

Summary
Environmental microbes oscillate between feast and famine and need to carefully manage utilization, storage and conversion of reserve products to exploitable sources of carbon and energy. Polyhydroxyalkanoates (PHAs) are storage polymers that serve bacteria as sources of food materials under physiological conditions of carbon demand. In order to obtain insights into the role of PHA depolymerase (PhaZ) and its relationship to a PHA polymerase (PhaC2) in the carbon management activity of Pseudomonas putida strain U, we created a polymerase hyperexpression strain and a depolymerase knockout mutant of this strain, and examined their synthesis of PHA and expression of their PHA genes. This study revealed that hyperexpression of PhaC2 led to the accumulation of higher amounts of PHA (44%wt) than in the wild-type strain (24%wt) after 24 h of cultivation, which then returned to wild-type levels by 48 h, as a result of elevated depolymerization. The phaZ mutant, however, accumulated higher levels of PHA than the parental strain (62%wt), which were maintained for at least 96 h. Transcriptional analysis of the pha cluster by RT-PCR revealed that PHA operon proteins, including depolymerase, are expressed from the beginning of the growth phase. Hyperexpression of the PhaC2 polymerase was accompanied by an increase in the expression of the PhaZ depolymerase and a decrease in expression of another PHA polymerase, PhaC1. This suggests tight regulatory coupling of PHA polymerase and depolymerase activities that act in synergy, and in concert with other PHA proteins, to provide dynamic PHA granule synthesis and remodelling that rapidly and sensitively respond to changes in availability of carbon and the physiological-metabolic needs of the cell, to ensure optimal carbon resource management.
Introduction
All free-living organisms practice carbon resource management to an extent that is possible. Whereas many animals and plants generally regulate carbon uptake to match metabolic needs, other organisms, particularly opportunistic environmental microbes subjected to widely fluctuating carbon availability, may assimilate all carbon that is capturable and manage its utilization through consumption and growth, on one hand, and conservation by conversion to storage polymers, on the other (Reis et al., 2003). Interconversions between readily metabolizable and more inert intracellular storage products are central to this. Even organisms that regulate carbon uptake exploit such interconversions for fine tuning of their carbon management, in order to optimize their cellular metabolic networks and their population-level ecophysiological processes.
Widely exploited storage products in the microbial world are polyhydroxyalkanoates (PHAs), wax esters and triacylglycerols, which are formed into characteristic intracellular hydrophobic granules (Haywood et al., 1990; Huisman et al., 1991; Timm and Steinbüchel, 1992; Kim et al., 2007; Arias and Bassas, 2010). Such storage products are typically produced under conditions of carbon availability when growth is otherwise restricted because of limitation of another essential nutrient, such as nitrogen or oxygen (Madison and Huisman, 1999; Luengo et al., 2003; Velázquez et al., 2007). Carbon is then channelled less towards metabolism and growth, and more towards storage products. Food reserves are subsequently mobilized for cellular maintenance under environmental conditions allowing no or slow growth, or for growth when conditions of nutrient balance return. It is to be expected that the competitive ability of microbes in changing environments is to some extent determined by how nimble they are in storing excess carbon, and especially in mobilizing their storage products, as a function of the metabolic status of the cell and the prevailing fluxes of nutrients needed for growth and development (Hoffmann and Rehm, 2005; Ciesielski et al., 2010). The opposing processes of storage product polymerization and depolymerization, their regulation, and the integration of their regulation in the metabolic and regulatory network hierarchy of the cell, are thus pivotal to optimal cellular function and organism-population competitivity.
A key issue in this context is: how does the cell assure optimal activities of opposing reactions while avoiding futile cycles? Part of the answer may lie in the spatial sequestration/compartmentalization of competing activities within the polymer granule, which is not simply a solid amorphous mass of polymer but rather a structured heterogeneous body containing polymerase(s) and depolymerase(s), other PHA-relevant enzymes, and structure-orchestrating proteins, like phasins (Fig. S1A) (Prieto et al., 1999a; York et al., 1995; Pötter and Steinbüchel, 2005; Neumann et al., 2008). But part of the answer also lies in the relative amounts of polymerase and depolymerase in the granule, which are determined to a large extent by their regulated production (Uchino et al., 2007; Ren et al., 2009a; de Eugenio et al., 2010a,2010b). Thus far, the factors controlling the processes of polymerization and depolymerization are still poorly understood; their characterization will not only lead to a better understanding of carbon management by the cell, and hence the biology of the system, but should also provide a basis for the development of improved systems for biotechnological production of storage polymers as alternatives to petrochemical-based polymers.
In the present study, we investigated regulatory interactions of the PhaC2 synthase-polymerase and the PhaZ depolymerase of Pseudomonas putida strain U (PpU), through the generation of hyperproduction and deletion mutants, employing a chromosomally integrated expression vector, and analysis of their transcription phenotypes and PHA accumulation properties.
Results
Hyperexpression of the PhaC2 PHA synthase results in increased PHA accumulation
We designed a bipartite, mini-transposon-based hyperexpression system for the PpU PhaC2 synthase, consisting of (i) a specialized mini-Tn5, pCNB1xylS/Pm::T7pol, expressing T7 polymerase from the XylS3-metylbenzoate (3-MB)-regulated promoter Pm; and (ii) a hybrid pUT-miniTn5-Tel-derivative expressing the phaC2 gene from the T7 polymerase promoter (Fig. S1B). The two mini-transposon components were separately and randomly inserted into the PpU chromosome. The best PHA producer was selected after two rounds of screening, involving semi-quantification of PhaC2 production by SDS-PAGE separation of cellular proteins and inspection of PHA granule formation by fluorescence microscopy of Nile Red-stained cells (data not shown). This strain was designated PpU 10–33.
Figure 1 and Table S1 show the growth and PHA yields of PpU 10–33 and its parental strain PpU over time in cultures in modified MM medium with sodium octanoate given in two pulses of 15 and 20 mM, the second pulse given at the time of induction. As can be seen, peak biomass and PHA levels were reached at 48 h (Fig. 1B, Table S1B). PHA levels in the hyperexpressing strain were around 50% higher than those in the parental strain at 24 h but were around 25% lower than those of the parental strain at 48 h and similar at 72 h, suggesting that an increase in PhaC2 causes a transient increase in PHA, which in turn provokes an increase in depolymerization activity until levels are normalized. Importantly, the PHA percentage of cellular dry weight (%wt) dropped precipitously after 48 h from 35% to 7%wt, in the case of PpU, and from 39% to 15%wt, in the case of PpU 10–33 induced cultures.
Figure 1.
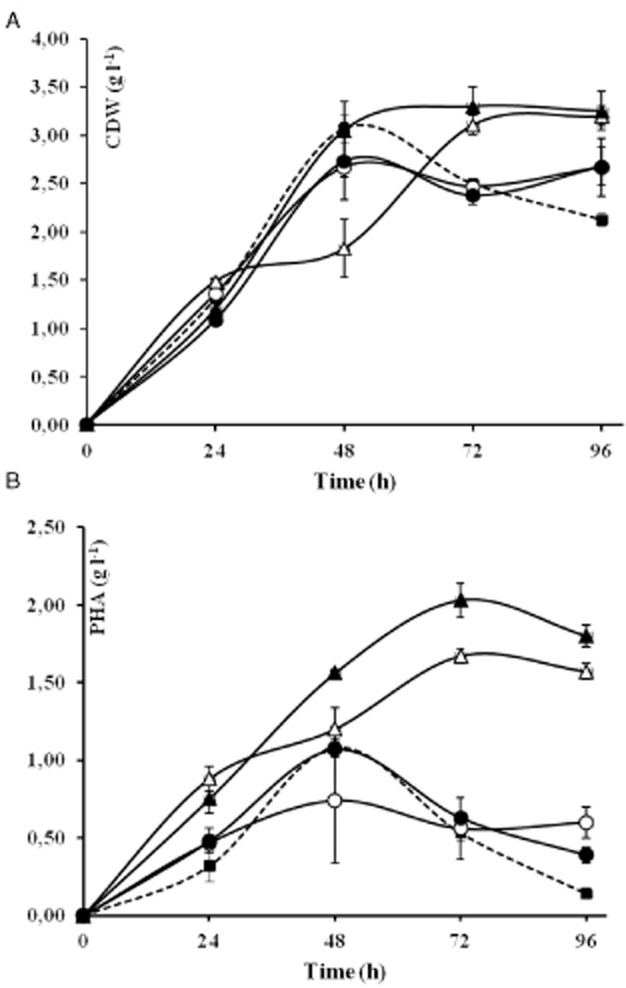
PHA production in P. putida U and the recombinant strains. Strains were cultured in modified MM with sodium octanoate 35 mM and were induced (I) with 0.5 mM 3-MB or not induced (NI). (A) shows the biomass yields and (B) PHA accumulation (g l−1) for PpU (filled squares), PpU 10–33 NI (open circles), PpU 10–33 I (filled circles), and PpU 10–33-ΔphaZ NI (open triangles) and I (filled triangles). Values are means of duplicates and/or triplicates and errors were calculated with the SEM function by using the GraphPad Prism statics program.
The reason why non-induced cultures of PpU 10–33 also showed a 50% increase in PHA accumulation over that of the wild-type strain at 24 h was not investigated further, but was assumed to reflect leakiness of the T7 promoter (also indicated by RT-PCR results; see below).
The highest biomass levels, 3.07 g l−1 in the case of PpU, and 2.67 g l−1 (uninduced, NI) and 2.73 g l−1 (induced, I) in the case of PpU 10–33 (Fig. 1A, Table S1A), and PHA accumulation, 1.08 g l−1, 0.74 g l−1 and 1.07 g l−1, respectively (Fig. 1B, Table S1B), were attained at 48 h of cultivation with both strains. After 48 h, biomass and PHA levels dropped, with PHA levels diminishing or falling more significantly than biomass levels. The PpU 10–33 strain gave higher yields of PHA, expressed as percentage of biomass, at almost all sampling times. The highest PHA yield measured in this experiment, 44%wt, was obtained in PpU 10–33 induced cells at 24 h, compared to 24%wt in PpU and 35%wt in uninduced PpU 10–33 cells (Table S1B). At 48 h, when the highest biomass yield was obtained, the highest absolute yield, 41% of cellular dry weight (CDW) of PHA, was obtained in uninduced cells of 10–33, compared with 35 in PpU and 40% wt in induced PpU 10–33 cultures. Thus, the effect of induction is seen primarily in relatively young cultures. Importantly, the percentage of PHA dropped precipitously after 48 h to 7%wt in the case of PpU and 15–22%wt in the case of PpU 10–33.
Inactivation of the PhaZ depolymerase results in higher PHA peak levels that are maintained over time
A phaZ deletion mutant of the PpU 10–33 strain, designated PpU 10–33 ΔphaZ, was created (see Experimental procedures) and subsequently assessed for PHA accumulation. As can be seen in Fig. 1 and Table S1, cultures of the mutant exhibited higher PHA levels (62%wt) and, in contrast to the situation with the PhaZ-producing strains, these levels were maintained until at least 96 h of cultivation. Thus, the ΔphaZ knockout phenotype suggests that the PhaZ depolymerase is a major determinant of PHA accumulation and maintenance in the cell.
In order to causally relate the phaZ gene mutation to the observed phenotype, and to rule out any indirect effects on expression of the pha cluster, the phaZ gene was PCR-amplified, cloned in the pBBR1MCS-5 plasmid vector, and introduced into the PpU 10–33-ΔphaZ strain. PHA production and maintenance in the complemented mutant, PpU 10–33-ΔphaZ pMC-phaZ, designated strain pMC-phaZ, was then assessed. Table 1 shows the biomass and PHA yields of the PpU 10–33 strain, its phaZ deletion mutant and the complemented derivative, after growth for 44 h in modified MM with sodium octanoate (20 mM). Biomass yields for the three stains were similar at about 2 g l−1 whereas PHA yields were 21%wt for the PpU 10–33 strain, 41%wt for its ΔphaZ mutant, and 5%wt for the complemented strain. The lower than wild-type levels of PHA in the complemented strain presumably reflects higher cellular depolymerase levels, resulting from the complementing gene being located on a multicopy vector.
Table 1.
Effect of phaZ gene deletion on PHA yields in PpU 10–33
| Strains | CDW (g l−1) | PHA (g l−1) | PHA (% wt) |
|---|---|---|---|
| PpU 10–33 | 2.11 | 0.45 | 21.0 |
| PpU 10–33- ΔphaZa | 2.18 | 0.90 | 41.0 |
| pMC-PhaZb | 1.98 | 0.10 | 5.0 |
Strains were cultivated in modified MM with 20 mM octanoate for 44 h, without the addition of 3-MB inducer.
Knockout strain of the phaZ.
Complemented strain.
PHA granule morphology is different in the constructs
Since hyperexpression of PhaC2 polymerase and inactivation of PhaZ depolymerase may entrain changes in the normal cellular stoichiometry and activity of PHA proteins, and associated proteins, other changes in phenotypes may result from these genetic manipulations. To assess this possibility, we compared the ultrastructure of the PHA granules in cells of the different constructs by transmission electron microscopy (TEM). Figure 2 shows that the PpU wild-type strain (Fig. 2A–C) contains one or two defined PHA granules per cell, distributed evenly within the cytoplasm, while the PpU 10–33 phaC2 hyperexpression strain (Fig. 2D–F) tends to contain one main granule with a morphology suggestive of the coalescence of smaller granules. This is particularly evident in the induced cultures, specifically during the mid-exponential growth phase. The phaZ deletion mutant tended to have multiple granules, some of which had irregular boundaries suggestive of granule fusion (Fig. 2G–I). The microscopic analysis also confirmed the results shown in Fig. 1B (Table S1B), namely that intracellular PHA accumulated in the PpU and PpU 10–33 strains starts to diminish after 48 h of cultivation, whereas the mutant lacking the depolymerase maintained accumulated PHA until the end of the experiment.
Figure 2.
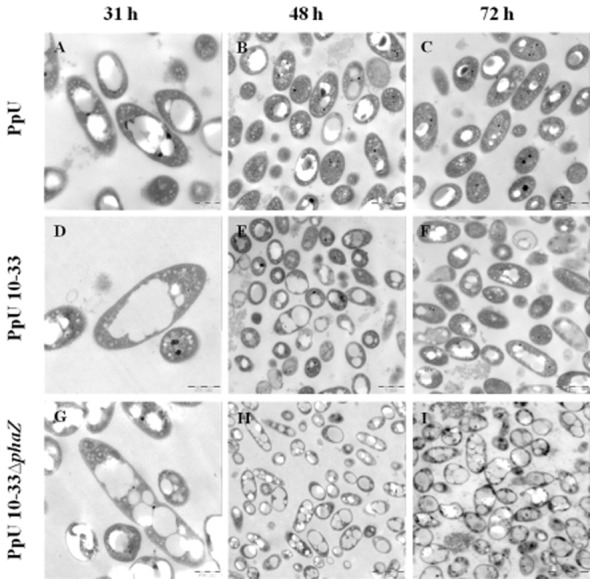
Electron micrographs of P. putida U wild-type and mutant cells. Transmission electron micrographs of thin sections of PpU (A–C); PpU 10–33 induced (D–F); PpU 10–33-ΔphaZ induced (G–I) cells. Strains were cultured in modified MM containing 35 mM sodium octanoate as a carbon source and sampled at 31 h (A, D and G), 48 h (B, E and H) and 72 h (C, F and I). Scale bars for (A, D and G), (B, C, E and F) and (H and I) are 500 nm, 1 μm and 2 μm respectively.
PHAs produced by different constructs are similar but not identical
Given that the two PHA synthases of PpU have slightly different substrate specificities, with PhaC2 exhibiting a preference for 3-hydroxyhexanoyl-CoA and PhaC1 biased towards 3-hydroxyoctanoyl-CoA (Arias et al., 2008), it was possible that hyperexpression of the PhaC2 polymerase in PpU 10–33 might alter the monomer composition and/or physicochemical properties of the polymer produced. Table 2 shows that PHAs produced during growth on sodium octanoate by PpU, PpU 10–33 and its phaZ deletion mutant had similar compositions, as determined by NMR, and were copolymers of P(3-hydroxyoctanoate-co-3-hydroxyhexanoate), composed of 3-hydroxyoctanoate (91.4–92.5% mol) and 3-hydroxyhexanoate (7.5–8.6% mol). Also, the glass transition temperature of the three polymers, Tg −35.9 to −40.8°C (Table 2), was in agreement with the Tg described previously for medium chain length (mcl)-PHAs, and they had similar melting temperatures (Tm, 59–61°C), indicating similar crystallinity grades.
Table 2.
Physicochemical properties of the PHAs obtained from the mutant strains
| Strains | Mw (kDa) | Mn (kDa) | PI | Tg (°C) | Tm (°C) | ΔHm (J g−1) | Td (°C) | Monomer composition (%mol) |
|
|---|---|---|---|---|---|---|---|---|---|
| 3-HHx | 3-HO | ||||||||
| PpU | 126.3 | 76.6 | 1.65 | −35.90 | 61.40 | 22.76 | 294.03 | 8.6 | 91.4 |
| PpU 10–33 NI | 132.9 | 75.7 | 1.76 | −35.92 | 59.68 | 28.42 | 294.93 | 7.5 | 92.5 |
| PpU 10–33 I | 141.1 | 74.9 | 1.88 | −37.16 | 59.21 | 24.89 | 294.04 | 8.4 | 91.6 |
| PpU10-33-ΔphaZ NI | 95.6 | 52.1 | 1.83 | −40.82 | 59.60 | 27.14 | 293.84 | 8.6 | 91.4 |
| PpU10-33-ΔphaZ I | 96.2 | 50.1 | 1.92 | −36.09 | 61.57 | 28.60 | 293.65 | 8.7 | 91.3 |
Experimental conditions as given in Table 1.
Mw, weight-average molecular weight; Mn, number-average molecular weight; PI, polydispersity index (Mw/Mn); Tg (°C), glass transition temperature; Tm (°C), melting temperature; Td (°C), decomposition temperature; ΔHm (J g−1), enthalpy of fusion; 3-HHx, 3-hydroxyhexanoate; 3-HO, 3-hydroxyoctanoate.
However, the polymers differed in length: the molecular weights (Mw and Mn values) of the polymers from the PpU parental strain and the PpU 10–33 (PhaC2 polymerase hyperexpressing construct) were similar, ranging from 126–142 and 74–77 kDa respectively, whereas those from the PhaZ knockout were considerably lower, 96 and 50 kDa respectively. Although this result may seem to be counter-intuitive, it is consistent with observations of Cai and colleagues (2009) and Solaiman and colleagues (2003), who also found lower polymer lengths in phaZ mutants. Thus, we cannot rule out the possibility that the depolymerase also plays a direct or indirect role in chain length determination and/or in PHA synthesis itself.
pha operon transcriptional activity indicates strong coupling of polymerization and depolymerization functions over the entire growth cycle
In order to investigate the relationship between PHA turnover and the hyperexpression of phaC2 and phaZ inactivation, we also carried out transcriptional analysis by relative RT-PCR of the pha cluster (Figs 3 and S2), as well as of the fadD1 and fadD2 genes (Fig. S3) in the three strains. Reference genes for the RT-PCR data normalization were gltA and proC2.
Figure 3.
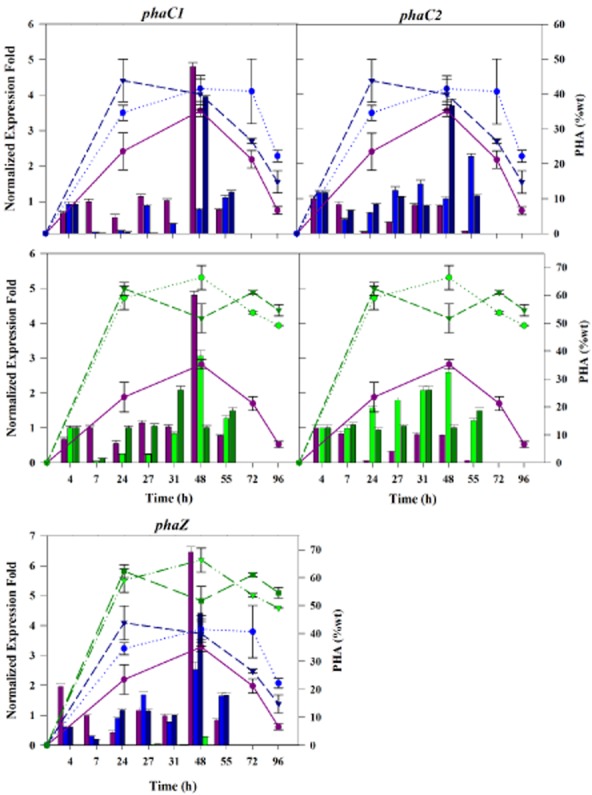
Expression of pha genes and PHA accumulation in P. putida U. Each panel shows normalized fold-increased in expression of the pha genes in PpU (violet), PpU 10–33 non induced (light blue) and PpU 10–33 induced (dark blue), PpU10-33-ΔphaZ uninduced (light green) and PpU10-33-ΔphaZ induced cells (dark green). The relative expression ratio of the target genes was calculated automatically with the CFX software using the standard error of the mean and the normalized expression method [ΔΔ(Ct)]. PHA content (% CDW) is showed with lines, using the same strain colour designation mentioned above. Experimental triplicates and independent biological duplicates were used for the calculations.
Polymerase/depolymerase gene transcripts
In the wild type, no major changes were detected in transcript levels of the two PHA polymerases, PhaC1 and PhaC2, during the first 24 h of cultivation (P > 0.1), and this was accompanied by a steady increase in PHA accumulation. However, a twofold increase (P < 0.001) in phaZ transcripts was measured at 4 h, corresponding to the onset of PHA production, which then fell back to lower levels. At 48 h, correlating with maximum levels of PHA accumulation, a rapid and substantive increase in the transcription of phaC1 was observed (4.5-fold, P < 0.0001) and, in parallel, a sixfold increase (P < 0.001) in phaZ transcriptional activity. This was followed by a rapid decrease in the PHA content (Fig. 3), and phaC1 and phaZ transcript levels. These results are indicative of a finely tuned coupling of phaC1 transcription and PHA accumulation, on one hand, and phaZ transcription and PHA mobilization, on the other.
In the case of the PpU 10–33 strain, expression of the phaC2 gene was, as expected, found to be higher than in the PpU parental strain throughout the cultivation period (P < 0.008) and especially at 48 h, when it peaked (3.5-fold increase, P < 0.0001). Interestingly, the expression of phaC1 in this strain was mostly lower than in PpU, especially in induced cultures at 7, 24 and 48 h, suggesting that hyperexpression of phaC2 negatively influences expression of phaC1 (Fig. 3). However, even though hyperexpression of phaC2 resulted in decreasing expression of phaC1, the combined cellular synthase activity resulted in an increased PHA production. Transcription levels of phaZ in PpU 10–33 tended to be similar to those in the parental strain, except at 24 h, when it was higher, correlating with the higher expression of phaC2 and in cultures older than 48 h in which it was also higher, consistent with the higher levels of PhaC2 and PHA. There is thus also a strong coupling of PhaC2 polymerase and depolymerase synthesis.
In the PpU 10–33-ΔphaZ strain, significantly higher transcription levels of phaC2 were observed throughout the cultivation period when compared with the wild type (P 0.0005-0.017), which is consistent with the higher PHA yields obtained (from 60%wt to 66%wt) (Fig. 3). In the case of phaC1 also higher levels were measured at 24 and 38 h, but only when phaC2 was induced (P < 0.0017). Thus, inactivation of phaZ not only prevents turnover and recycling of synthesized PHA, but also allows higher transcription levels of the PHA polymerases.
Phasin gene transcripts
As can be seen in Fig. S2A, transcription of the phaF and phaI phasin genes in all strains increased between 4 and 7 h, when PHA started to accumulate, then temporarily decreased before spiking at 27–31 h, in the case of the PpU parental strain and the phaZ deletion mutant, and 48 h, in the case of the PpU 10–33 phaC2 hyperexpression strain, when PHA accumulation was highest. Thereafter, transcription of the phasin genes decreased to low levels. Noteworthy are the much larger increases in transcription in the PpU parental strain and phaZ deletion mutant, compared to the PpU 10–33 hyperexpression strain (in the case of the phaZ mutant, but not the PpU 10–33 strain, this correlates with higher PHA levels), and the higher post-peak transcription levels of the PpU 10–33 mutant compared with the PpU parental strain (correlating this time with a higher PHA content). Interestingly, phasin gene expression in both the PpU wild-type strain and the phaZ deletion mutant spiked at 24 h well before the spiking of transcription of the phaC1/2 polymerase and phaZ depolymerase genes at 48 h, whereas transcriptional activity change was more gradual in the 10–33 hyperexpression strain and tended to follow more or less PHA accumulation. The results with the wild-type strain are consistent with phasin synthesis being in phase with, but in advance of, polymer synthesis, presumably to guide correct assembly of the polymer granule.
The PhaD pha gene cluster regulator
The transcription of phaD, which encodes a key regulator of the pha gene cluster, was similar in PpU and PpU 10–33 strains (except for the earliest sampling time, 4 h), but was expressed at higher levels in the phaZ depolymerase mutant throughout the cultivation period, consistent with the higher levels of expression of the phaC2 polymerase and the phaF and phaI phasin genes, which PhaD regulates (Klinke et al., 2000; Sandoval et al., 2007; de Eugenio et al., 2010b).
Acetyl-CoA synthase gene activities
As can be seen in Fig. S3, the transcription of the fadD2 acetyl-CoA synthase gene exhibited a remarkable spike at 48 h, especially in the PpU wild-type strain, paralleling the sharp peak in phaZ transcription at the time of maximum PHA accumulation. This peak was much lower in the phaZ deletion mutant, which is unable to depolymerize accumulated PHA, so it would seem that the FadD2 enzyme plays a role in the recycling of monomers released by PHA depolymerization. These results corroborate those of Olivera and colleagues, which indicated that FadD2 plays a fundamental role when the PHA depolymerization process dominates in P. putida U (Olivera et al., 2001a). In contrast to the dramatic expression profile of fadD2, fadD1 largely followed the expression profile of phaD, with higher levels at the times of initiation of PHA accumulation and maximal accumulation. Thus, FadD1 seems to be the main acetyl-CoA synthase involved in maintaining a constant supply of activated PHA precursors (see also García et al., 1999; Olivera et al., 2001a; Ruth et al., 2008; Hume et al., 2009; Ren et al., 2009a).
The analysis of possible transcription promoters using Softberry, PromScan and the PDBG online informatics tools, revealed that all pha genes are preceded by potential promoters and more specifically, that both σ70 and σ54 potential promoters were found upstream of the phaC1 gene. A search for transcriptional terminators, using the Arnold informatics tool, also disclosed the presence of rho-independent terminators downstream of the phaC1, phaZ, phaC2 and phaD genes, as well as the existence of a REP sequence (enterobacteriaceae repetitive extragenic palindromic region) downstream of phaD which may also act as a transcription terminator.
Discussion
Manipulation of gene expression is widely used to investigate cellular physiology, on one hand, and to increase product yields in biotechnology, on the other (Kraak et al., 1997; Prieto et al., 1999b; Olivera et al., 2001b; Conte et al., 2006; Kim et al., 2006; Ren et al., 2009b; Arias and Bassas, 2010). Manipulation strategies may be unsuccessful inter alia because the real rate limiting factor has not been addressed by the strategy, often because of genetic or physiological redundancies, which can compensate for knockout/knockdown/hyperexpression strategies. When successful, they often lead to perturbation of the carefully harmonized cellular metabolic network, and may ultimately select compensatory changes in the network (Diederich et al., 1994). An example of quasi-redundancy is the existence of multiple PHA polymerases in many bacteria; for instance in P. putida U there are two PHA synthases, PhaC1 and PhaC2. Thus far, manipulations of the expression of the PHA polymerase and depolymerase genes have given inconsistent results. Several attempts to increase PHA yield through hyperexpression of the PhaC1 polymerase (Kraak et al., 1997; Conte et al., 2006; Kim et al., 2006; Ren et al., 2009b) have had only limited success, because of either instability of the plasmid constructs, or poor inducibility of the cloned gene. Other attempts, involving deletion of phaZ genes are more contradictory since, although in some cases the lack of PhaZ resulted in higher yields and extended accumulation of PHA during the growth cycle (García et al., 1999; Sandoval et al., 2005; Cai et al., 2009), in others it did not (Huisman et al., 1991 and Solaiman et al., 2003). In this work, the stable chromosomal integration of a T7 RNA polymerase-based phaC2 hyperexpression construct led to significant increases in PHA yields. However, the elevated yields were transitory and the PHA content of cells declined dramatically after peaking at around 48 h of cultivation. This was shown to be due to the action of the PhaZ depolymerase: deletion of the phaZ gene in the PhaC2 hyperexpression strain resulted in higher PHA yields than the wild type and the PhaC2 hyperexpressing strains over the entire growth curve, and in maintenance of peak levels for at least 96 h. Complementation of the phaZ deletion resulted in lower PHA levels than the parental and hyperexpression strains, and faster PHA mobilization after 48 h. These results are in concordance with the observation of Sandoval and colleagues (2005) that PHA accumulation was not detectable in a b-oxidation mutant strain hyperexpressing PhaZ. Thus, PhaZ is clearly a rate-limiting factor in both PHA synthesis and maintenance, and would seem to be a non-redundant function in P. putida U. It is therefore a prime target for engineering strains for commercial production of PHA.
The implication that the PhaZ depolymerase functions not only once PHA accumulation peaks but also from the onset of PHA synthesis, was investigated by analysis of phaZ transcription by RT-PCR. This analysis revealed that indeed the phaZ gene is expressed throughout the growth curve (with an increase at the outset of PHA accumulation), as are the phaC1 and phaC2 genes, and its expression is correlated, in the parental strain, with the phaC1 transcriptional level and, in the PpU 10–33 strain, with that of phaC2. The results presented here are thus consistent with a strong functional coupling of PHA polymerization and depolymerization activities, and with the notion that PHA polymerization and depolymerization occur simultaneously in and on the PHA granule, from the outset of granule formation, presumably constituting a key functional element of dynamic granule structuring and remodelling, and interaction with the cellular environment.
Though it was not the purpose of this study to investigate the relationships of PHA production and expression of relevant genes other than phaC2 and phaZ, two observations made here would seem to merit brief comment. One is that upregulation of phaC2 seemed to result in downregulation of phaC1, suggesting that, although the two PHA polymerases have non-identical substrate spectra, and are thus not entirely functionally redundant, their combined cellular levels seem to be subject to a regulatory network setting upper synthesis thresholds. Interestingly, both polymerases were expressed at higher levels in the phaZ knockout mutant, correlating with the higher PHA accumulation, and indicating that such a regulatory network must integrate depolymerization parameters. Secondly, the changes in levels of PhaC2 and PhaZ that we have engineered in this study, not only caused changes in PHA accumulation levels but also in PHA granule morphology. In particular, upregulation of phaC2 resulted in larger, more amorphous granules. Since granule number and size (Pieper-Fürst et al., 1995; Wieczorek et al., 1995; Grage et al., 2009), as well as distribution in the cytoplasm and segregation (Ren et al., 2010; Galán et al., 2011), are orchestrated by the major PHA-associated proteins, the phasins (Steinbüchel et al., 1995; Prieto et al., 1999a; York et al., 1995; de Eugenio et al., 2010b), it would seem that upregulation of PhaC2 creates an imbalance in PHA : phasin ratios, leading to altered phasin-determined granule morphology. Such an imbalance is also indicated by the RT-PCR expression results obtained in this study: although higher expression levels of the phasins generally followed increases in PHA accumulation, but peaked prior to the peaking of expression of the PHA synthases (at 31 and 48 h respectively), peak phasin expression levels in the phaC2-hyperexpressing strain were lower than in the wild type. We also cannot rule out a more direct effect of any possible phasin-like activity of PhaC2 on granule morphology, though this seems unlikely in the light of the findings of Arias and colleagues (2008). It thus seems likely that this altered ratio of phasin : synthase/PHA is responsible for the fused granule morphology (see also the observations of Sandoval et al., 2007). In any case, these results suggest that, if a direct coupling of phasins and PHA accumulation in general, and phasins and PhaC2 synthesis in particular, exists, it may be incomplete.
In summary, the picture that emerges from this and previous studies (Ren et al., 2010; de Eugenio et al., 2010a) is that of a highly dynamic PHA granule, resulting from tight regulatory coupling of polymerization : depolymerization processes that rapidly and sensitively respond to changes in availability of carbon and other nutrients, and the physiological-metabolic needs of the cell, to ensure optimal carbon resource management. This involves continual and coordinated expression of polymerases and depolymerase, and their constant remodelling of the PHA granule, throughout the growth and stationary periods. Avoidance of futile cycles of synthesis : degradation appears to be achieved more through the control of enzyme activity, perhaps through interactions with other PHA proteins, such as the phasins, and probably through spatial partitioning of the polymerases and depolymerase, than through modulation of gene expression. In terms of the commercial production of PHA, we have shown that the PhaZ depolymerase is a key yield-limiting parameter, and that its inactivation, in combination with hyperexpression of PhaC1/2 synthases, is pivotal to obtaining high PHA yields. Our use of chromosomally integrated genetic constructs resulted in stable strains and phenotypes that obviated the need for costly antibiotic selection pressure for plasmid maintenance. Also our finding that the T7-based expression system was leaky, and did not require the addition of inducer, is a further cost-saving aspect for production.
Experimental procedures
Microorganisms and vectors
Bacterial strains, mutants and plasmids used in this work are summarized in Table S2.
Culture media conditions
Unless otherwise stated, Escherichia coli and P. putida strains were cultured in Luria–Broth (LB) and incubated at 37°C and 30°C respectively. Where required, supplements were added to media as follows: rifampicin (Rf, 20 μg ml−1 in solid and 5 μg ml−1 in liquid media), kanamycin (Km, 25 μg ml−1 in solid and 12.5 μg ml−1 in liquid media), ampicillin (Ap, 100 μg ml−1), tellurite (Tel, 100 μg ml−1), gentamicin (Gm, 30 μg ml−1), chloramphenicol (Cm, 30 μg ml−1), isopropyl-β-d-thiogalactopyranosid (IPTG, 70 μM) and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (XGal, 34 μg ml−1).
DNA manipulations
All genetic procedures were performed as described by Sambrook and Russell (2001). Genomic and plasmid DNA extraction, DNA purification from agarose gel and PCR cleaning were carried out using the corresponding Qiagen kits (QIAGEN, Hilden, Germany), according to the manufacturers' instructions. All DNA modifying enzymes were purchased from NEB (Massachusetts, USA). Polymerase chain reactions (PCR) were performed in an Eppendorf Vapo.protect Thermal Cycler (Eppendorf, Hamburg, Germany). The 50 μl PCR reaction mixture consisted of 2 μl of DNA (50 μg ml−1), 1× PCR buffer, 2 mM MgCl2 (PROMEGA, Wisconsin, USA), 0.2 μM of each primer (Eurofins mgw Operon, Ebersberg, Germany), 0.2 mM dNTPs (Amersham, GE HealthCare, Amersham, UK), and 1.25 U GoTaq Hot Start Polymerase (PROMEGA). PCR cycling conditions consist of: an initial step at 96°C/10 min, followed by 30 cycles of 96°C/30 s, 60°C/30 s and 72°C/1 min, with a final extension at 72°C/5 min. Plasmid transfer to Pseudomonas strains (pCNB1mini-Tn5 xylS/Pm::T7pol or pUTminiTn5-Tel-phaC2) was made by triparental conjugation experiments (Selvaraj and Iyer, 1983; Herrero et al., 1990) and transconjugants clones were confirmed by PCR.
PCR reactions for sequencing were performed in 10 μl reaction mixture consisted of 6–12 ng of the purified PCR product, 2 μl BigDye Ready Reaction Mix (Applied Biosystems, Foster City, CA, USA), 1 μl of BigDye buffer and 1 μl of the specific primer (2 μM). The cycling conditions included: an initial step at 96°C/1 min, followed by 25 cycles of 96°C/20 s, 52–58°C/20 s and 60°C/4 min, with a final extension step at 60°C/1 min. Nucleotide sequences were determined using the dideoxy-chain termination method (Big Dye Terminator v3.1 Kit, Applied Biosystems) and the ABI PRISM 3130 Genetic Analyser (Applied Biosystems).
Identification of potential transcriptional promoter regions and terminators was made using the Softberry, (http://linux1.softberry.com/berry.phtml), PromScan (http://molbiol-tools.ca/promscan/), BDGP online (http://www.fruitfly.org/seq_tools/promoter.html); and Arnold (http://rna.igmors.u-psud.fr/toolbox/arnold/index.php) bioinformatics tools.
Design and construction of the phaC2 hyperexpression strain PpU 10–33
PpU 10–33 is a P. putida strain U derivative in which phaC2 gene expression is driven by the T7 polymerase promoter-T7 polymerase system (Fig. S1B). It consists of two chromosomally integrated cassettes: one containing the phaC2 gene expressed from the T7 polymerase promoter, and another containing the T7 polymerase gene expressed from the Pm promoter and regulated by the cognate benzoate/toluate-inducible XylS regulator derived from the TOL plasmid. The phaC2 cassette was constructed as follows: The phaC2 gene of P. putida strain U was excised from the pBBR1MCS-3-phaC2 plasmid (Arias et al., 2008), cloned into the pUC18NotI/T7 vector (Herrero et al., 1993), and the correct orientation of the gene confirmed by sequencing. The phaC2 gene and the T7 promoter were then transferred as a cassette into the pUTminiTn5-Tel vector (Sánchez-Romero et al., 1998). First, the miniTn5 derivative pCNB1 xylS/Pm::T7pol was transferred to P. putida U by filter-mating and selected by the Km selection marker (Harayama et al., 1989; Herrero et al., 1993). Since integration of the transposon in the genome is essentially random, and different sites of insertion can markedly influence transcription levels of inserted genes, a pool of approximately 100 transconjugants was prepared for the second transfer. A 5 ml LB culture of this pool was incubated for 3 h (30°C, 180 rpm), and used as recipient to transfer the pUTmini-Tn5-Tel-T7phaC2 construct. Transconjugants were readily scored by the black colour they display when they transform the tellurite (selection marker), and subsequently confirmed by PCR. The final recipients varying in insertion sites of both cassettes were subsequently scored for PhaC2 levels and PHA, the best selected and designated PpU 10–33.
Knockout of phaZ in PpU 10–33 and complementation
Deletion of the phaZ gene was accomplished by using a method involving a double-recombination event and selection of the required mutant by expression of the lethal sacB gene (Quant and Hynes, 1983; Donnenberg and Kaper, 1991). First, a DNA fragment containing the ORFs adjacent to the phaZ gene, encoding the PhaC1 and PhaC2 synthases, was synthesized by GENEART AG (Life Technologies, California, USA) and subsequently sub-cloned into the pJQ200SK vector containing the GmR and SacB selection markers. The hybrid plasmid was then introduced by triparental mating into the PpU 10–33 strain. Transconjugants, in which the plasmid was integrated into the chromosome by a single cross-over, were selected on Gm + Km + Tel-containing plates and confirmed by PCR. Deletion mutants resulting from the second recombination were subsequently selected on LB plates containing 10% sucrose, scored for sensitivity to Gm, and further analysed by PCR to confirm the position and extent of the deletion. One deletion mutant was selected and designated PpU 10–33-ΔphaZ.
For complementation of the deletion mutant, the phaZ gene (921 bp) was amplified by PCR and cloned into the pBBR1MCS-5 vector. Transconjugants were selected for their Gm resistance and further confirmed by PCR.
RNA manipulations
Samples (3 ml) were taken from cultures through the growth phase (4, 7, 24, 27, 31, 48 and 55 h) and immediately mixed with an equal volume of RNA protect Buffer (QIAGEN). After incubation for 5 min at room temperature, suspensions were centrifuged at 5000 g (Allegra 25R, Beckman Coulter, California, USA), the supernatant fluids discarded, and the pellets placed at −80°C. Total RNA was extracted using the RNeasy mini kit (QIAGEN) with DNAse treatment, according to the manufacturer's protocol. RNA was then eluted in 100 μl of RNase-free water, and kept at −80°C. The integrity of the RNA was assessed by electrophoresis in formaldehyde agarose gels and the concentration and purity determined spectrophotometrically (Spectrophotometer ND-100, peQlab-biotechnologie GmbH, Erlangen, Germany). For cDNA synthesis all reagents were purchased from Invitrogen (Life Technologies) and reactions performed according to manufacturer's protocols, in 20 μl using 10 μg of total RNA and random primers. Samples in which Superscript III RT was not added were used as negative controls. After cDNA synthesis, the remaining RNA was precipitated by addition of 1/5 volume of 1 M NaOH and incubated at 65°C for 10 min, followed by 10 min at 25°C and neutralized with the same volume of 1 M KCl. The resultant cDNA was then purified using the PCR purification kit (QIAGEN) and the concentration and purity measured spectrophotometrically. cDNAs were diluted with DEPC (diethylpyrocarbonate)-treated water to 100 ng μl−1 and kept at 4°C.
Relative RT-PCR assay
The MIQE guidelines for the experimental design of RT-PCR were followed (Bustin et al., 2009). Oligonucleotides used for the RT-PCR assays (Eurofins mgw Operon, Germany) were designed with the help of the Primer3 (http://frodo.wi.mit.edu/primer3/) and Oligo Calc (http://www.basic.northwestern.edu/biotools/oligocalc.html) bio-informatic tools and are summarized in Table S3. First, optimal PCR conditions, annealing temperature and primer concentrations were established using a standard set of samples (genomic DNA) as templates. Primer specificity was determined by melt curve analysis and gel visualization of the amplicon bands (not longer than 300 bp). Primer efficiency was determined in a dose-response assay using as a template a pool of cDNAs in a fourfold dilutions series (in triplicates) and standard curves determined for each set of primers, using the CFX96™ real-time PCR detection system (Bio-Rad, California, USA) and the CFX Manager software (Bio-Rad). In all cases, efficiencies were measured in the range between 89% and 100%. The choice of appropriate reference genes for data normalization, was determined using the geNorm method existing in the CFS software, taking into consideration the target stability for the different experimental conditions and time points. Several candidate genes, including those encoding ‘housekeeping’ (16 s rDNA, rpsL), general metabolism (gltA, gap-1, proC1, proC2), cell division (mreB, ftsZ) and signalling (ffH) functions, were tested and from these, gltA and proC2 were selected as the reference since they showed a coefficient variance and M value of around 0.5–1. For relative RT-PCR, experimental triplicates were performed and samples without cDNA were used as negative controls. PCR reactions contained 12.5 μl of iQ™ SYBR Green Supermix (2×) (Bio-Rad), 1 μl forward primer (10 μM), 1 μl reverse primer (10 μM), 2 μl of cDNA (1/10 diluted), in 20 μl final volume. The PCR cycling conditions were: 50°C/2 min and 95°C/10 min, followed by 40 cycles of 95°C/15 s, 60°C/30 s and 72°C/30 s, with a final extension at 72°C/10 min. Fluorescence was measured at the end of each cycle. For the melting curve, an initial denaturation step at 95°C/10 min was followed by a reduction in temperature to 65°C, and then temperature increases in increments of 0.5°C/5 s up to 95°C, with continual signal acquisition.
All RT-PCR results are means of biological duplicates (from independent experiments) and experimental triplicates (of measurements), with inclusion of an internal calibrator in each plate for data normalization. For the statistical analysis, the relative expression ratio of the target genes was calculated automatically with the CFX software (Bio-Rad), using the standard error of the mean and the normalized expression method [ΔΔ(Ct)]. Values are expressed as Normalized fold-increases in expression. Two tailed unpaired student's t-test has been used for the statistical analysis using the GraphPad Prims5 program. P-values were calculated for each observation made along the test regarding differences in gene expression.
Culture conditions for PHA production
3-methylbenzoate (3-MB) was used to induce the Pm promotor, via its cognate XylS activator, to mediate expression of T7 polymerase gene, which in turns triggers hyperexpression of the phaC2 synthase. In order to determine optimal conditions for PhaC2 expression/PHA synthesis in PpU 10–33, the concentrations of the 3-MB inducer (from 0.2–3 mM), the times of induction expressed as culture density (OD550 0.4–1.5), and concentrations of carbon sources in the medium, were varied and yields measured. Two-litre Erlenmeyer flasks containing 400 ml of mineral medium (MM) (Martínez-Blanco et al., 1990) plus 0.1% yeast extract, 15 mM sodium octanoate and appropriate antibiotics were inoculated with a colony suspension from a fresh overnight culture (incubated at 30°C) on an MM agar plate containing 20 mM succinate. Flasks were then incubated at 30°C in a rotary shaker (Multitron, INFORS AG, Switzerland) at 180 rpm. Once the cultures reached an OD550 of 0.8, the culture was split into two (1 l Erlenmeyer flasks containing 200 ml) and 3-MB added to a final concentration of 0.5 mM to one of both flasks. At the same time a second pulse of sodium octanoate (20 mM) was added. For the wild-type control strain, the procedure was the same but without the induction. Samples were collected every 24 h and the biomass (cellular dry weight, CDW), PHA, OD550, Nile red staining and NH4+ concentration were determined. For CDW determination, samples were dried at 80°C for 24 h and expressed in g l−1 of original culture. Values are means of duplicates of at least two independent experiments.
Fluorescence and transmission electron microscopy
One millilitre of culture was mixed with 2 drops of a Nile red solution in dimethylsulfoxide (0.25 mg ml−1) and centrifuged at 16 000 g (Centrifuge 5417R; Eppendorf, Hamburg, Germany), at 4°C, 5 min. Pellets were washed twice with 2 ml MgCl2 10 mM, and 5–10 μl of the cell suspension mounted on a microscopic slide (Bassas, 2010). The presence and morphology of PHA granules were visualized with a ZEISS Axio Imager A1 epiflourescence microscope equipped with a Cy3 filter (EX BP 550/25, BS FT 570, EM BP 605/70) (ZEISS, Jena, Germany) and the AxioVision rel 4.6.3 software (ZEISS Imaging Solutions GmbH, Jena, Germany).
For transmission electron microscopy, bacteria were fixed in growth medium containing 2% glutaraldehyde plus 5% formaldehyde at 4°C and processed as described previously (Bassas, 2010). Ultrathin sections were then examined in a TEM910 transmission electron microscope (Carl Zeiss, Oberkochen, Germany) at an acceleration voltage of 80 kV and images were taken at calibrated magnifications using a line replica and recorded digitally with a Slow-Scan CCD-Camera (ProScan, 1024 × 1024, Scheuring, Germany) with ITEM-Software (Olympus Soft Imaging Solutions, Münster, Germany).
PHA extraction and purification
Culture samples were centrifuged at 6500 g for 15 min at 4°C (Allegra 25R, Beckman Coulter), and pellets washed twice in distilled water and lyophilized (Lyophilizer alpha 1–4 LSC, Christ, Germany) at −59°C at 0.140 mbar. Five millilitres of samples was taken along the growth phase to monitor the PHA production and were lyophilized. The lyophilized biomass was extracted with 10 ml chloroform for 3 h at 80°C (Bassas-Galia et al., 2012). PHA content (%wt) is defined as the percentage of the CDW represented by PHA.
PHA analysis
For 1H and 13C-NMR analysis, 10–20 mg of polymer was dissolved into 0.7 ml of deuterated chloroform (CDCl3) and NMR spectra recorded as described elsewhere (Bassas-Galia et al., 2012) Chemical shifts are given in ppm relative to the signal of the solvent (1H: 7.26, 13C 77.3) and coupling constants in Hz. Standard Bruker pulse programs were used throughout. For determination of the polymers molecular weight and analysis of their thermal properties the protocol previously described by Bassas-Galia and colleagues (2012) and Cheema and colleagues (2012) was followed. Briefly, average molecular weights were determined by gel permeation chromatography in a HPLC system (Waters 2695 Alliance separations Module, Waters, USA) equipped with a Styragel HR5E column and a 2414 differential refractive index detector. Elution was with tetrahydrofuran (THF) at 45°C at a flow rate of 0.5 ml min−1 (isocratic). Sample concentration and injection volume were 0.5 mg ml−1 and 50 μl respectively. The calibration curve was obtained using a polystyrene standards kit (Fluka) in the Mw range of 10 000–700 000 g mol−1 (Sigma-Aldrich, Missouri, USA). The thermal properties were determined by differential scanning calorimetry (DSC), using 10–20 mg of the purified polymer for analysis. DSC analyses were performed with a DSC-30 (Mettler Toledo instruments, NY, USA). All data were acquired by STARe System acquisition and processing software (Mettler Toledo).
Acknowledgments
We are deeply indebted to José María Luengo Rodríguez (Universidad de León, Spain) for providing the P. putida U strain and pBBR1MCS-3-phaC2, and Victor de Lorenzo (CNB-CSIC, Madrid) for the miniTn5 derivatives plasmids and invaluable advice. We also thank Manfred Rohde for assistance with electron microscopy, and Agnes Waliczek, Susanne Müller and Annette Krüger, for technical support. The presented study arose from a larger project within the scope of a patent value fund. In this context we acknowledge the financial support by the Dritte Patentportfolio Beteiligungsgesellschaft mbH & Co. KG (Germany) and the overall project coordination provided by Clou Partners (Germany).
Conflict of interest
All authors confirm they do not have any conflicting interests.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Fig. S1. Genetic organization of the pha cluster and the expression system used for the hyperexpression of PhaC2 in PpU 10–33.
Fig. S2. Expression of the phaF, phaI and phaD genes and PHA accumulation in P. putida U.
Fig. S3. Expression of fadD1 and fadD2 genes and PHA accumulation in P. putida U.
Table S1. Biomass and PHA production in P. putida U and the recombinant strains.
Table S2. Strains, mutants and plasmids used in this work.
Table S3. List of oligonucleotides used for the RT-PCR assay in this study.
References
- Arias S, Bassas M. Rediscovering biopolymers. In: Timmis KN, editor. Handbook of Hydrocarbon and Lipid Microbiology: Consequences of Microbial Interactions with Hydrocarbons Oils, Fats and Lipids. Berlin, Germany: Springer Verlag; 2010. pp. 2967–2980. [Google Scholar]
- Arias S, Sandoval A, Arcos M, Cañedo ML, Naharro G, Luengo JM. Poly-3-hydroxyalkanoate synthases from Pseudomonas putida U: substrate specificity and ultrastructural studies. Microb Biotechnol. 2008;1:170–176. doi: 10.1111/j.1751-7915.2007.00016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassas M. Isolation and analysis of storage compounds. In: Timmis KN, editor. Handbook of Hydrocarbon and Lipid Microbiology: Experimental Protocols and Appendices. Berlin, Germany: Springer Verlag; 2010. pp. 3725–3741. [Google Scholar]
- Bassas-Galia M, Nogales B, Arias S, Rohde M, Timmis KT, Molinari G. Plant original Massilia isolates producing polyhydroxybutyrate, including one exhibiting high yields from glycerol. J Appl Microbiol. 2012;112:443–454. doi: 10.1111/j.1365-2672.2011.05228.x. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- Cai L, Yuan MQ, Liu F, Jian J, Chen GQ. Enhanced production of medium-chain-length polyhydroxyalkanoates (PHA) by PHA depolymerase knockout mutant of Pseudomonas putida KT2442. Bioresour Technol. 2009;100:2265–2270. doi: 10.1016/j.biortech.2008.11.020. [DOI] [PubMed] [Google Scholar]
- Cheema S, Bassas-Galia M, Sarma PM, Lal B, Arias S. Exploiting metagenomic diversity for novel polyhydroxyalkanoate synthases: production of a terpolymer poly(3-hydroxybutyrate-co-3-hydroxyhexanoate-co-3-hydroxyoctanoate) with a recombinant Pseudomonas putida strain. Bioresour Technol. 2012;103:322–328. doi: 10.1016/j.biortech.2011.09.098. [DOI] [PubMed] [Google Scholar]
- Ciesielski S, Pokoj T, Klimiuk E. Cultivation-dependent and -independent characterization of microbial community producing polyhydroxyalkanoates from raw-glycerol. J Microbiol Biotechnol. 2010;20:853–861. doi: 10.4014/jmb.0909.09038. [DOI] [PubMed] [Google Scholar]
- Conte E, Catara V, Greco S, Russo M, Alicata R, Strano L, et al. Regulation of polyhydroxyalkanoate synthases (phaC1 and phaC2) gene expression in Pseudomonas corrugata. Appl Microbiol Biotechnol. 2006;72:1054–1062. doi: 10.1007/s00253-006-0373-y. [DOI] [PubMed] [Google Scholar]
- Diederich L, Roth A, Messer W. A versatile plasmid vector system for the regulated expression of genes in Escherichia coli. Biotechniques. 1994;16:916–923. [PubMed] [Google Scholar]
- Donnenberg MS, Kaper JB. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect Immun. 1991;59:4310–4317. doi: 10.1128/iai.59.12.4310-4317.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Eugenio LI, Escapa IF, Morales V, Dinjaski N, Galán B, García JL, Prieto MA. The turnover of medium-chain-length polyhydroxyalkanoates in Pseudomonas putida KT2442 and the fundamental role of PhaZ depolymerase for the metabolic balance. Environ Microbiol. 2010a;12:207–221. doi: 10.1111/j.1462-2920.2009.02061.x. [DOI] [PubMed] [Google Scholar]
- de Eugenio LI, Galán B, Escapa IF, Maestro B, Sanz JM, García JL, Prieto MA. The PhaD regulator control the simultaneous expression of the pha genes involved in polyhydroxyalkanoates metabolism and turnover in Pseudomonas putida KT2442. Environ Microbiol. 2010b;12:1591–1603. doi: 10.1111/j.1462-2920.2010.02199.x. [DOI] [PubMed] [Google Scholar]
- Galán B, Dinjaski N, Maestro B, de Eugenio LI, Escapa IF, Sanz JM, et al. Nucleoid-associated PhaF phasin drives intracellular location and segregation of polyhydroxyalkanoate granules in Pseudomonas putida KT2442. Mol Microbiol. 2011;79:402–418. doi: 10.1111/j.1365-2958.2010.07450.x. [DOI] [PubMed] [Google Scholar]
- García B, Olivera ER, Minambres B, Fernández-Valverde M, Cañedo LM, Prieto MA, et al. Novel biodegradable aromatic plastics from a bacterial source. Genetic and biochemical studies on a route of the phenylacetyl-CoA catabolon. J Biol Chem. 1999;274:29228–29241. doi: 10.1074/jbc.274.41.29228. [DOI] [PubMed] [Google Scholar]
- Grage K, Jahns AC, Parlane N, Palanisamy R, Rasiah IA, Atwood JA, Rehm BH. Bacterial polyhydroxyalkanoate granules: biogenesis, structure, and potential use as nano-/micro-beads in biotechnological and biomedical applications. Biomacromolecules. 2009;10:660–669. doi: 10.1021/bm801394s. [DOI] [PubMed] [Google Scholar]
- Harayama S, Rekik M, Wubbolts M, Rose KR, Leppik RA, Timmis KN. Characterization of five genes in the upper-pathway operon of TOL plasmid pWW0 from Pseudomonas putida and identification of the gene products. J Bacteriol. 1989;171:5048–5055. doi: 10.1128/jb.171.9.5048-5055.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haywood GW, Anderson AJ, Ewing DF, Dawes EA. Accumulation of a polyhydroxyalkanoate containing primarily 3-hydroxydecanoate from simple carbohydrate substrates by Pseudomonas sp. strain NCIMB 40135. Appl Environ Microbiol. 1990;56:3354–3359. doi: 10.1128/aem.56.11.3354-3359.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero M, de Lorenzo V, Timmis KN. Transposon vector containing non-antibiotic selection markers for cloning and stable chromosomal insertion of foreign DNA in gram-negative bacteria. J Bacteriol. 1990;172:6557–6567. doi: 10.1128/jb.172.11.6557-6567.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero M, de Lorenzo V, Ensley B, Timmis KN. A T7 RNA polymerase-based system for the construction of Pseudomonas strains with phenotypes dependent on TOL-meta pathway effecters. Gene. 1993;134:103–106. doi: 10.1016/0378-1119(93)90181-2. [DOI] [PubMed] [Google Scholar]
- Hoffmann N, Rehm BH. Nitrogen-dependent regulation of medium-chain length polyhydroxyalkanoate biosynthesis genes in Pseudomonads. Biotechnol Lett. 2005;27:279–282. doi: 10.1007/s10529-004-8353-8. [DOI] [PubMed] [Google Scholar]
- Huisman GW, Wonink E, Meima R, Kazemier B, Terpstra P, Witholt B. Metabolism of poly(3-hydroxyalkanoates) by Pseudomonas oleovorans. Identification and sequences of genes and function of the encoded proteins in the synthesis and degradation of PHA. J Biol Chem. 1991;266:2191–2198. [PubMed] [Google Scholar]
- Hume AR, Nikodinovic-Runic J, O'Connor KE. FadD from Pseudomonas putida CA-3 is a true long-chain fatty acyl coenzyme A synthetase that activates phenylalkanoic and alkanoic acids. J Bacteriol. 2009;191:7554–7565. doi: 10.1128/JB.01016-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DY, Kim HW, Chung MG, Rhee YH. Biosynthesis, modification, and biodegradation of bacterial medium-chain-length polyhydroxyalkanoates. J Microbiol. 2007;45:87–97. [PubMed] [Google Scholar]
- Kim TK, Jung YM, Vo MT, Shioya S, Lee YH. Metabolic engineering and characterization of phaC1 and phaC2 genes from Pseudomonas putida KCTC1639 for overproduction of medium-chain-length polyhydroxyalkanoate. Biotechnol Prog. 2006;22:1541–1546. doi: 10.1021/bp0601746. [DOI] [PubMed] [Google Scholar]
- Klinke S, de Roo G, Witholt B, Kessler B. Role of phaD in accumulation of medium-chain-length poly(3-hydroxyalkanoates) in Pseudomonas oleovorans. Appl Environ Microbiol. 2000;66:3705–3710. doi: 10.1128/aem.66.9.3705-3710.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraak MN, Kessler B, Witholt B. In vitro activities of granule bound poly-(R)-3-hydroxyalkanoate) polymerase C1 of Pseudomonas oleovorans. Development of an activity test for medium-chain-length-Poly(3-hydroxyalkanoate) polymerases. Eur J Biochem. 1997;250:432–439. doi: 10.1111/j.1432-1033.1997.0432a.x. [DOI] [PubMed] [Google Scholar]
- Luengo JM, García B, Sandoval A, Naharro G, Olivera ER. Bioplastics from microorganisms. Curr Opin Microbiol. 2003;6:251–260. doi: 10.1016/s1369-5274(03)00040-7. [DOI] [PubMed] [Google Scholar]
- Madison LL, Huisman GW. Metabolic engineering of poly(3-hydroxyalkanoates): from DNA to plastic. Microbiol Mol Biol Rev. 1999;63:21–53. doi: 10.1128/mmbr.63.1.21-53.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Blanco H, Reglero A, Rodríguez-Aparicio LB, Luengo JM. Purification and biochemical characterization of phenylacetyl-CoA ligase from Pseudomonas putida. A specific enzyme for the catabolism of phenylacetic acid. J Biol Chem. 1990;265:7084–7090. [PubMed] [Google Scholar]
- Neumann L, Spinozzi F, Sinibaldi R, Rustichelli F, Pötter M, Steinbüchel A. Binding of the major phasin, PhaP1, from Ralstonia eutropha H16 to poly(3-hydroxybutyrate) granules. J Bacteriol. 2008;190:2911–2919. doi: 10.1128/JB.01486-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera ER, Carnicero D, García B, Miñambres B, Moreno MA, Cañedo L, et al. Two different pathways are involved in the beta-oxidation of n-alkanoic and n-phenylalkanoic acids in Pseudomonas putida U: genetic studies and biotechnological applications. Mol Microbiol. 2001a;39:863–874. doi: 10.1046/j.1365-2958.2001.02296.x. [DOI] [PubMed] [Google Scholar]
- Olivera ER, Carnicero D, Jodra R, Miñambres B, García B, Abraham GA, et al. Genetically engineered Pseudomonas: a factory of new bioplastics with broad applications. Environ Microbiol. 2001b;3:612–618. doi: 10.1046/j.1462-2920.2001.00224.x. [DOI] [PubMed] [Google Scholar]
- Pieper-Fürst U, Madkour MH, Mayer F, Steinbüchel A. Identification of the region of a 14-kilodalton protein of Rhodococcus ruber that is responsible for the binding of this phasin to polyhydroxyalkanoic acid granules. J Bacteriol. 1995;177:2513–2523. doi: 10.1128/jb.177.9.2513-2523.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pötter M, Steinbüchel A. Poly(3-hydroxybutyrate) granule-associated proteins: impacts on poly(3-hydroxybutyrate) synthesis and degradation. Biomacromolecules. 2005;6:552–560. doi: 10.1021/bm049401n. [DOI] [PubMed] [Google Scholar]
- Prieto MA, Bühler B, Jung K, Witholt B, Kessler B. PhaF, a polyhydroxyalkanoate-granule-associated protein of Pseudomonas oleovorans GPo1 involved in the regulatory expression system for pha genes. J Bacteriol. 1999a;181:858–868. doi: 10.1128/jb.181.3.858-868.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto MA, Kellerhals MB, Bozzato GB, Radnovic D, Witholt B, Kessler B. Engineering of stable recombinant bacteria for production of chiral medium-chain-length poly-3-hydroxyalkanoates. Appl Environ Microbiol. 1999b;65:3265–3271. doi: 10.1128/aem.65.8.3265-3271.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quant J, Hynes MF. Versatile suicide vectors which allow direct selection for gene replacement in Gram negative bacteria. Gene. 1983;127:15–21. doi: 10.1016/0378-1119(93)90611-6. [DOI] [PubMed] [Google Scholar]
- Reis MA, Serafim LS, Lemos PC, Ramos AM, Aguair FR, Van Loosdrecht MC. Production of polyhydroxyalkanoates by mixed microbial cultures. Bioprocess Biosyst Eng. 2003;25:377–385. doi: 10.1007/s00449-003-0322-4. [DOI] [PubMed] [Google Scholar]
- Ren Q, de Roo G, Ruth K, Witholt B, Zinn M, Thöny-Meyer L. Simultaneous accumulation and degradation of polyhydroxyalkanoates: futile cycle or clever regulation? Biomacromolecules. 2009a;10:916–922. doi: 10.1021/bm801431c. [DOI] [PubMed] [Google Scholar]
- Ren Q, de Roo G, Witholt B, Zinn M, Thöny-Meyer L. Overexpression and characterization of medium-chain-length polyhydroxyalkanoate granule bound polymerases from Pseudomonas putida GPo1. Microb Cell Fact. 2009b;19:8–60. doi: 10.1186/1475-2859-8-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Q, de Roo G, Witholt B, Zinn M, Thöny-meyer L. Influence of growth stage on activities of polyhydroxyalkanoate (PHA) polymerase and PHA depolymerase in Pseudomonas putida U. BMC Microbiol. 2010;10:254. doi: 10.1186/1471-2180-10-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruth K, de Roo G, Egli T, Ren Q. Identification of two acyl-CoA synthetases from Pseudomonas putida GPo1: one is located at the surface of polyhydroxyalkanoates granules. Biomacromolecules. 2008;9:1652–1659. doi: 10.1021/bm8001655. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning. A Laboratory Manual. Cold Spring Harbor, NY, USA: CSHL Press; 2001. [Google Scholar]
- Sánchez-Romero JM, Díaz-Orejas R, de Lorenzo V. Resistance to tellurite as a selection marker for genetic manipulations of Pseudomonas strains. Appl Environ Microbiol. 1998;64:4040–4046. doi: 10.1128/aem.64.10.4040-4046.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval A, Arias-Barrau E, Bermejo F, Cañedo L, Naharro G, Olivera ER, Luengo JM. Production of 3-hydroxy-n-phenylalkanoic acids by a genetically engineered strain of Pseudomonas putida. Appl Microbiol Biotechnol. 2005;67:97–105. doi: 10.1007/s00253-004-1752-x. [DOI] [PubMed] [Google Scholar]
- Sandoval A, Arias-Barrau E, Arcos M, Naharro G, Olivera ER, Luengo JM. Genetic and ultrastructural analysis of different mutants of Pseudomonas putida affected in the poly-3-hydroxy-n-alkanoate gene cluster. Environ Microbiol. 2007;9:737–751. doi: 10.1111/j.1462-2920.2006.01196.x. [DOI] [PubMed] [Google Scholar]
- Selvaraj J, Iyer VN. Suicide plasmid vehicles for insertion mutagenesis in Rhizobium mellioti and related bacteria. J Bacteriol. 1983;156:1292–1300. doi: 10.1128/jb.156.3.1292-1300.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaiman DK, Ashby RD, Foglia TA. Effect of inactivation of poly(hydroxyalkanoates) depolymerase gene on the properties of poly(hydroxyalkanoates) in Pseudomonas resinovorans. Appl Microbiol Biotechnol. 2003;62:536–543. doi: 10.1007/s00253-003-1317-4. [DOI] [PubMed] [Google Scholar]
- Steinbüchel A, Aerts K, Babel W, Follner C, Liebergesell M, Madkour MH, et al. Considerations on the structure and biochemistry of bacterial polyhydroxyalkanoic acid inclusions. Can J Microbiol. 1995;41:94–105. doi: 10.1139/m95-175. [DOI] [PubMed] [Google Scholar]
- Timm A, Steinbüchel A. Cloning and molecular analysis of the poly(3-hydroxyalkanoic acid) gene locus of Pseudomonas aeruginosa PAO1. Eur J Biochem. 1992;209:15–30. doi: 10.1111/j.1432-1033.1992.tb17256.x. [DOI] [PubMed] [Google Scholar]
- Uchino K, Saito T, Gebauer B, Jendrossek D. Isolated poly(3-hydroxybutyrate) (PHB) granules are complex bacterial organelles catalyzing formation of PHB from acetyl coenzyme A (CoA) and degradation of PHB to acetyl-CoA. J Bacteriol. 2007;189:8250–8256. doi: 10.1128/JB.00752-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velázquez F, Pflüger K, Cases I, de Eugenio LI, de Lorenzo V. The phosphotransferase system formed by PtsP, PtsO, and PtsN proteins controls production of polyhydroxyalkanoates in Pseudomonas putida. J Bacteriol. 2007;189:4529–4533. doi: 10.1128/JB.00033-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek R, Pries A, Steinbüchel A, Mayer F. Analysis of a 24-kilodalton protein associated with the polyhydroxyalkanoic acid granules in Alcaligenes eutrophus. J Bacteriol. 1995;177:2425–2435. doi: 10.1128/jb.177.9.2425-2435.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York GM, Stubbe J, Sinskey AJ. The Ralstonia eutropha PhaR protein couples synthesis of the PhaP phasin to the presence of polyhydroxybutyrate in cells and promotes polyhydroxybutyrate production. J Bacteriol. 2002;184:59–66. doi: 10.1128/JB.184.1.59-66.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Genetic organization of the pha cluster and the expression system used for the hyperexpression of PhaC2 in PpU 10–33.
Fig. S2. Expression of the phaF, phaI and phaD genes and PHA accumulation in P. putida U.
Fig. S3. Expression of fadD1 and fadD2 genes and PHA accumulation in P. putida U.
Table S1. Biomass and PHA production in P. putida U and the recombinant strains.
Table S2. Strains, mutants and plasmids used in this work.
Table S3. List of oligonucleotides used for the RT-PCR assay in this study.