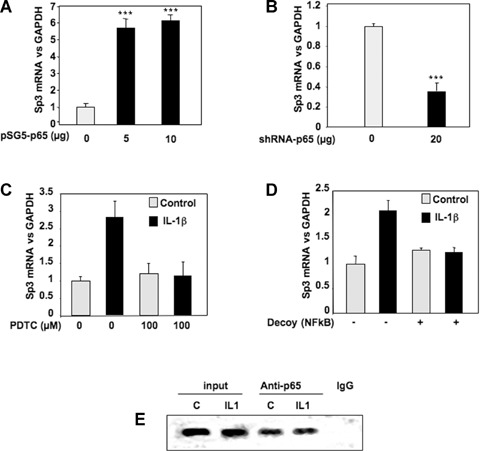
NFκB induces the expression of the repressor Sp3. A, B: Subconfluent chondrocytes were transfected with increased amounts of pSG5-p65 (A) or with 20 μg of shRNA-p65 vector (B). Thereafter, media were replaced with DMEM + 2% FCS for 24 hrs, and Sp3 mRNA levels were analysed and expressed as relative expressions versus GAPDH. C: Subconfluent cultures of chondrocytes were treated with IL-1β (1 ng/ml) for 48 hrs in the presence or in the absence of the NFκB inhibitor, PDTC. Histograms represent the relative Sp3 mRNA levels versus GAPDH signal. D: HAC were transfected with an NFκB consensus sequence (NFκB decoy). Then, they were incubated in DMEM + 2% FCS in the presence or not of IL-1R (1 ng/ml) for 24 hrs, and real-time RT-PCR analysis of Sp3 gene expression was performed. E: HAC were fixed with formaldehyde, lysed and then digested with enzyme restriction. In vivo cross-linked chromatin was then precipitated independently using anti-p65 antibody or IgG. The recovered immunoprecipitated DNA was then used for PCR with specific primers for the Sp3 promoter. The amplicons were analysed by elec-trophoresis on a 3% agarose gel.
Discussion
Loss or reduced expression of TGFβ receptors has been implicated in TGFβ resistance leading to a variety of diseases, including tumours or degenerative pathologies. We have recently established that IL-1β down-regulates TβRII expression through NFκB pathway, leading to the decrease of cell responsiveness to TGFβ. The loss of TβRII expression in chondrocytes is due to decreased activity of the TβRII promoter. In the present study, we attempt to determine the mechanism of TβRII transcriptional repression. Transient transfection of deleted or mutated TβRII promoter-luciferase constructs showed that the Sp1 site located at position −25 was responsible for the repression of TβRII promoter activity upon IL-1β treatment. Mobility shift assays and ChIP showed that IL-1β enhanced the binding activity of Sp1-like factors. This increased binding activity was found to be due to higher levels of Sp3 factor in IL-1β-stimulated chondrocytes and contribute to the reduced TβRII expression, leading to resistance to TGFβ.
Numerous studies have described that TβRII expression could be modulated by trans factor of Sp1 family [11–13]. TβRII promoter contains two consensus sites for Sp1, the first in the core promoter (located at position −25) and the second in upstream NRE2 (sited at position −147). Recently, two other regulatory sites binding to Sp1 family factors have been identified at position –102 and −59 [14]. Sp gene family members Sp1, Sp2 and Sp4 are generally known as activators of gene transcription, whereas Sp3 can act both as a repressor and activator of gene transcription [15]. Sp1 is necessary for the transcription of TATA-less genes, such as TGFβ receptors genes, through its interaction with the TFIID to direct the start site of transcription [16]. Of interest is the fact that the promoters of other TGFβ family members contain Sp1 sites. TGFβ1 and TβRI genes have been shown to contain functionally significant Sp1 motifs in their promoters [17, 18].
In this work, we showed that IL-1β induces binding of a complex, containing at least Sp1 and Sp3, on the Sp-1 site at position −25. Since we also demonstrated that IL-1β enhances Sp3 expression, whereas it does not modify that of Sp1, we propose that Sp3 binding to the Sp1 site at −25 is increased in the presence of IL-1β to the prejudice of Sp1 binding. The 115-κD Sp3 protein is the biologically active form, and the inactive 68–70-κD species present in Western blots arise as a result of differential internal translational initiation [19]. However, as opposed to direct promoter repression, the Sp3-derived 68-70-κD species can bind GC elements and thus act as inhibitors of Sp1-mediated gene activation.
A similar mechanism involving modifications of Sp1/Sp3 ratio has been reported to regulate the expression of the other TGFβ receptor, TβRI [20]. Moreover, the roles of Sp1 and Sp3 seem to be important in chondrocyte metabolism. For instance, previous works in our laboratory showed that variations in Sp1/Sp3 ratio, which could be induced by IL-1β or TGFβ, were implicated in the regulation of type II collagen expression in rabbit articular chon-drocytes [21, 22].
Previously, we demonstrated that IL-1β-induced down-regulation of TβRII expression was dependant on de novo protein synthesis and involved NFκB/p65 pathway. Unexpectedly, EMSA experiments presented here, showed that this transcriptional factor is not able by itself to bind to the region −47/−15 of the human TβRII promoter, which mediates IL-1β effect, suggesting an indirect action of NFκB. However, chromatin immunopre-cipitation demonstrates that p65 is recruited to the TβRII core promoter and that IL-1β decreased its binding, which seems to be mediated through protein–protein interaction with Sp1 or Sp3, as proven by immunoprecipitation and immunoblotting. The physical interaction between NFκB and Sp1 has already been observed in various cell types and may be mediated by the amino-terminal region of p65 and the zinc finger region of Sp1 [23, 24]. Conversely, to our knowledge, the interaction between Sp3 and p65 has never been described. Since p65 is subject to multiple post-translational modifications (phosphorylation, acetylation), which modulate its affinity for others proteins or DNA [25], we suggest that IL-1β by modifying p65 state, reduces the protein–protein interactions with Sp1 and Sp3, and consequently decreases p65 binding on TβRII core promoter. Nevertheless, we cannot neglect the possibility that the decrease of p65–Sp3 interaction is due to modification of Sp3 by acetylation or sumoylation. However, the precise role of NFκB binding remains unclear.
The Sp3 promoter was recently sequenced [26]. We performed a computational analysis (TF Bind [27]), which revealed the presence of numerous putative NFκB binding sites. Here, we showed that p65 is able to up-regulate Sp3 as IL-1β did and can bind to Sp3 promoter as shown by our ChIP experiment. Moreover, when used, cycloheximide partially prevented IL-1β-induced Sp3. Upon IL-1β stimulation, NFκB accumulates from two pathways: activation of pre-existing protein (i.e. degradation of lκB) and neosyn-thesis by transcription/traduction. Cycloheximide only inhibits the second, resulting to a less potent induction of Sp3. Therefore, Sp3 seems to be the required protein for TβRII down-regulation by IL-1β. Interestingly, experiments where Sp1 was silenced or its binding inhibited (decoy, inhibitor, mutation) revealed a decreased transcriptional activity of TβRII without IL-1β. Taken together, these data permit to propose a mechanism of TβRII regulation by IL-1β (Fig. 9). First, Sp1 transcription factor is required for constitutive expression. IL-1β induces both activation and transcription of NFκB, which induces Sp3 transcription. The latter, in turn, competes with Sp1 for the same site at the −25 position and therefore down-regulates TβRII. In addition, since p65 was shown to physically interact with Sp1 and Sp3 in the nucleus, it is not excluded that NFκB could be recruited to the same site by protein–protein interaction.
9.
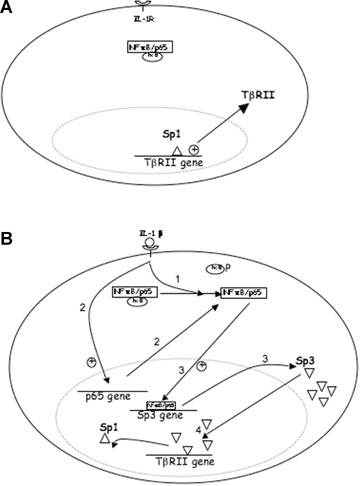
Sp1/Sp3 ratio controls TβRII expression under IL-βtreatment. A: In the constitutive state, Sp1 ensures the basal transcription of TβRII. B: Under IL-1β exposure, NFκB accumulates by both activation (1) (after lκB phosphorylation) and transcription/translation (2) of p65 and then up-regulates Sp3 (3), which in turn competes with Sp1 (4) to down-regulate TβRII. (Alternatively, NFκB might interfere in this mechanism, since it is able to interact with Sp1 and Sp3).
In conclusion, we previously demonstrated that the loss of responsiveness to TGFβ under IL-1β is a result of TβRII expression down-regulation. Here, we bring new insights into mechanisms that control TβRII expression. Indeed, TβRII expression is sensitive to Sp1/Sp3 ratio. After IL1-β stimulation, Sp3 is up-regulated through NFκB activation/transcription and thus competes with Sp1, which normally ensure constitutive expression of TβRII, and leads to reduction of the receptor expression. These data bring new insight in the growing knowledge about the interference between two separate transduction pathways and is consistent with the reduced sensitiveness of chondrocytes observed in osteoarthritis for example. These observations are of importance in all inflammatory processes. Manipulation of Sp1/Sp3 activity may be a possible mechanism to induce TβRII expression in order to restore sensitivity to the growth effects of TGFβ.
Acknowledgments
We thank Dr Jalinot (Laboratoire de Biologie Moleculaire et Cellulaire, ENS, Lyon, France), Dr Suske (Institut fur Molekularbiologie and tumor-forschung, Marburg, Germany) and Dr Kim (National Cancer Institute, Bethesda, MD, USA) for providing pEVR2-p65, pEVR2-Sp1, pCMV-Sp3 and TßRII promoter constructs, respectively. C. Baugé was a recipient of a fellow from French Ministry of Education and Research (MENRT).
References
- 1.Lin HY, Wang XF, Ng-Eaton E, Weinberg RA, Lodish HF. Expression cloning of the TGF-beta type II receptor, a functional transmembrane serine/threonine kinase. Cell. 1992;68:775–85. doi: 10.1016/0092-8674(92)90152-3. [DOI] [PubMed] [Google Scholar]
- 2.Cheifetz S, Hernandez H, Laiho M, Ten Dijke P, Iwata KK, Massague J. Distinct transforming growth factor-beta (TGF-beta) receptor subsets as determinants of cellular responsiveness to three TGF-beta iso-forms. J Biol Chem. 1990;265:20533–8. [PubMed] [Google Scholar]
- 3.Sankar S, Mahooti-Brooks N, Centrella M, McCarthy TL, Madri JA. Expression of transforming growth factor type III receptor in vascular endothelial cells increases their responsiveness to transforming growth factor beta 2. J Biol Chem. 1995;270:13567–72. doi: 10.1074/jbc.270.22.13567. [DOI] [PubMed] [Google Scholar]
- 4.Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, Wang XF, Massague J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–14. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- 5.Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370:341–7. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 6.Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, Tamaki K, Hanai J, Heldin CH, Miyazono K, Ten Dijke P. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997;16:5353–62. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bae HW, Geiser AG, Kim DH, Chung MI, Burmester JK, Sporn MB, Roberts AB, Kim SE. Characterization of the promoter region of the human transforming growth factor-β type II promoter gene. J Biol Chem. 1995;270:29460–8. doi: 10.1074/jbc.270.49.29460. [DOI] [PubMed] [Google Scholar]
- 8.Lu T, Tian L, Han Y, Vogelbaum M, Stark GR. Dose-dependent cross-talk between the transforming growth factor-β and interleukin-1 signaling pathways. PNAS. 2007;104:4365–70. doi: 10.1073/pnas.0700118104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baugé C, Legendre F, Leclercq S, Ellissalde JM, Pujol JP, Galera P, Boumediene K. Interleukin-1ß impairs TGFß signalling in articular chondrocytes by down-regulating TßRII receptor and up-regulating inhibitory Smad7. Arthritis Rheum. 2007;56:3020–32. doi: 10.1002/art.22840. [DOI] [PubMed] [Google Scholar]
- 10.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–89. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Zhong X, Li W, Brattain MG, Banerji S. The role of Spl in the differential expression of the transforming growth fac-tor-p receptor type II in human breast ade-nocarcinoma MCF-7 cells. J Biol Chem. 2000;275:12231–6. doi: 10.1074/jbc.275.16.12231. [DOI] [PubMed] [Google Scholar]
- 12.Ammanamanchi S, Brattain MG. 5-AzaC treatment enhances expression of transforming growth factor-β receptors through down-regulation of Sp3. J Biol Chem. 2001;276:32854–9. doi: 10.1074/jbc.M103951200. [DOI] [PubMed] [Google Scholar]
- 13.Ammanamanchi S, Brattain MG. Sp3 is a transcriptional repressor of transforming growth factor-β receptors. J Biol Chem. 2001;276:3348–52. doi: 10.1074/jbc.M002462200. [DOI] [PubMed] [Google Scholar]
- 14.Jennings R, Alsarraj M, Wright KL, Munoz-Antonia T. Regulation of the human transforming growth factor β type II receptor gene promoter by novel Spl sites. Oncogene. 2001;20:6899–909. doi: 10.1038/sj.onc.1204808. [DOI] [PubMed] [Google Scholar]
- 15.Suske G. The Sp-family of transcription factors. Gene. 1999;238:291–300. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- 16.Pugh BF, Tjian R. Mechanism of transcriptional activation by Sp1: evidence for coac-tivators. Cell. 1990;61:1187–97. doi: 10.1016/0092-8674(90)90683-6. [DOI] [PubMed] [Google Scholar]
- 17.Geiser AG, Busam KJ, Kim SJ, Lafyatis R, O’Reilly MA, Webbink R, Roberts AB, Sporn MB. Regulation of the transforming growth factor-beta 1 and -beta 3 promoters by transcription factor Sp1. Gene. 1993;129:223–8. doi: 10.1016/0378-1119(93)90272-5. [DOI] [PubMed] [Google Scholar]
- 18.Ji C, Casinghino S, McCarthy TL, Centrella M. Multiple and essential Sp1 binding sites in the promoter for transforming growth factor-beta type I receptor. J Biol Chem. 1997;272:21260–7. doi: 10.1074/jbc.272.34.21260. [DOI] [PubMed] [Google Scholar]
- 19.Kennett SB, Udvadia AJ, Horowitz JM. Sp3 encodes multiple proteins that differ in their capacity to stimulate or repress transcription. Nucleic Acids Res. 1997;25:3110–7. doi: 10.1093/nar/25.15.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ammanamanchi S, Brattain MG. Restoration of transforming growth factor-β signalling through receptor RI by histone deacetylase activity inhibition in breast cancer cells. J Biol Chem. 2004;279:32620–5. doi: 10.1074/jbc.M402691200. [DOI] [PubMed] [Google Scholar]
- 21.Chadjichristos C, Ghayor C, Herrouin JF, Ala-Kokko L, Suske G, Pujol JP, Galera P. Down-regulation of human type II collagen gene expression by transforming growth factor-beta 1 (TGF-beta 1) in articular chondrocytes involves SP3/SP1 ratio. J Biol Chem. 2002;277:43903–17. doi: 10.1074/jbc.M206111200. [DOI] [PubMed] [Google Scholar]
- 22.Chadjichristos C, Ghayor C, Kypriotou M, Martin G, Renard E, Ala-Kokko L, Suske G, De Crombrugghe B, Pujol JP, Galera P. Sp1 and Sp3 transcription factors mediate interleukin-1 beta down-regulation of human type II collagen gene expression in articular chondrocytes. J Biol Chem. 2003;278:39762–72. doi: 10.1074/jbc.M303541200. [DOI] [PubMed] [Google Scholar]
- 23.Perkins ND, Agranoff AB, Pascal E, Nabel GJ. An interaction between the DNA-binding domains of RelA (p65) and Sp1 mediates human immunodeficiency virus gene activation. Mol Cell Biol. 1994;14:6570–83. doi: 10.1128/mcb.14.10.6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuang PP, Berk JL, Rishikof DC, Foster JA, Humphries DE, Ricupero DA, Goldstein RH. NF-kappaB induced by IL-1beta inhibits elastin transcription and myofibroblast phenotype. Am J Physiol Cell Physiol. 2002;283:C58–65. doi: 10.1152/ajpcell.00314.2001. [DOI] [PubMed] [Google Scholar]
- 25.Chen LF, Greene WC. Shaping the nuclear action of NFκB. Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 26.Tapias A, Monasterio P, Ciudad CJ, Noe V. Characterization of the 5′-flanking region of the human transcription factor Sp3 gene. Biochim Biophys Acta. 2005;1730:126–36. doi: 10.1016/j.bbaexp.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 27.Tsunoda T, Takagi T. Estimating transcription factor bind ability on DNA. Bioinformatics. 1999;15:622–30. doi: 10.1093/bioinformatics/15.7.622. [DOI] [PubMed] [Google Scholar]