

Abstract
Sterol metabolites are critical signaling molecules that regulate metabolism, development, and homeostasis. Oxysterols, bile acids, and steroids work primarily through cognate sterol-responsive nuclear hormone receptors to control these processes through feed-forward and feedback mechanisms. These signaling pathways are conserved from simple invertebrates to mammals. Indeed, results from various model organisms have yielded fundamental insights into cholesterol and bile acid homeostasis, lipid and glucose metabolism, protective mechanisms, tissue differentiation, development, reproduction, and even aging. Here, we review how sterols act through evolutionarily ancient mechanisms to control these processes.
1. INTRODUCTION
Sterols are important for innumerable biological processes essential to reproduction and survival. The major source of sterols in mammals, cholesterol, is both synthesized de novo and acquired from the diet, and plays a crucial role in modulating cell membrane dynamics (1). In addition, cholesterol is metabolized into many physiologically important molecules, including the triumvirate of oxysterols, bile acids, and steroid hormones. It has become increasingly clear that sterols act in key signaling events to regulate both metabolic and developmental processes. Accordingly, the synthesis and metabolism of cholesterol is tightly regulated, reflecting the essential nature of these molecules.
Oxysterols, bile acids, and steroid hormones have distinct structures and functions. Oxysterols are oxygenated forms of cholesterol produced through both autoxidation and enzymatic reactions (2). Enzymatically derived oxysterols are important intermediates in steroid and bile acid synthesis, and they are considered indicators of endogenous cholesterol levels. Bile acids (BAs) are highly oxidized amphipathic molecules composed of a hydroxylated sterol core and a side chain carboxylic acid moiety (3). Produced in the liver and stored in the gall bladder as bile, they are required for dietary fat absorption. Finally, cholesterol is also metabolized into steroid hormones, including the classical steroids: mineralocorticoids, glucocorticoids, androgens, estrogens, and progestins. These molecules, derived from the common precursor pregnenolone in the adrenal glands and gonads, control many aspects of physiology, including ion balance, stress response, and various aspects of development and reproduction (4). Related molecules, the secosteroids, have a broken B ring structure catalyzed by photolysis in the skin. The most important secosteroid is 1,25-dihydroxyvitamin D3 [1,25-(OH)2D3], which acts to modulate calcium homeostasis as well as various cell growth, differentiation, and developmental processes (5).
Nuclear hormone receptors (NHRs) are transcription factors that regulate gene transcription in response to lipophilic ligands and are major players in sterol regulation and homeostasis. These receptors are highly conserved from invertebrates to mammals, characterized by having conserved paired zinc-finger DNA-binding domains as well as C-terminal ligand-binding domains that bind ligands and dock coregulators (6). Oxysterols, BAs, and steroid hormones activate NHRs to exert many of their effects. In particular, mammalian NHRs include a subcluster of evolutionarily related, metabolically active receptors, which partition cholesterol into oxysterol, BA, and steroid hormone pathways, and coordinate energy homeostasis, reproduction, and survival. These include the liver X receptor (LXR), farnesoid X receptor (FXR), pregnane X receptor (PXR), constitutive androstane receptor (CAR), and the vitamin D receptor (VDR), which act together with the heterologous binding partner retinoid X receptor (RXR) to transactivate target gene expression. In addition, model invertebrate organisms, such as Drosophila melanogaster and Caenorhabditis elegans, have conserved counterparts (Table 1). In Drosophila, the ecdysone receptor (EcR), activated by 20-hydroxyecdysone (20E), is responsible for morphological changes during larval development and for directing the life cycle (7). Fly DHR96 binds cholesterol and participates in cholesterol, lipid, and perhaps ecdysteroid metabolism (8, 9). In C. elegans, the NHR DAF-12 is activated by BA-like dafachronic acids (DAs) and regulates developmental progression, controlling components of the heterochronic circuit and the decision to undergo normal reproductive development or enter the dauer diapause, a state of arrested development (10). In particular, invertebrate studies have revealed unique insights into roles of sterol signaling on development, reproduction, and life span. Across phyla, sterol-activated NHRs link energy homeostasis with developmental programs in response to changing nutritional and environmental status. By acting as metabolic and environmental sensors, they govern the choice of programs geared toward survival and somatic endurance versus growth and reproduction. Here we review the literature on vertebrate and invertebrate receptors governing sterol homeostasis, and take a comparative approach to illuminate how the different systems inform one another regarding aspects of metabolism, physiology and organismal homeostasis, and what questions remain to be addressed.
Table 1.
The Sterol-Sensing Nuclear Hormone Receptors
| NHR | Systematic Name | Physiological Role(s) | Notable Ligandsa | Refs |
|---|---|---|---|---|
| Liver X Receptor (LXR) | NR1H3 (LXRα) NR1H2 (LXRβ) |
Cholesterol sensor and a key regulator of cholesterol, bile acid, and fatty acid homeostasis, in addition to roles in processes of differentiation and immunity. | Oxysterols, including 24(S),25-epoxycholesterol, 22(R)-hydroxycholesterol, 24(S)-hydroxycholesterol Bile Acids (LXRα), including Hyodeoxycholic Acid, Taurohyodeoxycholic Acid, and Cholestenoic Acid Synthetic Ligands include T0901317, GW3965 |
(12–17, 183) |
| Farnesoid X Receptor-α (FXRα) | NR1H4 | Bile acid sensor and master regulator of bile acid metabolism as well as cholesterol and fatty acid homeostasis. Also involved in hepatoprotection, liver regeneration, and inflammation. | Bile Acids, including Chenodeoxycholic Acid, Cholic Acid, Deoxycholic Acid, Lithocholic Acid Synthetic Ligands include GW4064 and 6E-CDCA |
(41) |
| Pregnane X Receptor (PXR) | NR1I2 | Xenobiotic receptor responsive to many compounds including bile acids, involved in hepatoprotection in addition to roles in the inflammatory response and metabolism. | Xenobiotics, including Pregnenolone-16α-carbonitrile (PCN), Phenobarbitol, Rifampicin, and Coumestrol Bile Acids, including Lithocholic and 3-keto-lithocholic Acid Various Steroid Hormones |
(56–57, 60) |
| Constitutive Androstane Receptor (CAR) | NR1I3 | Xenobiotic receptor involved in hepatoprotection as well as liver regeneration and various metabolic processes. | Xenobiotics, including Phenobarbitol, 1,4-Bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP), and 6-(4-Chloro-phenyl)imidazo[2,1b][1,3]thiazole-5-carbaldehyde O-3,4-dichlorobenzyl) oxime (CITGO) Various Steroid Hormones Lithocholic Acid (in vivo) |
(60–62) |
| Vitamin D Receptor (VDR) | NR1I1 | Primarily known as the master regulator of calcium homeostasis, but also involved in differentiation, immunity, and metabolic processes. | 1α,25-dihydroxy-vitamin D3 (Calcitriol) and various synthetic analogs Lithocholic Acid |
(5, 64–65) |
| Drosophila Ecdysone Receptor (EcR) | CG1765 | The Drosophila molting receptor, responsible for key processes of larval molting and metamorphosis. | 20-Hydroxyecdysone | (7) |
| Drosophila Hormone Receptor 96 (DHR96) | CG11783 | Proposed cholesterol sensor of the fly, regulating cholesterol and fatty acid metabolism. | Cholesterol | (8–9) |
| C. elegans DAF-12 | CE27584 | Regulator of the C. elegans dauer diapause decision, DAF-12 governs key developmental and metabolic events and is also required for gonadal longevity. | Dafachronic acids: 25(S),25-3-keto-4-Cholestenoic Acid and 25(S),26-3-keto-7-(5α)-Cholestenoic Acid Bile acid: 24(S)-Cholestenoic Acid |
(10,181, 182) |
Not a comprehensive list
2. STEROL-MEDIATED CONTROL OF MAMMALIAN ENERGY HOMEOSTASIS
2.1 Oxysterol Activation of the Liver X Receptor Regulates Cholesterol Metabolism
In mammalian cells, cholesterol levels are tightly controlled through regulation of endogenous biosynthesis, absorption from the diet, storage, catabolism to other sterols, and elimination. One component of this regulation includes the well-studied sterol regulatory element binding proteins (SREBPs), involved in regulation of cholesterol and fatty acid metabolism (11). Other key components include the LXRs, LXRα and LXRβ, which maintain cholesterol homeostasis through transcriptional control of genes involved in sterol dynamics and transport (Table 1) (12). LXRα is expressed in the liver and many other metabolically active tissues, including adipocytes, gonads, intestine, macrophages, and kidneys, whereas LXRβ is expressed in most tissues (12). These receptors are activated by oxysterols, the most potent of which are 22(R)-hydroxycholesterol, 24(S)-hydroxycholesterol, and 24(S),25-epoxycholesterol (12, 13). Other compounds that reportedly activate LXRs include isoprenoid precursors of cholesterol, BAs, and high concentrations of phytosterols and D-glucose (14–17).
Clear evidence of a role in cholesterol homeostasis came from analysis of LXRα knockouts, which display severe hepatomegaly owing to an accumulation of liver cholesterol esters when fed a high-cholesterol diet, as well as defects in BA metabolism (18). In mice, the LXRs positively regulate transcription of cholesterol 7α-hydroxylase (CYP7A1), which carries out the rate-limiting step in BA biosynthesis (18, 19). The consequent secretion of BAs provides one avenue for removal of excess cholesterol from the liver, the loss of which could explain several phenotypes of LXRα knockouts. More recent studies suggest that LXRα regulates other aspects of BA metabolism, including promotion of BA conjugation, which facilitates BA elimination by conversion into more hydrophilic compounds (20).
In addition to BA production, LXRs also regulate cholesterol homeostasis by controlling transcription of genes involved in cholesterol transport and storage. In response to oxysterols, several members of the ATP-binding cassette (ABC) transporter superfamily are upregulated, stimulating cholesterol efflux as well as resecretion of absorbed sterols into the intestine (21). Further suppression of dietary cholesterol absorption by LXR occurs through downregulation of the intestinal sterol transporter Niemann-Pick C1-like 1 (NPC1L1) (22). Additionally, LXR promotes expression of the cholesteryl ester transferase protein CETP, indirectly facilitating uptake of cholesteryl esters, storage forms of cholesterol, by the liver (23). Several cholesterol-accepting apolipoproteins and associated enzymes are upregulated by LXR, including hepatic apolipoprotein A-IV (ApoAIV) and macrophage apolipoprotein E, which promote cholesterol transfer to high-density lipoprotein (HDL) particles and have protective roles in atherogenesis (24). Accordingly, LXR agonists have antiatherogenic effects in mice, making LXR a target for treatment of this disease (25).
LXR also impacts subcellular cholesterol distribution through regulation of Niemann-Pick C1 (NPC1), which transfers sterols from the endosomal/lysosomal compartments to endoplasmic reticulum and plasma membrane. Mutations in NPC1 and NPC2 in humans cause Niemann-Pick type C (NPC) disease, a rare autosomal recessive syndrome in which sterols accumulate in late endosomal/lysosomal compartments, leading to progressive neurodegeneration and premature death. LXR activation with nonsteroidal agonists leads to upregulation of NPC1 (26, 27). Importantly, treatment with an LXR agonist delayed the demise of NPC1-null mice, whereas LXRβ loss accelerated disease progression (28). This suggests that Niemann-Pick pathology could partially arise from failure to activate LXR and that NPC1 normally facilitates production of bioactive LXR ligands (29).
Aside from BA synthesis, LXR may also divert cholesterol toward steroid hormone production. In the adrenal gland, LXR regulates the steroidogenic acute regulatory protein (StAR) and cytochrome P450 11A1 (CYP11A1) (30, 31). Catalyzing the rate-limiting step in steroidogenesis, StAR transports cholesterol from the outer to the inner mitochondrial membrane, and CYP11A1 functions downstream, converting cholesterol to pregnenolone. Conflicing reports have shown both increased and decreased expression of StAR and CYP11A1 in the adrenal gland upon treatment of mice with different synthetic LXR agonists, suggesting agonist-specific effects (30, 31). The regulation of StAR by LXR complements other studies showing oxysterols stimulate StAR expression in specific cell types (32). Paradoxically, LXR knockouts display elevated levels of plasma glucocorticoids, which might be caused by derepression of StAR (30, 31). Further studies are needed to elucidate the role of LXR in the adrenal steroid production.
Evidence suggests that LXR may also influence gonadal steroidogenesis. Both LXRα and LXRβ are expressed in ovaries and testes, and LXR deficiency is associated with decreased fertility in mice of both sexes, although only LXRβ affects male fertility (33). LXR agonists reportedly inhibit luteal cell production of progesterone in the ovary and downregulate both StAR and CYP11A1 (34). Nevertheless, mice lacking LXR have unaltered progesterone or estradiol levels, but they are defective in resumption of meiosis during oocyte maturation, which may partially explain decreased fertility (35). LXRαβ knockouts also exhibit an attenuated response to meiosis resumption by gonadotropins. Interestingly, a precholesterol metabolite that induces meiotic resumption, follicular-fluid meiosis-activating sterol, weakly activates LXR, correlating with a possible role of LXR in this process (36). In addition, LXR may impact fertility through activities in the placenta and uterus, although further in vivo studies are needed to support these functions (33). In males, loss of LXRβ leads to impaired Sertoli cell function and decreased spermatogenesis attributed to accumulation of testicular cholesterol droplets (37, 38). LXRαβ-null mice display decreased androgen production, and treatment with the T0901317 agonist leads to increased testosterone in the testes of wild-type mice and increased expression of StAR (39). Thus, LXRs may act to couple sterol availability with reproductive function and fertility, although whether LXR influences reproductive development itself remains undetermined. Interestingly, the C. elegans DAF-12 homolog of LXR regulates aspects of reproductive development, which we discuss later, and it will be interesting to see whether its mammalian counterparts are involved in similar processes.
2.2 Bile Acid Sensors Coordinate Cholesterol and Bile Acid Homeostasis
The synthesis of bile acids from cholesterol is critical to cholesterol homeostasis and essential to dietary lipid absorption. Moreover, BAs themselves are toxic and therefore must be tightly regulated. The BA pool is maintained through control of synthesis, recycling in enterohepatic circulation, catabolism, and excretion. Synthesis occurs in the liver through two pathways, the classical neutral pathway and the acidic pathway, involving similar steps: initiation by 7α-hydroxylation, ring structure modifications, oxidation and shortening of the side chain, and amino acid conjugation to facilitate export to the gall bladder (40). Upon food ingestion, the gall bladder secretes BA-containing bile into the intestine, where the BAs act upon fat. From there, most are reabsorbed by the intestine and return to the liver through the portal vein (3).
The BA sensor farnesoid X receptor-α (FXRα) is the master regulator of BA physiology. Several endogenous BAs bind and activate FXRα, most potently chenodeoxycholic acid (CDCA) and its conjugated derivatives (41). FXRα is expressed at high levels in tissues where bile salts are transported, including the liver, intestine, and kidney. Other compounds that activate FXRα with lower affinity include farnesol and related metabolites, androsterone, polyunsaturated fatty acids, and oxysterols as well as other intermediates in BA synthesis, although their physiological roles are less defined. Consistent with a role in BA homeostasis, mice lacking FXRα display severe alterations in BA metabolism (42–44). Notably, when challenged with dietary cholic acid, they experience hepatotoxicity and wasting. Plasma BA levels are elevated, along with cholesterol and triglycerides, whereas gall bladder levels are reduced relative to wild-type mice. FXRα regulates expression of genes involved in almost every aspect of BA physiology, including promotion of BA conjugation to facilitate secretion (e.g., UDP glucuronosyltransferase UGT2B4), export into the bile canaliculli (through transporters including the bile salt export pump BSEP and the ABC transporters MRP2/ABCB4 and MDR3/ABCC2), modulation of import into the liver from circulation (e.g., the sodium taurocholate cotransporting polypeptide NTCP and the organic anion transporter polypeptides OATPs), and control of reabsorption in the intestine (the intestinal BA-binding protein I-BABP, apical sodium-dependent BA transporter ASBT, and organic solute transporter OSTα/β), whereas activation of fibroblast growth factor (mouse FGF15/human FGF19) controls gall bladder filling to regulate bile release into the intestine (45, 46). Additionally, FXRα acts in coordination with PXR, CAR, and VDR in hepatoprotection from BA toxicity through regulation of genes involved in BA detoxification, including hydroxylation, export, and sulfation (47).
A series of elegant experiments over the past decade has yielded molecular insights into how BAs regulate their own synthesis through feedback repression (40). In response to BA excess, FXRα acts to repress BA synthesis primarily through downregulation of cholesterol 7α-hydroxylase (CYP7A1) and sterol 12α-hydroxylase (CYP8B1) (45), cytochrome P450 enzymes essential for BA synthesis, in a manner opposing regulation by LXRα. Repression is achieved by upregulation of the small heterodimer partner (SHP), a noncanonical nuclear receptor, lacking a DNA-binding domain, that works as a transcriptional repressor. SHP inactivates several NHRs responsible for either CYP7A1 or CYP8B1 expression, including LXR, liver receptor homolog-1 (LRH-1, NR5A2), and hepatocyte nuclear factor-4α (HNF4α) (48–50). FXRα also orchestrates a hormonal feedback mechanism originating in the intestine and circulating back to the liver, which involves fibroblast growth factor (mouse FGF15/human FGF19) (51–53). In the ileum, FXRα directly activates FGF15/19, which binds to cognate coreceptors in the liver: FGFR4, a receptor tyrosine kinase, and β-klotho, a single-pass transmembrane protein. Downstream, MAP kinase cascades repress hepatic CYP7A1 expression. Thus, FXRα acts in opposition to LXR in the regulation of BA synthesis (Figure 1A). There is currently intense research aimed at uncovering further details of these fascinating signaling networks.
Figure 1. Pleiotropic actions of mammalian sterol-sensing nuclear hormone receptors (NHRs) on energy homeostasis.
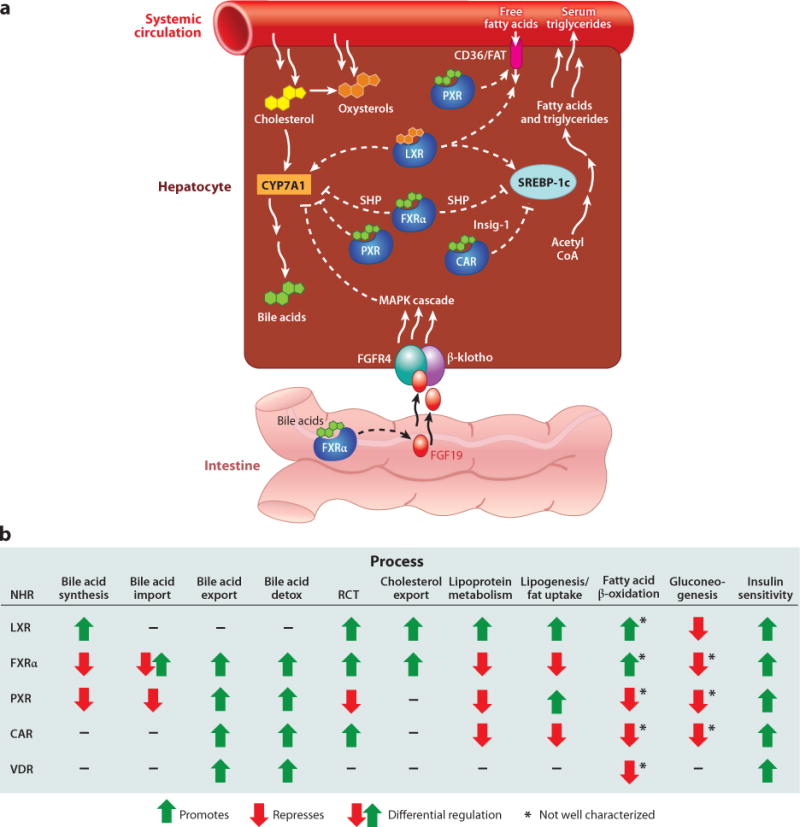
(A) Differential regulation of select components of bile acid (BA) synthesis and lipogenesis by the sterol-sensing NHRs. LXR and FXRαact in an opposing manner upon expression of the CYP7A1 cholesterol 7α-hydroxylase, impacting BA synthesis, and the SREBP-1c transcription factor, which regulates fatty acid and triglyceride synthesis. LXR and PXR also positively regulate the fatty acid translocase CD36/FAT, promoting uptake of free fatty acids. (B) Comparison of the roles of sterol-sensing NHRs in various metabolic processes demonstrates the pleiotropic actions of these receptors. CAR, constitutive androstane receptor; FGF19, fibroblast growth factor 19 (mouse FGF15); FXRα, farnesoid X receptor-α; Insig-1, an endoplasmic reticulum-associated membrane protein that prevents proteolytic activation of SREBP; LXR, liver X receptor; PXR, pregnane X receptor; SHP, small heterodimer partner; RCT, reverse cholesterol transport; VDR, vitamin D receptor.
FXRα also mediates aspects of cholesterol transport, including reverse cholesterol transport and efflux. FXRα-null mice display elevated plasma levels of low-density lipoprotein, very low-density lipoprotein (VLDL), and HDL-associated cholesterol and triglycerides (43, 54). FXRα loss also leads to decreased cholesterol efflux, owing in part to decreased expression of ABC transporters involved in the elimination of hepatic cholesterol as well as resecretion into the intestine. FXRα also upregulates phospholipid transfer proteins and differentially regulates several apolipoproteins to favor overall reduction of plasma lipids. Accordingly, treatment with FXRα ligands reduces serum HDL and VLDL cholesterol as well as triglyceride levels (55).
Best known for its roles in xenobiotic metabolism, PXR has a flexible ligand-binding domain that binds structurally diverse hydrophobic compounds, including several BAs (56, 57). PXR is expressed in overlapping tissues with LXR and FXRα, including the liver, intestine, and kidneys. PXR has also been implicated in feedback regulation of BA synthesis through repression of CYP7A1, but in a FXRα- and SHP-independent manner (57). In contrast to FXRα, PXR positively regulates lipoprotein levels, as PXR activation leads to increased levels in wild-type but not PXR-null animals (58). In vitro studies suggest this may be partly mediated by downregulation of ABCA1 and SR-B1, which would result in reduced HDL uptake and efflux (59). Thus, PXR and FXRα both act to inhibit BA synthesis, but in opposition to influence lipoprotein distribution.
Another xenobiotic receptor, CAR, impacts several metabolic pathways and is activated by many of the same ligands as PXR (60). Although not directly binding BAs, CAR is activated in vivo by treatment with lithocholic acid (61). CAR is highly expressed in the liver and at lower levels in the intestine and stomach (62). Similar to FXRα, CAR activation decreases plasma lipoprotein levels in wild-type but not CAR−/− mice. Unlike FXRα-null mice, CAR knockouts exhibit normal levels of serum lipoproteins, unless fed a high fat diet (63).
VDR is also activated by lithocholic acid and regulates some of the same xenobiotic detoxification genes as CAR and PXR (64). VDR is expressed in a wide variety of tissues, where it exerts its many physiological effects (5, 65). However, relatively little is known about the effect of VDR on cholesterol metabolism. VDR-null mice demonstrate decreased levels of serum cholesterol and triglycerides relative to wild-type, and upon feeding a high-fat diet, show slower growth rates and accumulate less fat (66), although these studies may be complicated by the use of high-calcium diets to avoid hypocalcemia. Additional studies are needed to investigate the possible roles of VDR as well as the other BA-activated receptors in modulating cholesterol and BA homeostasis as well as to learn how they might interact to regulate these processes.
In addition to NHRs, BAs activate several other pathways, including mitogen-activated protein kinase (MAPK) and G protein–coupled receptor signaling. BAs activate all three MAPK signaling pathways (ERK, JNK, and p38/MAPK) (67, 68), and in particular, the JNK pathway may mediate FGF15/19 repression of BA synthesis. BAs also activate AKT, a key protein kinase involved in the insulin signaling pathway, possibly through induction of Gαi receptors. One G protein–coupled receptor activated by BAs is TGR5, which mediates BA-protective processes and promotes insulin secretion (69, 70). Accordingly, BA treatment attenuates diet-induced obesity and insulin resistance in mice and also induces mitochondrial activity and energy expenditure in human skeletal muscle and murine brown adipose tissue. This is dependent upon TGR5 and the thyroid hormone-activating type 2 iodothyronine deiodinase, further implicating BAs in regulating energy mobilization.
2.3 Sterol-Sensing Nuclear Hormone Receptors Modulate Lipid Metabolism
In concert with cholesterol homeostasis, both LXRs also regulate fatty acid metabolism, primarily through regulation of SREBP-1c, a master regulator of fatty acid and triglyceride biosynthesis (71). Excess cholesterol or synthetic ligands of LXR induce SREBP-1c expression and fatty acid synthesis, which is absent in LXRαβ-null mice. These animals are resistant to diet-induced obesity, display increased metabolic rates and decreased lipogenesis, and fail to induce SREBP-1c expression in response to cholesterol or insulin (71, 72). The LXRs also upregulate the carbohydrate-responsive element-binding protein (ChREBP), which independently regulates lipogenic gene expression in response to glucose (73). Furthermore, the LXRs directly upregulate enzymes involved in fatty acid synthesis and lipoprotein remodeling, including fatty acid synthase, lipoprotein lipase, and the fatty acid transfer protein, CD36 (74, 75). These studies demonstrate a link between cholesterol uptake and fatty acid synthesis, implicating LXR as a key regulator in this balance.
In contrast, the LXRs may facilitate fatty acid β-oxidation in muscle and adipocytes by upregulating pyruvate dehydrogenase kinase 4 (PDK4) (76, 77). PDK4 inhibits the activity of the pyruvate dehydrogenase complex, which represses glycolysis in favor of fatty acid oxidation, shifting the balance of energy derived from glucose to fat. LXRα also reportedly promotes hepatic peroxisomal β-oxidation of very long-chain fatty acids, possibly to counter increased triglyceride levels occurring upon its activation, but this remains unclear (78).
FXRα regulates lipid metabolism in a manner opposite that of LXR (Figure 1A). FXRα-null mice suffer from increased levels of plasma triglycerides, lipoproteins, and cholesterol, in addition to a fatty liver (54). Notably, activation of FXRα represses SREBP-1c expression via SHP, downregulates genes involved in lipoprotein metabolism, and lowers triglyceride levels (55). Similarly in humans, oral intake of chenodeoxycholic acid decreases plasma triglycerides (79). Furthermore, activation of FXRα leads to decreased secretion of VLDL from the liver and, together with the repression of SREBP-1c and fatty acid synthesis, indicates the importance of FXRα in the repression of hepatic lipogenesis (80). In short, the phenotypes of FXRα-null mice can be described as proatherogenic, and the triglyceride- and cholesterol-lowering effects of FXRα agonists make them attractive therapeutic candidates. Finally, FXRα may also influence β-oxidation in a manner similar to LXR by inducing PDK4, possibly through upregulation of the peroxisome proliferator-activated receptor α (PPARα), an important regulator of fatty acid oxidation (81).
Other BA-activated receptors also impact lipid metabolism, although the underlying mechanisms are not fully understood. For PXR, activation is generally associated with enhanced lipogenesis and suppressed fatty acid β-oxidation. Expression of constitutively activated PXR leads to increased triglyceride levels, hepatomegaly, fatty liver, and lipogenesis, but in an SREBP-1c-independent manner (82). Instead, activated PXR stimulates expression of fatty acid translocase CD36 and several other lipogenic enzymes. PXR also directly transactivates PPARγ, a key regulator of adipogenesis and lipogenesis (75). Conversely, PXR represses PPARα expression as well as inhibits forkhead transcription factor FoxA2, repressing rate-limiting genes in β-oxidation (82, 83).
Activation of CAR in mice also appears to modulate genes involved in fatty acid β-oxidation but not in CAR-null animals, although conflicting reports describe either repression or induction of this process, making additional studies necessary (84–87). In contrast, both PXR and CAR can suppress lipogenesis through activation of Insig-1, an endoplasmic reticulum-associated membrane protein that prevents proteolytic activation of SREBP (88).
VDR may also inhibit β-oxidation as this occurs at elevated rates in the white adipose tissue of VDR-null mice (66). Increased expression of several uncoupling proteins (UCPs) was noted in these animals. Accordingly, treatment of primary brown adipocyte cultures with 1,25-(OH)2D3 downregulated UCP expression. The UCPs dissipate energy as heat in brown adipose tissue during the process of adaptive thermogenesis, the regulated production of heat in response to environmental stimuli, and thus presumably lead to higher energy expenditure in the absence of VDR.
2.4 Glucose Metabolism and the Hepatic Fasting Response
Several sterol-activated NHRs also impact energy homeostasis through regulation of glucose metabolism. As discussed above, LXRs regulate SREBPs, which promote the conversion of glucose to fatty acids. LXR agonists improve glucose tolerance in a mouse model of diet-induced obesity in the liver and adipose tissue, and suppress hepatic gluconeogenesis (89). In support of these findings, LXRαβ-null mice fail to suppress gluconeogenesis in response to agonist treatment, and mice deficient in LXRβ display reduced glucose tolerance (90, 91). Furthermore, in response to glucose or synthetic agonists the LXRs also promote insulin secretion by pancreatic β-cells, although no changes in insulin sensitivity have been detected in the absence of the LXRs (91, 92). High concentrations of D-glucose and D-glucose-6-phosphate also reportedly activate the LXRs, but the physiological relevance is unclear (17).
In response to fasting or starvation, the liver modulates several metabolic processes to maintain blood glucose levels: Glycogenosis and gluconeogenesis increase glucose levels, fatty acid oxidation provides energy for gluconeogenesis and substrates for ketogenesis, and attenuation of lipogenesis shifts fatty acids from storage to utilization. LXRα knockouts exhibit an impaired response to fasting, with delayed mobilization of hepatic glycogen stores (93). Upon refeeding, induction of hepatic lipogenesis is also reduced, secondary to reduced SREBP activity. Interestingly, LXRαβ-null mice display cholesterol-dependent elevation of thyroid hormone levels and a corresponding increased energy expenditure through upregulation of UCPs (72). Thus, LXR may coordinate the balance between energy accretion and mobilization depending upon dietary conditions.
FXRα influences glucose metabolism through several targets, including genes involved in gluconeogenesis, glycolysis, and glycogen synthesis (94, 95). FXRα-null mice display impaired glucose tolerance and insulin resistance, although reports conflict on the glucose levels and altered expression of gluconeogenic genes in these mice (95–97). In response to fasting, FXRα-null mice fail to induce gluconeogenesis to the same extent as wild-type, suggesting FXRα is required for its upregulation (96). By contrast, FXRα agonists suppress gluconeogenic genes in FXRα- and SHP-dependent manners (97). Knockout mice also display decreased insulin sensitivity in peripheral white adipose and muscle tissues (95). It is interesting to note that FXRα is expressed in adipose tissue and may promote preadipocyte differentiation. Moreover, treatment of adipocytes with an FXRα agonist enhances glucose uptake in response to insulin (95, 98). Similar to LXR, FXRα agonists also improve glucose tolerance in a diabetic mouse model and promote insulin secretion in pancreatic β-cells (95, 99). As might be expected, FXRα-deficient mice exhibit defective fasting responses, characterized by transient hypoglycemia and impaired hepatic glucose production (100). Mutants also fail to lower serum triglycerides in response to fasting, possibly because of defects in fat utilization. Finally, fasting FXRα knockouts have an increased susceptibility to enter torpor (101), a state of decreased physiological activity induced by stress, characterized by reduced body temperature and metabolic rates to conserve energy. By inference, FXRα may promote adaptive thermogenesis, the regulated production of heat, through increased UCP expression. These data suggest that FXRα, like LXR, plays a role in the regulation of energy homeostasis and mobilization in response to changing nutritional conditions. Speculatively, torpor might be analogous to diapause states of arrest in invertebrates, occuring in response to stress and in which altered metabolism and energy conservation are hallmarks. As we will discuss in detail later, the invertebrate sterol-sensing NHRs play a key role in regulating these states.
PXR and CAR also appear to influence glucose metabolism and the fasting response. Treatment of mice with corresponding agonists represses hepatic expression of gluconeogenic enzymes as well as improves insulin sensitivity in both diabetic animal models and human subjects (60, 84, 87, 102). Provision of PXR agonists reportedly decreases serum glucose levels in fasting wild-type mice and decreases expression of β-oxidation and ketogenesis components (83). Interestingly, CAR and its target genes are induced during the fasting response, and CAR-deficient mice exhibit a defective response (103, 104). CAR may act in an NHR network that governs the fasting response, as two NHRs, PPARα and HNF4α, activate CAR in this context. In addition, dramatic weight loss occurs in CAR-deficient animals during long-term caloric restriction, presumably from reduced degradation of the thyroid hormone and a resultant dysregulation of metabolic rates (103). Accordingly, fasting or treatment with a CAR agonist induces thyroid hormone-metabolizing enzymes, depending upon CAR. CAR may thus function to attenuate the fasting response by dampening gluconeogenesis, fatty acid β-oxidation, and energy utilization.
Finally, although little is known about its roles in glucose homeostasis, VDR deficiency is associated with decreased insulin production and a corresponding increased risk for diabetes in both animal models and humans (66, 105). In a similar manner, vitamin D deficiency leads to decreased glucose tolerance and pancreatic insulin secretion. These effects may arise from misregulation of pancreatic β-cell insulin secretion and glycolysis by altered calcium concentrations. As mentioned above, VDR may repress adaptive thermogenesis through downregulation of UCPs, although its association with the hepatic fasting response and torpor has not been studied.
In summary, the mammalian sterol-sensing NHRs have pleiotropic roles governing energy homeostasis and control the balance between energy accretion and mobilization (Figure 1B). Through the control of key metabolic pathways, they have an important impact on organismal health and disease.
3. STEROL-ACTIVATED NUCLEAR HORMONE RECEPTORS MODULATE INFLAMMATION AND IMMUNITY
The immune system naturally functions to protect the animal throughout life, yet when dysregulated can lead to immunosuppression or inflammation, with deleterious consequences. LXR plays a central role in regulating inflammation and innate immunity in several contexts. In response to bacterial infection, LPS, or cytokine exposure, the inflammatory response is induced in macrophages, resulting in expression of interleukins and chemokines. Oxysterol activation of LXR inhibits expression of several of these genes, including iNOS, IL-6 and IL-1βin macrophages derived from wild-type but not LXR-null mice (106). After challenge with LPS, LXR knockouts reportedly display an enhanced systemic inflammatory response and increased hepatic expression of inflammatory mediators, although conflicting studies of primary macrophages from these animals describe decreased expression (107, 108). The antiinflammatory effects of LXR are proposed to stem largely from suppression of NF-κB signaling (107). Recent studies reveal that sumoylated LXR stabilizes NCoR corepressor complexes residing at promoters of NF-κB dependent genes, thereby preventing their expression (109). Similarly, derepression of the hepatic inflammatory response is seen in FXRα-null mice, and ligand activation of FXRα, PXR, or PPARγ also results in transrepression of NF-κB targets via sumoylation and NCoR stabilization. Thus, these receptors share a common mechanism by which they modulate the inflammatory response (110–114). In addition, the heterodimeric partner of these receptors, RXR, is regulated by inflammatory mediators, possibly an important aspect of the common involvement of sterol-sensing NHRs in this response (115). Corresponding with a role in innate immunity, LXR-deficient animals are susceptible to intracellular Listeria monocytogenes infection, while FXRα-null animals display increased intestinal bacterial levels and susceptibility to development of colitis (114, 116). Moreover, the coupling of xenobiotic receptors with immunity may help explain how compounds such as polyphenols have beneficial effects on inflammation and metabolism (117).
Sterol-sensing NHRs are also involved in regulating acquired immunity. Mice deficient in LXRβ display age-associated lymphoid hyperplasia and T cells derived from these mice exhibit enhanced proliferation after antigenic stimulus (118). Conversely, activation of LXRβ inhibits the proliferation of cultured lymphocytes, by driving ABC-mediated cholesterol efflux, which in turn inhibits the cell cycle (119). Similarly, PXR-deficient mice also display increased T lymphocyte proliferation, while PXR activation has the opposite effect (120). Lastly, VDR also modulates the immune response, targeting multiple cell types of the immune system, extensively reviewed elsewhere (121). It will be interesting to see whether corresponding invertebrate receptors also influence immunity.
4. DEVELOPMENTAL ROLES OF MAMMALIAN STEROL-SENSING NUCLEAR HORMONE RECEPTORS
4.1 Oxysterols and the Liver X Receptor Regulate Differentiation
In addition to modulation of energy homeostasis, oxysterols promote differentiation in a variety of tissues. Among these processes, the best described role of oxysterols is the regulation of skin keratinocyte differentiation. Keratinocytes comprise the majority of cells in the epidermis, arising from a stem cell population located in the basal proliferative layer. These cells divide and differentiate into progeny that migrate upward in a continuous process of renewal, eventually dying and forming a cornified outer layer. Oxysterol ligands of LXR induce expression of keratinocyte differentiation markers (e.g., involucrin) and lipid synthesis while suppressing proliferation, depending upon LXRβ, the primary isoform found in the epidermis of the mouse (122). This is due in part to upregulation of components of the AP-1 protein complex (including c-Fos and jun-D), which regulate genes required for keratinocyte differentiation. LXRβ−/− mice display thin epidermal layers and have reduced levels of proliferating keratinocytes, which are refractory to differentiation after topical treatment with oxysterols. In addition, skin from these mice has similarities in gene expression profiles to photoaged and chronologically aged skin, and treatment with LXR agonists reduces UV-induced skin thickening and wrinkle formation in hairless mice (123). Thus, LXR activation may protect against age-related skin damage, making it a promising antiaging target.
Recent evidence reveals that oxysterols and LXR regulate dopaminergic neurogenesis (124). In LXRαβ-deficient mice, the number of progenitor neurons in the ventral midbrain is reduced, leading to a decrease in dopaminergic neurons. Accordingly, overexpression of LXRβ or treatment with oxysterol agonists enhances neurogenesis, although this is not seen upon treatment of LXR-deficient progenitor cells. Loss of LXR leads to defective cell-cycle progression of progenitor cells at the G2/M phase and a corresponding failure of differentiation (Figure 2A). Although LXRs have been implicated in regulation of several cell-cycle genes, the mechanism by which LXR exerts cell-cycle control of neural progenitors, and how it acts specifically on this subset of neurons, remains unclear. In addition, LXRβ and double knockout mice exhibit age-associated lipid accumulation in the brain, an abnormal blood-brain barrier, and neural degeneration (125, 126). The LXRs thus appear to both promote neurogenesis during early development as well as prevent neural degeneration with age.
Figure 2. Sterols act through conserved pathways to mediate developmental processes.
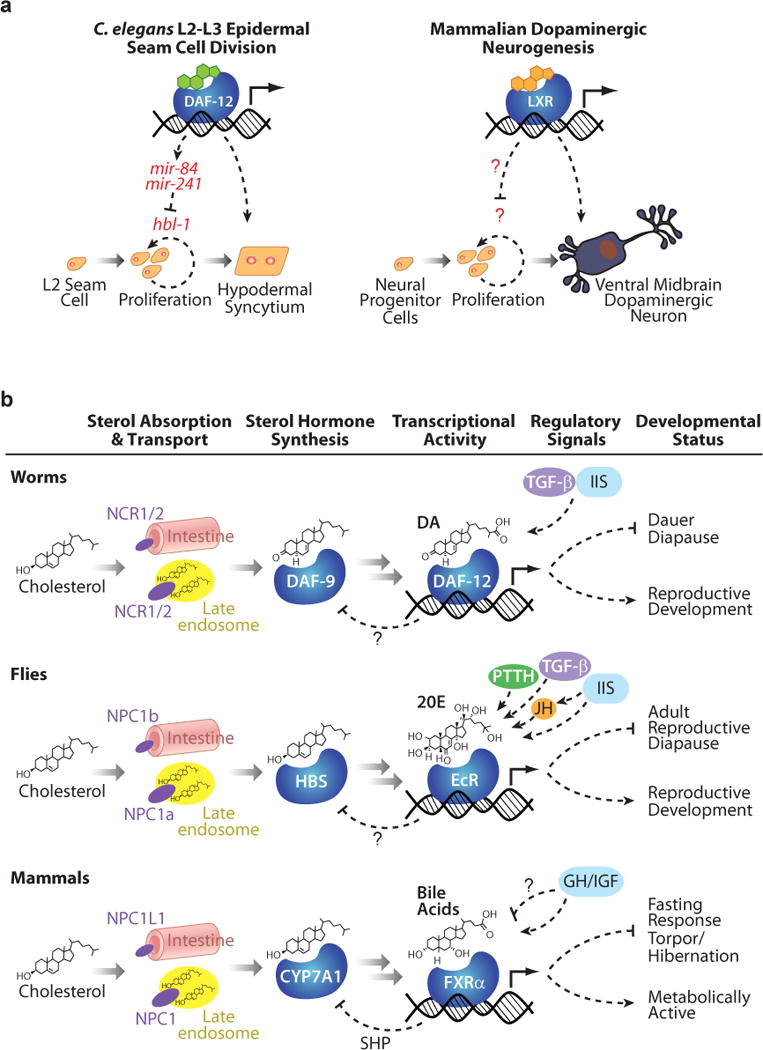
(A) In specific contexts, both C. elegans DAF-12 and the mammalian liver X receptor (LXR) act to inhibit proliferation of precursor cells to promote terminal differentiation. (B) Across phyla, similar mechanisms of cholesterol absorption and transport, sterol biosynthesis, and transcriptional regulation by sterol-sensing nuclear hormone receptors mediate decisions between energy-conserving states and normal reproductive development. Upstream endocrine networks, notably insulin/insulin-like growth factor-1 signaling, impact the production of sterol ligands, and thus activities of these receptors, although many mechanistic details remain to be uncovered. CYP7A1, a cytochrome P450 which acts at the rate-limiting step in mammalian BA synthesis; DA, dafachronic acid; DAF-9, C. elegans homolog of mammalian BA synthetic enzyme CYP27A1 required for DA production; DAF-12, C. elegans DA-activated nuclear hormone receptor; EcR, ecdysone receptor; GH, growth hormone; HBS, hormone biosynthesis; IGF, insulin-like growth factor; IIS, insulin/IGF-I Signaling; JH, juvenile hormone; mir-84, -241, targets of DAF-12 that are involved in developmental timing and seam cell differentiation during the L2-L3 stage in C. elegans; NCR1/2, C. elegans homologs of the Niemann-Pick C1 and Niemann-Pick C1-Like-1 sterol transporters; NPC1a, Drosophila homolog of Niemann-Pick C1; NPC1b, Drosophila homolog of Niemann-Pick C1-Like-1; NPC1, mammalian Niemann-Pick C1 sterol transporter; NPC1L1, mammalian Niemann-Pick C1-Like-1 sterol transporter; PTTH, prothoraciotropic hormone, TGF-β, transforming growth factor β.
Oxysterols also function in the osteogenic differentiation of multipotent bone marrow stromal cells. When exposed to 20(S)-hydroxycholesterol in combination with 22(S)- or 22(R)-hydroxycholesterol, a strong induction of osteogenic gene expression and cell matrix mineralization are seen (127, 128). This is reportedly mediated through activation of hedgehog signaling (129, 130), while additional studies suggest that activation of LXR may actually inhibit this signaling (131). Further studies in vivo are needed to clarify roles of oxysterols and LXR in osteogenesis.
4.2 Bile Acid-Activated Nuclear Hormone Receptors Promote Liver Regeneration
The liver is unique in its ability to undergo growth and repair after injury to restore normal size, followed by the return of cells to quiescence. Regeneration is induced by several signaling events, including secretion of cytokines and growth factors such as transforming growth factor α (TGF-α), IL-6, and tumor necrosis factor α (132). BAs, acting through FXRα, play an important role in the regeneration process. Notably, diets enriched with 0.2% cholic acid accelerate regeneration, and diets containing a BA-sequestering resin impede it (133). These effects are not observed in FXRα-null mice in which liver regeneration is retarded. Thus, BAs, acting through FXRα, are regeneration-promoting signals, which are normally required for this process. One mechanism underlying regeneration is the activation of Forkhead Box transcription factor M1b (FoxM1b), which regulates cell-cycle progression during liver regeneration and is a direct target of FXRα (134). Interestingly, hepatic FoxM1b expression declines with age, whereas increased expression of FoxM1b or activation of FXRα restores regenerative potential (135). FXRα may thus be a target for therapeutics aimed at enhancing liver regeneration after transplant or surgery and during aging. Paradoxically, FXRα knockout mice are more susceptible to spontaneous liver tumors (41). It is thought that FXRα normally plays a role in hepatoprotection from BA toxicity, damage, oxidative stress and inflammation, which when absent could contribute to tumorigenesis. One potential mechanism by which some of these effects might be mediated is through upregulation of CAR, which can be tumorogenic in the presense of agonists such as phenobarbitol (136–138).
Other BA-responsive receptors also appear to promote liver regeneration. PXR activation stimulates hepatocyte proliferation and liver growth, and PXR-null mice display impaired regeneration after partial hepatectomy (139, 140). Similarly, CAR−/− mice exhibit delayed hepatocyte proliferation after partial hepatectomy, and vitamin D deficiency compromises hepatic regeneration (133, 141). Whether these receptors are specifically regulated by BAs for this process is currently unknown. In addition to liver regeneration, VDR has been implicated in diverse developmental processes governing calcium homeostasis, bone biology, nervous system and skeletal development, immunity, skin differentiation, and hair follicle cycling, which are extensively reviewed elsewhere (5).
5. MAMMALIAN STEROL-SENSING NUCLEAR HORMONE RECEPTORS AND LIFE SPAN
Aging entails a decline in metabolic, tissue, and organismal homeostasis. As major regulators of these processes, sterol-sensing NHRs are bound to play a role. Few studies have examined their effects on life span, although a growing body of evidence suggests they impact aging and age-related diseases. For example, LXR may play a role in ameliorating Alzheimer’s disease (AD). In an amyloid precursor protein transgenic mouse model, loss of LXRα or LXRβ increased amyloid plaque formation, a key component of AD pathogenesis; however, treatment with an LXR agonist decreased plaque formation and improved cognitive ability, possibly owing to increased cholesterol flux (142, 143). Interestingly, different human LXR polymorphisms associate with either increased risk of AD or increased life span, rendering LXR a potential target for treatment of age-related pathologies (144, 145).
Vitamin D production also declines with age, and VDR influences aging (146). Mice deficient in VDR exhibit decreased longevity and phenotypes reminiscent of premature aging, including skin wrinkling, alopecia, muscle atrophy, osteoporosis, decreased immunity, and deficiencies in hearing and balance. Knockout of 25-hydroxyvitamin D3 1α-hydroxylase, required for vitamin D synthesis, results in similar phenotypes (147). Mice deficient in either the transmembrane protein Klotho or FGF-23, which modulate vitamin D homeostasis, also display progeroid phenotypes (148). Interestingly, these mutants have extremely high circulating levels of vitamin D, or hypervitaminosis D, and their phenotypes are rescued by vitamin D-deficient diets or genetic ablation of VDR. In contrast, transgenic mice overexpressing Klotho exhibit increased longevity and resistance to oxidative stress, possibly through decreased insulin/insulin-like growth factor I signaling (IIS) (149). The adverse effects of both hypo- and hypervitaminosis D reveal that the appropriate balance of vitamin D is essential to avoid age-related pathologies.
Finally, sterol-sensing NHRs may invoke protective mechanisms that extend survival, and perhaps life span. Xenobiotic metabolism protects the animal through elimination of toxic compounds, and several studies suggest that upregulation of these pathways may be beneficial and correlate with longevity. In C. elegans, transcriptional profiles of long-lived daf-2 insulin/IGF-1 receptor mutants revealed upregulation of numerous genes involved in xenobiotic detoxification, postulated to act as a means of longevity assurance (150). Similar upregulation has been reported in several long-lived mouse models, including calorie-restricted mice, Snell dwarf mice, and Little mice (151, 152). In Little mice, deficient in growth hormone releasing hormone receptor, this is associated with increased hepatotoxin resistance, partially dependent upon FXRα (153). Interestingly, these mice display increased BA levels, and treatment of wild-type mice with cholic acid induces a similar profile of xenobiotic detoxification genes. These studies suggest that BAs may act through FXRα to induce the xenobiotic response and impact life span. In the nematode C. elegans, BA-like steroids acting through the FXRα homolog, DAF-12, promote increased longevity in the absence of the germline, discussed in detail below (154). Microarray studies indicate that genes involved in xenobiotic metabolism are indeed altered in long-lived daf-12(rh273) ligand-binding domain mutants (155). Conceivably, the conserved pathways of BA signaling may stimulate xenobiotic protection and enhance longevity, although causal support for this hypothesis remains to be found (Figure 2B).
6. REQUIREMENT OF STEROLS BY INVERTEBRATE MODEL ORGANISMS
Work in both C. elegans and D. melanogaster has revealed critical roles for sterols in these model organisms. As cholesterol auxotrophs, these animals must obtain exogenous cholesterol from the diet. In C. elegans, cholesterol depletion for one generation leads to defects in molting, growth, gonadal morphogenesis, and germline proliferation and differentiation, and depletion for additional generations leads to larval arrest (156). Similarly in the fly, larvae grown in cholesterol-deficient conditions also arrest early in larval development because cholesterol is required for molting, growth, and reproductive development (157). The striking phenotypes seen upon cholesterol depletion in both worms and flies demonstrates that although these organisms are unable to produce cholesterol, it is an essential component of the diet required for normal development.
Although obviously important molecules, relatively little is known about the sterol metabolome of either nematodes or insects. Both harbor sterol metabolites derived from dietary cholesterol, some of which are also found in mammals, such as 7-dehydrocholesterol (158, 159). The ecdysteroids and derivatives have been extensively studied in insects, but oxysterols and BAs have not yet been identified. In the worm, the BA-like DAs work through the DAF-12 NHR to regulate dauer diapause, developmental timing, fat metabolism, and life span (10). However, the full spectrum of BAs and their functions remain unexplored, and no oxysterols are yet described. As these endogenous molecules regulate aspects of metabolism, development and even longevity through conserved signaling pathways, understanding the sterol metabolome of these organisms could potentially lead to identification of novel mediators of health and life span.
7. CONSERVED STEROL-SIGNALING PATHWAYS MEDIATE DEVELOPMENTAL PROCESSES
A key role of sterols in C. elegans and D. melanogaster is the regulation of molting and developmental timing events. Nematodes and arthropods develop through larval stages separated by molts when the exoskeleton is shed and replaced. In Drosophila, the cholesterol derivative 20E regulates molting and morphogenesis during larval and pupal development (7). Ecdysteroid pulses during early larval stages induce molting, and later pulses initiate key morphological events of the prepupa and pupa. Mutations in genes of ecdysone synthesis confer severe larval arrest phenotypes, demonstrating the importance of this hormone in development (159). The effects of 20E are mediated by a heterodimer complex of NHRs, the ecdysone receptor (EcR) and ultraspiracle (USP) [a homolog of mammalian RXR], which initiate transcription of genes critical for life cycle progression (7). The synthesis of ecdysone itself is also hormonally regulated by IIS, TGF-β and prothoraciotropic hormone signaling, which are subject to environmental and nutritional cues, coupling external signals with molting and growth (7, 160) (Figure 2B).
A number of downstream NHRs are activated by the ecdysone cascades in a stage-specific manner to control specific molting programs, including Drosophila hormone receptor 3 (DHR3), Fushi tarazu-F1 (Ftz-F1), and DHR38. DHR38, a relative of the mammalian NGF-1B class of NHRs, forms heterodimers with USP/RXR and is regulated by a combination of ecdysteroid and RXR agonists (161). Interestingly, DHR38 lacks a conventional ligand-binding pocket and is thought to be ligand regulated through other structural interactions.
Recent studies suggest that DHR96, the Drosophila homolog of DAF-12, binds cholesterol and regulates cholesterol homeostasis and triglyceride metabolism similar to vertebrate LXR (8). Mutants arrest as second instar larvae in low-cholesterol conditions and accumulate cholesterol upon provision of excess. Moreover, numerous genes show cholesterol-dependent regulation through DHR96, although critical evidence confirming cholesterol as a bona fide ligand is lacking. DHR96 targets include the fly ortholog of NPC1-L1, implicated in intestinal sterol absorption, and homologs of NPC2. Mutations in the NPC1 or NPC2 genes lead to defective sterol trafficking and decreased ecdysteroid titers (162). Although developmental arrest phenotypes of these mutants can be rescued with 20E, those of DHR96 mutants cannot, suggesting that DHR96 has a more global role in sterol homeostasis. DHR96 also impacts fat metabolism, as mutants are starvation sensitive and have reduced triglycerides, whereas overexpressors are starvation resistant and have increased triglycerides (9). These phenotypes arise partly from regulation of a gastric lipase implicated in dietary fat absorption. The similarities between DHR96 and LXR are striking examples of conservation of cholesterol and lipid homeostasis mechanisms.
Although sterols are implicated in C. elegans molting, edysteroids have not been identified, and homologs of EcR and USP are not present. Nevertheless, components tied to sterol metabolism or transport impact this process. Intriguingly, mutations in apl-1, a homolog of amyloid precursor protein implicated in AD, functions in nematode molting and may work through cell nonautonomous mechanisms via release of its ectodomain (163). The low-density lipoprotein receptor-like protein LRP-1 is required for molting, and loss of proteins homologous to NPC1 and NPC1L1, thought to be involved in sterol transport, induces sensitivity to cholesterol depletion (164, 165). Importantly, two NHRs, NHR-23 and NHR-25, promote molting and are orthologous to the ecdysone-regulated DHR-3 and Ftz-F1 in Drosophila (166, 167). Despite the discovery of these components and others, the active hormones, signaling cascades, transport mechanisms, and cognate receptors governing molting remain unidentified.
An important developmental process regulated by sterols in both C. elegans and Drosophila is diapause, a stress-resistant state of arrested development and reproductive quiescence induced by stressful conditions, possibly analogous to mammalian torpor and hibernation. In flies, adult reproductive diapause (ARD) is important for overwintering (168). C. elegans can enter diapause at multiple points including larval L1, L3, and adult stages in response to starvation cues, but the L3 dauer diapause has been most intensively studied (10). Diapause has a major impact on organismal life history, significantly increasing overall life span. Upon return of favorable conditions, animals exit diapause and resume reproductive growth, revealing enormous phenotypic plasticity in response to environmental and nutritional conditions. This physiology may reflect a trade-off between programs geared toward growth and reproduction versus extended survival.
In Drosophila, ARD is characterized by arrested ovarian development, decreased metabolism, increased lipid deposition, resistance to stress, and increased longevity (169). A product of sesquiterpenoid biosynthesis, juvenile hormone (JH) regulates many physiological processes, including metamorphosis, all major aspects of reproduction, and ARD. In response to insulin-like signals, JH is produced in the corpora allata gland and is downregulated during ARD. Removal of the corpora allata during adulthood induces ARD, whereas the JH analog methoprene stimulates diapause recovery. Flies in ARD suppress ovarian ecdysteroid synthesis as well as vitellogenesis in the fat body. Reduced insulin signaling may also be responsible in promoting diapause: heteroallelic mutations in the insulin/IGF-I receptor provoke phenotypes reminiscent of ARD, including increased stress resistance and longevity, suppression of JH and ecdysteroid biosynthesis, and arrest of vitellogenesis and reproduction. Moreover, methoprene reverses several phenotypes of these mutants, including longevity. Animals heterozygous for mutations in EcR display increased longevity, revealing that 20E also has life-shortening effects (170). Both JH and ecdysone can restore vitellogenesis of animals in diapause, implying both hormones stimulate diapause exit (169). USP, the RXR-like heterodimeric partner of EcR, has actually been proposed as a receptor for JH, although this remains controversial (171). Overall, JH and 20E evidently act together to regulate a trade-off between reproduction and longevity, promoting increased reproductive capacity at the expense of long life.
A recent discovery suggests that C. elegans also has ARD, induced by fasting under crowded conditions (172). Upon removal from food during the mid-L4 stage, resultant adults halt reproduction while harboring two arrested embryos inside the uterus, trim back the germline, and undergo gonadal regression. Worms in ARD can survive weeks of starvation yet when returned to favorable conditions will rejuvenate the germline and other tissues, produce small broods, and live a normal life span. Little is known of the physiologic and molecular events underlying ARD except that it depends partly on the nuclear receptor NHR-49, which functions similarly to mammalian PPARα in regulating fat metabolism, β-oxidation, and response to food deprivation (173). It will be interesting to see if C. elegans ARD displays metabolic and molecular features comparable to Drosophila ARD or mammalian torpor.
Best understood in C. elegans is the dauer diapause, a developmental program executed during the third larval stage in response to stressful conditions of high temperature, low food, sterol depletion, or crowding during early larval development (10). As in the diapausing female fly, dauer larvae display increased lipid accumulation, stress resistance, and longevity. In addition, dauer larvae undergo dramatic morphological changes, including remodeling of the neural system and pharynx, bodily constriction, and secreting a thickened stress-resistant cuticle and cuticle plug over the mouth.
Early evidence for a sterol-derived hormone regulating dauer formation came from the observation that depletion of cholesterol for more than one generation induced dauer formation, revealing that cholesterol promotes nondauer reproductive growth (156, 174). Furthermore, defects in putative sterol transport proteins induced dauer formation, including mutations in ncr-1 and ncr-2, homologs of both NPC1L1 and NPC1, as well as mrp-1, a multidrug-resistant protein (165, 175). Perhaps the most critical insights arose, however, from systematic genetic analyses of mutations in genes that govern dauer formation, called daf genes, which revealed that the dauer decision is mediated by an endocrine network, including IIS, cGMP, TGF-β, serotonergic, and steroid hormone signaling (10, 176).
Among these genes, daf-12 encodes an NHR related to mammalian LXRs, FXRα, and VDR, which ultimately mediates the dauer decision (177). daf-12 null mutants are dauer defective and cannot enter the dauer diapause. Genetic epistasis placed daf-12 downstream of IIS, cGMP, and TGF-β signaling, as daf-12 null mutations suppressed dauer-constitutive (Daf-c) mutations in all three pathways (176, 178, 179). In contrast, mutations in the daf-12 ligand-binding domain confer Daf-c phenotypes, suggesting that loss of the DAF-12 ligand induces dauer diapause (177). Accordingly, animals deficient in DAF-9, a cytochrome P450 related to mammalian steroidogenic enzymes, also constitutively form dauers, which can recover into sterile, stress-resistant adults that are long-lived (174, 180). daf-12 null mutations completely suppress daf-9 phenotypes, suggesting that a hormone produced by DAF-9 acts upon DAF-12 to promote reproductive development and prevent dauer formation.
Indeed, biochemical analysis revealed that DAF-9 produces BA-like 3-keto steroids called ∆4- and ∆7-DA, which work as high-affinity ligands for DAF-12, with ∆7-DA being more potent (181). A related compound, 25(S)-cholestenoic acid, binds DAF-12 as well as its mammalian relative LXR, albeit with lower affinity (182, 183). It is unknown if different ligands have distinct physiological roles and transcriptomes. DA rescues all known daf-9 phenotypes including constitutive dauer formation, gonadal morphogenesis defects, and longevity. Importantly, by “deorphanizing” DAF-12, these studies identified the first ligands for a NHR in the worm and provided evidence that BAs regulate animal development and longevity.
The current paradigm for dauer formation suggests that, in favorable conditions, environmental and internal signals are integrated via neurosensory processing, resulting in stimulation of IIS and TGF-β signaling (10). These pathways converge in downstream endocrine tissues to induce production of DAs, which activate DAF-12 throughout the body, promoting reproductive development. In unfavorable conditions, DAs are not produced, and DAF-12 works in a corepressor complex with DIN-1, the homolog of mammalian SHARP, to repress reproductive programs and specify dauer formation and longevity (184). This working model has served as a paradigm for understanding how environmental cues are converted to global hormonal signals that regulate an organismal decision between reproduction and survival, but many questions remain. What is the nature of the hormone biosynthetic pathway? How is it regulated by environmental and signaling inputs? How are inputs integrated to mediate the dauer decision? What are the downstream outputs for stress resistance and longevity?
Further studies have begun to elucidate the DA biosynthetic pathways. On the basis of sterols and hormone biosynthetic genes identified in the worm, a branched biosynthetic pathway has been proposed, starting with dietary cholesterol and ending with the ∆7- and ∆4-DAs (185). The first step in the ∆7 branch is carried out by DAF-36, a Rieske-like oxygenase, proposed to work as a cholesterol ∆7-desaturase, converting cholesterol to 7-dehydrocholesterol. Remarkably, the Drosophila homolog Neverland works similarly in the first steps of ecdysteroid biosynthesis (186). Although mammalian homologs of daf-36 do not exist, the first modification in BA synthesis also occurs with hydroxylation by CYP7A1 at the 7 position, suggesting that this residue may play a critical and conserved role in initialization. Downstream, a proposed 5α-reduction to lathosterol and further 3β-dehydrogenation to lathosterone might be achieved by several conserved enzymes present in the worm. HSD-1, a 3β-hydroxysterol dehydrogenase homolog, is proposed to work in the ∆4 branch, converting cholesterol to 4-cholestene-3-one, although biochemical evidence remains to be found (187). The last step in both of the ∆4 and ∆7 pathways is catalyzed by DAF-9, which carries out successive side chain oxidations to create the carboxylic acid moiety, much like its mammalian counterpart CYP27A1 in BA biosynthesis (181). The conservation of these components in nematodes, flies, and mammals suggests an ancient origin for the partition of sterols toward steroid and BA production (Figure 2B).
Another similarity to mammals is the importance of feedback regulation by NHRs. As mentioned above, mammalian LXR promotes feed-forward synthesis of BAs, and FXRα regulates negative feedback through modulation of CYP7A1 expression. Similarly, DAF-12 regulates DA synthesis through both feed-forward and feedback mechanisms by modulating hypodermal expression of DAF-9/CYP27A1 (188, 189). DAF-9 is expressed constitutively in a pair of neuroendocrine cells, called XXXL/R, from early larval development onward and in the hypodermis from mid-L2 at the point of commitment to reproductive growth. On the basis of DAF-9::GFP expression studies, it is inferred that tonic levels of DA released from XXXL/R are amplified and distributed in the hypodermis upon commitment to reproductive growth. In particular, hypodermal DAF-9 expression is dependent upon daf-12 and DA. Although modestly expressed in wild-type animals, this expression is absent in daf-12 and daf-9 null mutants, whereas such expression is strongly upregulated upon moderately diminished DA production, occurring with mutations in IIS, TGF-β, and daf-36/Rieske (185, 188, 189). These observations suggest that when tonic DA levels are absent, DAF-12 represses or fails to activate hypodermal DAF-9, driving dauer development. When levels are low, DAF-12 amplifies hypodermal DAF-9 in a process of positive feedback, promoting reproductive development, whereas when tonic levels are high, DAF-12 decreases hypodermal DAF-9 expression through negative feedback. This complex regulation is thought to tightly control DA levels in a switch-like mechanism, thus ensuring an all-or-none dauer decision and maintaining proper hormone levels for reproductive development. How DAF-12 regulates DAF-9 in this switch, directly or through target gene expression, and what mechanisms mediate communication between DAF-9 expressing tissues remains unknown. Interestingly, like daf-12 null animals, FXRα-null mice display decreased CYP27A1 levels and fail to downregulate CYP27A1 upon BA provision, demonstrating defective feedback mechanisms and the conservation of this regulation (43).
Most nematode species are capable of entering similar diapause states, including pathogenic nematodes. Notably, these states often correspond to the infective stage; thus, targeting entry and exit from this stage is of interest in creating antihelminthic therapeutics. DAF-12 signaling is remarkably conserved in several parasitic nematodes, and DA treatment of the infective stages of several species, including the canine hookworm Ancylostoma caninum, the bacteriovorous beetle parasite Pristionchus pacificus, as well as the human pathogen Strongyloides stercoralis, promotes diapause exit (190, 191). In S. stercoralis, ∆7-DA prevents the initial formation of infective larvae, suggesting that selective DAF-12 modulators could be used to treat these infections. Evidently DAF-12 signaling has also been co-opted to regulate the dimorphic trait of mouth denticles in P. pacificus (192). Upon starvation, these nematodes are predisposed to develop mouth structures used for predation, a switch regulated by DAF-12/DA signaling, revealing an ancient role of this pathway in coupling environmental cues to developmental plasticity. Additionally, a unique feature of nematode sterol metabolism is the production of 4-methyl sterols, an activity ascribed to the STRM-1 methyltransferase thought to partition sterols away from DA biosynthesis (193). Mammals lack this activity, opening the possibility of targeting this enzyme in antihelminthics.
In addition to the dauer decision, DAF-12 regulates the heterochronic circuit, a regulatory hierarchy that specifies the temporal identity of each stage during larval development. DAF-12 generally regulates progression from larval stage L2 to L3, and various daf-12 mutants repeat L2 programs during the L3 stage in various tissues, most notably affecting the division patterns of epidermal seams, larval stem cells that undergo specific asymmetric divisions at each stage (194). Several direct transcriptional targets of DAF-12 mediate these events, including members of the let-7 family of microRNAs: mir-84 and -241 (195). These microRNAs specify L3 stage programs by downregulating hbl-1/HUNCHBACK, which encodes a zinc finger protein that specifies the earlier L2 stage (196). It is proposed that in the presence of DA, DAF-12 upregulates these microRNAs, which in turn repress hbl-1 and L2 programs, thereby promoting the L3 fate (195). Conversely, in the absence of DA, these microRNAs are repressed and animals enter the dauer diapause. Thus, DAs regulate the L2/L3 transition as well as the L2/L3 dauer transition through modulation of DAF-12 activity. By working at the convergence of dauer and heterochronic pathways, DAF-12 couples environmental and global hormonal signals to developmental progression in specific cells and tissues. The role of DAF-12 in this process may be analogous to that of mammalian LXR in promoting developmental progression in dopaminergic neurogenesis or skin differentiation described above (Figure 2A). Interestingly, let-7 homologs in mammals and flies act in differentiation and metamorphosis, but it remains to be seen whether DAF-12 homologs regulate let-7 microRNAs to control these processes (197, 198).
Similar to mammalian and Drosophila homologs, DAF-12 has also been implicated in lipid metabolism. Mutations in daf-9/CYP27A1 lead to increased fat storage in L2 stage larvae preparing for dauer (referred to as L2d), dependent upon DAF-12 and its corepressor DIN-1/SHARP (174, 184). Other Daf-c mutants, such daf-2 insulin/IGF-I receptor mutants, also display increased fat deposition, but in a daf-12-independent manner. Surprisingly, under reproductive growth conditions, mutations in din-1 alone also lead to increased fat accumulation, dependent upon daf-12. Conceivably, the DAF-12/DIN-1 corepressor complex suppresses fat storage in reproductive conditions but promotes fat storage during dauer-inducing conditions. Microarray studies indicate that genes involved in lipid metabolism are misregulated in daf-12 mutants, but additional molecular and biochemical studies are needed to clarify this role (150).
8. DAF-12 MODULATES C. ELEGANS LONGEVITY IN MULTIPLE CONTEXTS
Aside from regulation of worm life span through larval dauer formation, DAF-12 also regulates adult longevity in various contexts. One such context is life span regulation in response to temperature. C. elegans and many other ectotherms live longer at lower temperatures and shorter at higher ones (199). Although shorter life at higher temperatures is typically ascribed to increased metabolic rates and consequent damage, evidence indicates that this process is regulated in the worm. Temperature sensation in C. elegans is mediated by the AFD neurons, a pair of bilateral neurons required for movement to temperatures previously associated with food (200). Loss of AFD function leads to an even shorter life span in response to increased temperature, depending upon daf-12, possibly owing to decreased DA production. Accordingly, DAF-9 expression in wild-type animals is increased at higher temperatures, depending upon AFD function, suggesting that thermal sensation by these neurons acts to oppose earlier death at high temperature by upregulating DA and DAF-12 activity. Conversely, at lower temperatures, daf-9 null mutants are longer-lived depending on daf-12 and din-1. DAF-12/DA signaling may thus act in response to temperature to catalyze homeostatic mechanisms that maintain metabolism, and hence life span, within normal bounds.
DAF-12/DA also regulates adult life span in response to signals from the reproductive system, termed gonadal longevity. Animals lacking germline stem cells, resulting from ablation by laser microsurgery or genetic manipulation, live up to 50–60% longer than wildtype (201). Longevity is not the result of sterility alone as animals lacking both somatic gonad and germline have normal life spans, suggesting that signals originating from these tissues act antagonistically to regulate longevity. Genetic analysis reveals that gonadal longevity depends on the hormone biosynthetic genes daf-36/Rieske, daf-9/CYP27A1, and daf-12, as well as daf-16/FOXO, a forkhead transcription factor that promotes longevity and extended survival in response to reduced IIS (174, 185, 201). Germline ablation further extends the long life of daf-2/InsR mutant animals, suggesting that the gonadal pathway and IIS regulate DAF-16/FOXO in parallel. As predicted, treatment of germline ablated daf-9 and daf-36 mutants with DA restores longevity but does not influence wild-type or daf-12 mutants, indicating that DA works through DAF-12 to promote longevity in animals without a germline (154). The longevity signal emanating from the somatic gonad likely involves DA itself because animals lacking the whole gonad (somatic gonad and germline) are not long-lived unless supplemented with DA (202).
What mechanisms lie downstream of DA signaling to influence life span? A molecular correlate of gonadal longevity is the translocation of DAF-16/FOXO into the nucleus of intestinal cells, presumably initiating target gene expression (203, 204). DA signaling and DAF-12 appear to facilitate nuclear localization and activity of DAF-16/FOXO because mutants abrogate nuclear localization (154, 203). However, DAF-16 nuclear localization alone is not sufficient for life span extension and requires DAF-12 as expression of a constitutively nuclear-localized DAF-16 does not increase life span of daf-12 germline-ablated animals. Interestingly, DAF-16/FOXO targets include fatty acid/cholesterol lipases whose overexpression extends life, whereas loss of function abrogates extension (205). This remarkably suggests that free fatty acids or cholesterol, through hormonal signals or metabolism, impact organismal life span. Although increased attention has unveiled some mechanisms involved in gonadal longevity, many questions remain. What are the repressive signals from the germline, and are there other components of the longevity-promting somatic gonad signal? How do the DA-controlled DAF-12 pathway and DAF-16 interact to promote gonadal longevity, and how are they regulated? What are the downstream targets of DAF-12 that are required for gonadal longevity?
Recent evidence suggests that gonadal longevity may be evolutionarily conserved. In Drosophila, ectopic misexpression of bag of marbles in the germline leads to germline stem cell loss and life span extension (206). Although little is known about the underlying molecular mechanisms, this may arise from impaired IIS because insulin-like peptides and binding proteins are misregulated. In mice, transplantation of ovaries from young mice to older, ovariectomized mice leads to an increased median life span compared with nontransplanted ovariectomized mice or fertile controls, suggesting that the gonad can also control mammalian longevity (207). Further studies should uncover the molecular mechanisms underlying these observations, which include the possibility of sterol signaling pathways. It will be interesting to see if the mammalian sterol-sensing NHRs are also components of this process to influence longevity. The increasing parallels between invertebrate and mammalian sterol signaling pathways make this an exciting possiblity.
SUMMARY POINTS.
Sterols are partitioned away from cholesterol and toward biosynthesis of oxysterols, bile acids, and steroid hormones.
Conserved sterol-sensing NHR-mediated pathways govern cholesterol partitioning through feed-forward and feedback circuits.
Sterol-sensing NHRs couple energy balance with protective and developmental processes, ultimately ensuring survival and reproductive success in the face of changing environments and nutritional states.
In vertebrates, sterol-sensing receptors regulate multiple metabolic pathways to impact energy homeostasis, including cholesterol, lipid, and glucose metabolism, as well as modulating the fasting response and possibly torpor.
Studies in mammals demonstrate protective roles of these receptors in immunity, hepatoprotection, liver regeneration, and metabolic homeostasis, as well as governing various differentiation processes and possibly influencing longevity.
Invertebrate model organisms have provided critical insights into the impact of sterols and corresponding signaling pathways on development, survival and life span.
FUTURE ISSUES.
The vertebrate sterol signaling pathways have illuminated various aspects of metabolism, whereas invertebrate studies have shed light on development and life span. The challenge for the future is to utilize comparative biology and evolutionary conservation to further elucidate the roles of sterol metabolites and their receptors as well as to answer several complementary questions:
What functions do vertebrate sterol receptors have in reproduction, differentiation, developmental timing, stress management, and longevity?
What is the invertebrate sterol metabolome? And how might their corresponding NHRs couple metabolism with development and reproduction?
To what extent does conservation exist between animal models and humans at molecular, mechanistic, and physiologic levels?
The answers to these and other questions are bound to yield fundamental insights into basic biology, with important implications for human health and disease.
References
- 1.Simons K, Ikonen E. Science. 2000;290:1721–6. doi: 10.1126/science.290.5497.1721. [DOI] [PubMed] [Google Scholar]
- 2.Gill S, Chow R, Brown AJ. Prog Lipid Res. 2008;47:391–404. doi: 10.1016/j.plipres.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Redinger RN. Am J Surg. 2003;185:168–72. doi: 10.1016/s0002-9610(02)01212-6. [DOI] [PubMed] [Google Scholar]
- 4.Hu J, Zhang Z, Shen WJ, Azhar S. Nutr Metab (Lond) 2010;7:47. doi: 10.1186/1743-7075-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dusso AS, Brown AJ, Slatopolsky E. Am J Physiol Renal Physiol. 2005;289:F8–28. doi: 10.1152/ajprenal.00336.2004. [DOI] [PubMed] [Google Scholar]
- 6.Magner DB, Antebi A. Trends Endocrinol Metab. 2008;19:153–60. doi: 10.1016/j.tem.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.King-Jones K, Thummel CS. Nat Rev Genet. 2005;6:311–23. doi: 10.1038/nrg1581. [DOI] [PubMed] [Google Scholar]
- 8.Horner MA, Pardee K, Liu S, King-Jones K, Lajoie G, et al. Genes Dev. 2009;23:2711–6. doi: 10.1101/gad.1833609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sieber MH, Thummel CS. Cell Metab. 2009;10:481–90. doi: 10.1016/j.cmet.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fielenbach N, Antebi A. Genes Dev. 2008;22:2149–65. doi: 10.1101/gad.1701508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Espenshade PJ, Hughes AL. Annu Rev Genet. 2007;41:401–27. doi: 10.1146/annurev.genet.41.110306.130315. [DOI] [PubMed] [Google Scholar]
- 12.Zhao C, Dahlman-Wright K. J Endocrinol. 2010;204:233–40. doi: 10.1677/JOE-09-0271. [DOI] [PubMed] [Google Scholar]
- 13.Janowski BA, Grogan MJ, Jones SA, Wisely GB, Kliewer SA, et al. Proc Natl Acad Sci U S A. 1999;96:266–71. doi: 10.1073/pnas.96.1.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forman BM, Ruan B, Chen J, Schroepfer GJ, Jr, Evans RM. Proc Natl Acad Sci U S A. 1997;94:10588–93. doi: 10.1073/pnas.94.20.10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song C, Hiipakka RA, Liao S. Steroids. 2000;65:423–7. doi: 10.1016/s0039-128x(00)00127-6. [DOI] [PubMed] [Google Scholar]
- 16.Plat J, Nichols JA, Mensink RP. J Lipid Res. 2005;46:2468–76. doi: 10.1194/jlr.M500272-JLR200. [DOI] [PubMed] [Google Scholar]
- 17.Mitro N, Mak PA, Vargas L, Godio C, Hampton E, et al. Nature. 2007;445:219–23. doi: 10.1038/nature05449. [DOI] [PubMed] [Google Scholar]
- 18.Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, et al. Cell. 1998;93:693–704. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- 19.Lehmann JM, Kliewer SA, Moore LB, Smith-Oliver TA, Oliver BB, et al. J Biol Chem. 1997;272:3137–40. doi: 10.1074/jbc.272.6.3137. [DOI] [PubMed] [Google Scholar]
- 20.Barbier O, Trottier J, Kaeding J, Caron P, Verreault M. Mol Cell Biochem. 2009;326:3–8. doi: 10.1007/s11010-008-0001-5. [DOI] [PubMed] [Google Scholar]
- 21.Kalaany NY, Mangelsdorf DJ. Annu Rev Physiol. 2006;68:159–91. doi: 10.1146/annurev.physiol.68.033104.152158. [DOI] [PubMed] [Google Scholar]
- 22.Duval C, Touche V, Tailleux A, Fruchart JC, Fievet C, et al. Biochem Biophys Res Commun. 2006;340:1259–63. doi: 10.1016/j.bbrc.2005.12.137. [DOI] [PubMed] [Google Scholar]
- 23.Luo Y, Liang CP, Tall AR. J Biol Chem. 2001;276:24767–73. doi: 10.1074/jbc.M100912200. [DOI] [PubMed] [Google Scholar]
- 24.Zelcer N, Tontonoz P. J Clin Invest. 2006;116:607–14. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levin N, Bischoff ED, Daige CL, Thomas D, Vu CT, et al. Arterioscler Thromb Vasc Biol. 2005;25:135–42. doi: 10.1161/01.ATV.0000150044.84012.68. [DOI] [PubMed] [Google Scholar]
- 26.Rigamonti E, Helin L, Lestavel S, Mutka AL, Lepore M, et al. Circ Res. 2005;97:682–9. doi: 10.1161/01.RES.0000184678.43488.9f. [DOI] [PubMed] [Google Scholar]
- 27.Dai XY, Ou X, Hao XR, Cao DL, Tang YL, et al. J Cardiovasc Pharmacol. 2008;51:467–75. doi: 10.1097/FJC.0b013e31816a5be3. [DOI] [PubMed] [Google Scholar]
- 28.Repa JJ, Li H, Frank-Cannon TC, Valasek MA, Turley SD, et al. J Neurosci. 2007;27:14470–80. doi: 10.1523/JNEUROSCI.4823-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang JR, Coleman T, Langmade SJ, Scherrer DE, Lane L, et al. J Clin Invest. 2008;118:2281–90. doi: 10.1172/JCI32561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cummins CL, Volle DH, Zhang Y, McDonald JG, Sion B, et al. J Clin Invest. 2006;116:1902–12. doi: 10.1172/JCI28400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nilsson M, Stulnig TM, Lin CY, Yeo AL, Nowotny P, et al. Mol Endocrinol. 2007;21:126–37. doi: 10.1210/me.2006-0187. [DOI] [PubMed] [Google Scholar]
- 32.Christenson LK, McAllister JM, Martin KO, Javitt NB, Osborne TF, Strauss JF., 3rd J Biol Chem. 1998;273:30729–35. doi: 10.1074/jbc.273.46.30729. [DOI] [PubMed] [Google Scholar]
- 33.Beltowski J, Semczuk A. Pharmacol Rep. 2010;62:15–27. doi: 10.1016/s1734-1140(10)70239-5. [DOI] [PubMed] [Google Scholar]
- 34.Drouineaud V, Sagot P, Garrido C, Logette E, Deckert V, et al. Mol Hum Reprod. 2007;13:373–9. doi: 10.1093/molehr/gam019. [DOI] [PubMed] [Google Scholar]
- 35.Steffensen KR, Robertson K, Gustafsson JA, Andersen CY. Mol Cell Endocrinol. 2006;256:9–16. doi: 10.1016/j.mce.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 36.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. Nature. 1996;383:728–31. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 37.Frenoux JM, Vernet P, Volle DH, Britan A, Saez F, et al. J Mol Endocrinol. 2004;33:361–75. doi: 10.1677/jme.1.01515. [DOI] [PubMed] [Google Scholar]
- 38.Robertson KM, Schuster GU, Steffensen KR, Hovatta O, Meaney S, et al. Endocrinology. 2005;146:2519–30. doi: 10.1210/en.2004-1413. [DOI] [PubMed] [Google Scholar]
- 39.Volle DH, Mouzat K, Duggavathi R, Siddeek B, Dechelotte P, et al. Mol Endocrinol. 2007;21:1014–27. doi: 10.1210/me.2006-0277. [DOI] [PubMed] [Google Scholar]
- 40.Russell DW. Annu Rev Biochem. 2003;72:137–74. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 41.Wang YD, Chen WD, Moore DD, Huang W. Cell Res. 2008;18:1087–95. doi: 10.1038/cr.2008.289. [DOI] [PubMed] [Google Scholar]
- 42.Kim I, Morimura K, Shah Y, Yang Q, Ward JM, Gonzalez FJ. Carcinogenesis. 2007;28:940–6. doi: 10.1093/carcin/bgl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Cell. 2000;102:731–44. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 44.Kok T, Hulzebos CV, Wolters H, Havinga R, Agellon LB, et al. J Biol Chem. 2003;278:41930–7. doi: 10.1074/jbc.M306309200. [DOI] [PubMed] [Google Scholar]
- 45.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Physiol Rev. 2009;89:147–91. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 46.Choi M, Moschetta A, Bookout AL, Peng L, Umetani M, et al. Nat Med. 2006;12:1253–5. doi: 10.1038/nm1501. [DOI] [PubMed] [Google Scholar]
- 47.Zollner G, Marschall HU, Wagner M, Trauner M. Mol Pharm. 2006;3:231–51. doi: 10.1021/mp060010s. [DOI] [PubMed] [Google Scholar]
- 48.Brendel C, Schoonjans K, Botrugno OA, Treuter E, Auwerx J. Mol Endocrinol. 2002;16:2065–76. doi: 10.1210/me.2001-0194. [DOI] [PubMed] [Google Scholar]
- 49.Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, et al. Mol Cell. 2000;6:507–15. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 50.Wang L, Lee YK, Bundman D, Han Y, Thevananther S, et al. Dev Cell. 2002;2:721–31. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 51.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, et al. Cell Metab. 2005;2:217–25. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 52.Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, et al. Genes Dev. 2003;17:1581–91. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ito S, Fujimori T, Furuya A, Satoh J, Nabeshima Y. J Clin Invest. 2005;115:2202–8. doi: 10.1172/JCI23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lambert G, Amar MJ, Guo G, Brewer HB, Jr, Gonzalez FJ, Sinal CJ. J Biol Chem. 2003;278:2563–70. doi: 10.1074/jbc.M209525200. [DOI] [PubMed] [Google Scholar]
- 55.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, et al. J Clin Invest. 2004;113:1408–18. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carnahan VE, Redinbo MR. Curr Drug Metab. 2005;6:357–67. doi: 10.2174/1389200054633844. [DOI] [PubMed] [Google Scholar]
- 57.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, et al. Proc Natl Acad Sci U S A. 2001;98:3369–74. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bachmann K, Patel H, Batayneh Z, Slama J, White D, et al. Pharmacol Res. 2004;50:237–46. doi: 10.1016/j.phrs.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 59.Sporstol M, Tapia G, Malerod L, Mousavi SA, Berg T. Biochem Biophys Res Commun. 2005;331:1533–41. doi: 10.1016/j.bbrc.2005.04.071. [DOI] [PubMed] [Google Scholar]
- 60.Wada T, Gao J, Xie W. Trends Endocrinol Metab. 2009;20:273–9. doi: 10.1016/j.tem.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 61.Zhang J, Huang W, Qatanani M, Evans RM, Moore DD. J Biol Chem. 2004;279:49517–22. doi: 10.1074/jbc.M409041200. [DOI] [PubMed] [Google Scholar]
- 62.Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD. Nature. 2000;407:920–3. doi: 10.1038/35038112. [DOI] [PubMed] [Google Scholar]
- 63.Masson D, Qatanani M, Sberna AL, Xiao R, Pais de Barros JP, et al. J Lipid Res. 2008;49:1682–91. doi: 10.1194/jlr.M700374-JLR200. [DOI] [PubMed] [Google Scholar]
- 64.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, et al. Science. 2002;296:1313–6. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 65.Nagpal S, Na S, Rathnachalam R. Endocr Rev. 2005;26:662–87. doi: 10.1210/er.2004-0002. [DOI] [PubMed] [Google Scholar]
- 66.Wong KE, Szeto FL, Zhang W, Ye H, Kong J, et al. Am J Physiol Endocrinol Metab. 2009;296:E820–8. doi: 10.1152/ajpendo.90763.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. J Lipid Res. 2009;50:1509–20. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim KM, Yoon JH, Gwak GY, Kim W, Lee SH, et al. Biochem Biophys Res Commun. 2006;342:1108–13. doi: 10.1016/j.bbrc.2006.02.072. [DOI] [PubMed] [Google Scholar]
- 69.Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Nat Rev Drug Discov. 2008;7:678–93. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 70.Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, et al. Cell Metab. 2009;10:167–77. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, et al. Genes Dev. 2000;14:2819–30. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kalaany NY, Gauthier KC, Zavacki AM, Mammen PP, Kitazume T, et al. Cell Metab. 2005;1:231–44. doi: 10.1016/j.cmet.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 73.Cha JY, Repa JJ. J Biol Chem. 2007;282:743–51. doi: 10.1074/jbc.M605023200. [DOI] [PubMed] [Google Scholar]
- 74.Tontonoz P, Mangelsdorf DJ. Mol Endocrinol. 2003;17:985–93. doi: 10.1210/me.2003-0061. [DOI] [PubMed] [Google Scholar]
- 75.Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, et al. Gastroenterology. 2008;134:556–67. doi: 10.1053/j.gastro.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 76.Kase ET, Wensaas AJ, Aas V, Hojlund K, Levin K, et al. Diabetes. 2005;54:1108–15. doi: 10.2337/diabetes.54.4.1108. [DOI] [PubMed] [Google Scholar]
- 77.Stenson BM, Ryden M, Steffensen KR, Wahlen K, Pettersson AT, et al. Endocrinology. 2009;150:4104–13. doi: 10.1210/en.2009-0676. [DOI] [PubMed] [Google Scholar]
- 78.Hu T, Foxworthy P, Siesky A, Ficorilli JV, Gao H, et al. Endocrinology. 2005;146:5380–7. doi: 10.1210/en.2005-0591. [DOI] [PubMed] [Google Scholar]
- 79.Bell GD, Lewis B, Petrie A, Dowling RH. Br Med J. 1973;3:520–3. doi: 10.1136/bmj.3.5879.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hirokane H, Nakahara M, Tachibana S, Shimizu M, Sato R. J Biol Chem. 2004;279:45685–92. doi: 10.1074/jbc.M404255200. [DOI] [PubMed] [Google Scholar]
- 81.Savkur RS, Bramlett KS, Michael LF, Burris TP. Biochem Biophys Res Commun. 2005;329:391–6. doi: 10.1016/j.bbrc.2005.01.141. [DOI] [PubMed] [Google Scholar]
- 82.Zhou J, Zhai Y, Mu Y, Gong H, Uppal H, et al. J Biol Chem. 2006;281:15013–20. doi: 10.1074/jbc.M511116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nakamura K, Moore R, Negishi M, Sueyoshi T. J Biol Chem. 2007;282:9768–76. doi: 10.1074/jbc.M610072200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ueda A, Hamadeh HK, Webb HK, Yamamoto Y, Sueyoshi T, et al. Mol Pharmacol. 2002;61:1–6. doi: 10.1124/mol.61.1.1. [DOI] [PubMed] [Google Scholar]
- 85.Kassam A, Winrow CJ, Fernandez-Rachubinski F, Capone JP, Rachubinski RA. J Biol Chem. 2000;275:4345–50. doi: 10.1074/jbc.275.6.4345. [DOI] [PubMed] [Google Scholar]
- 86.Maglich JM, Lobe DC, Moore JT. J Lipid Res. 2009;50:439–45. doi: 10.1194/jlr.M800226-JLR200. [DOI] [PubMed] [Google Scholar]
- 87.Dong B, Saha PK, Huang W, Chen W, Abu-Elheiga LA, et al. Proc Natl Acad Sci U S A. 2009;106:18831–6. doi: 10.1073/pnas.0909731106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roth A, Looser R, Kaufmann M, Blattler SM, Rencurel F, et al. Mol Pharmacol. 2008;73:1282–9. doi: 10.1124/mol.107.041012. [DOI] [PubMed] [Google Scholar]
- 89.Laffitte BA, Chao LC, Li J, Walczak R, Hummasti S, et al. Proc Natl Acad Sci U S A. 2003;100:5419–24. doi: 10.1073/pnas.0830671100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stulnig TM, Steffensen KR, Gao H, Reimers M, Dahlman-Wright K, et al. Mol Pharmacol. 2002;62:1299–305. doi: 10.1124/mol.62.6.1299. [DOI] [PubMed] [Google Scholar]
- 91.Gerin I, Dolinsky VW, Shackman JG, Kennedy RT, Chiang SH, et al. J Biol Chem. 2005;280:23024–31. doi: 10.1074/jbc.M412564200. [DOI] [PubMed] [Google Scholar]
- 92.Sugden MC, Holness MJ. Biochem Soc Trans. 2008;36:891–900. doi: 10.1042/BST0360891. [DOI] [PubMed] [Google Scholar]
- 93.Oosterveer MH, van Dijk TH, Grefhorst A, Bloks VW, Havinga R, et al. J Biol Chem. 2008;283:25437–45. doi: 10.1074/jbc.M801922200. [DOI] [PubMed] [Google Scholar]
- 94.Stayrook KR, Bramlett KS, Savkur RS, Ficorilli J, Cook T, et al. Endocrinology. 2005;146:984–91. doi: 10.1210/en.2004-0965. [DOI] [PubMed] [Google Scholar]
- 95.Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, et al. J Biol Chem. 2006;281:11039–49. doi: 10.1074/jbc.M510258200. [DOI] [PubMed] [Google Scholar]
- 96.Zhang Y, Lee FY, Barrera G, Lee H, Vales C, et al. Proc Natl Acad Sci U S A. 2006;103:1006–11. doi: 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ma K, Saha PK, Chan L, Moore DD. J Clin Invest. 2006;116:1102–9. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rizzo G, Disante M, Mencarelli A, Renga B, Gioiello A, et al. Mol Pharmacol. 2006;70:1164–73. doi: 10.1124/mol.106.023820. [DOI] [PubMed] [Google Scholar]
- 99.Renga B, Mencarelli A, Vavassori P, Brancaleone V, Fiorucci S. Biochim Biophys Acta. 2010;1802:363–72. doi: 10.1016/j.bbadis.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 100.Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk T, Grefhorst A, et al. FEBS Lett. 2005;579:4076–80. doi: 10.1016/j.febslet.2005.06.033. [DOI] [PubMed] [Google Scholar]
- 101.Cariou B, Bouchaert E, Abdelkarim M, Dumont J, Caron S, et al. FEBS Lett. 2007;581:5191–8. doi: 10.1016/j.febslet.2007.09.064. [DOI] [PubMed] [Google Scholar]
- 102.Venkatesan N, Davidson MB, Simsolo RB, Kern PA. Metabolism. 1994;43:348–56. doi: 10.1016/0026-0495(94)90103-1. [DOI] [PubMed] [Google Scholar]
- 103.Maglich JM, Watson J, McMillen PJ, Goodwin B, Willson TM, Moore JT. J Biol Chem. 2004;279:19832–8. doi: 10.1074/jbc.M313601200. [DOI] [PubMed] [Google Scholar]
- 104.Ding X, Lichti K, Kim I, Gonzalez FJ, Staudinger JL. J Biol Chem. 2006;281:26540–51. doi: 10.1074/jbc.M600931200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Palomer X, Gonzalez-Clemente JM, Blanco-Vaca F, Mauricio D. Diabetes Obes Metab. 2008;10:185–97. doi: 10.1111/j.1463-1326.2007.00710.x. [DOI] [PubMed] [Google Scholar]
- 106.Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Nat Med. 2003;9:213–9. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- 107.Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, et al. Cell. 2005;122:707–21. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Patel R, Patel M, Tsai R, Lin V, Bookout AL, et al. J Clin Invest. 2011;121:431–41. doi: 10.1172/JCI41681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, et al. Mol Cell. 2007;25:57–70. doi: 10.1016/j.molcel.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, et al. Nature. 2005;437:759–63. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Hepatology. 2008;48:1632–43. doi: 10.1002/hep.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moreau A, Vilarem MJ, Maurel P, Pascussi JM. Mol Pharm. 2008;5:35–41. doi: 10.1021/mp700103m. [DOI] [PubMed] [Google Scholar]
- 113.Hu G, Xu C, Staudinger J. J Pharmacol Exp Ther. 2010 doi: 10.1124/jpet.110.171744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. J Immunol. 2009;183:6251–61. doi: 10.4049/jimmunol.0803978. [DOI] [PubMed] [Google Scholar]
- 115.Zimmerman TL, Thevananther S, Ghose R, Burns AR, Karpen SJ. J Biol Chem. 2006;281:15434–40. doi: 10.1074/jbc.M508277200. [DOI] [PubMed] [Google Scholar]
- 116.Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, et al. Cell. 2004;119:299–309. doi: 10.1016/j.cell.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 117.Rahman I, Biswas SK, Kirkham PA. Biochem Pharmacol. 2006;72:1439–52. doi: 10.1016/j.bcp.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 118.Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, et al. Cell. 2008;134:97–111. doi: 10.1016/j.cell.2008.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Martinez-Botas J, Ferruelo AJ, Suarez Y, Fernandez C, Gomez-Coronado D, Lasuncion MA. Biochim Biophys Acta. 2001;1532:185–94. doi: 10.1016/s1388-1981(01)00125-1. [DOI] [PubMed] [Google Scholar]
- 120.Dubrac S, Elentner A, Ebner S, Horejs-Hoeck J, Schmuth M. J Immunol. 2010;184:2949–57. doi: 10.4049/jimmunol.0902151. [DOI] [PubMed] [Google Scholar]
- 121.Baeke F, Takiishi T, Korf H, Gysemans C, Mathieu C. Curr Opin Pharmacol. 2010;10:482–96. doi: 10.1016/j.coph.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 122.Schmuth M, Jiang YJ, Dubrac S, Elias PM, Feingold KR. J Lipid Res. 2008;49:499–509. doi: 10.1194/jlr.R800001-JLR200. [DOI] [PubMed] [Google Scholar]
- 123.Chang KC, Shen Q, Oh IG, Jelinsky SA, Jenkins SF, et al. Mol Endocrinol. 2008;22:2407–19. doi: 10.1210/me.2008-0232. [DOI] [PubMed] [Google Scholar]
- 124.Sacchetti P, Sousa KM, Hall AC, Liste I, Steffensen KR, et al. Cell Stem Cell. 2009;5:409–19. doi: 10.1016/j.stem.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 125.Wang L, Schuster GU, Hultenby K, Zhang Q, Andersson S, Gustafsson JA. Proc Natl Acad Sci U S A. 2002;99:13878–83. doi: 10.1073/pnas.172510899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Andersson S, Gustafsson N, Warner M, Gustafsson JA. Proc Natl Acad Sci U S A. 2005;102:3857–62. doi: 10.1073/pnas.0500634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kha HT, Basseri B, Shouhed D, Richardson J, Tetradis S, et al. J Bone Miner Res. 2004;19:830–40. doi: 10.1359/JBMR.040115. [DOI] [PubMed] [Google Scholar]
- 128.Richardson JA, Amantea CM, Kianmahd B, Tetradis S, Lieberman JR, et al. J Cell Biochem. 2007;100:1131–45. doi: 10.1002/jcb.21112. [DOI] [PubMed] [Google Scholar]
- 129.Dwyer JR, Sever N, Carlson M, Nelson SF, Beachy PA, Parhami F. J Biol Chem. 2007;282:8959–68. doi: 10.1074/jbc.M611741200. [DOI] [PubMed] [Google Scholar]
- 130.Corcoran RB, Scott MP. Proc Natl Acad Sci U S A. 2006;103:8408–13. doi: 10.1073/pnas.0602852103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kim WK, Meliton V, Park KW, Hong C, Tontonoz P, et al. Mol Endocrinol. 2009;23:1532–43. doi: 10.1210/me.2008-0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fausto N, Campbell JS, Riehle KJ. Hepatology. 2006;43:S45–53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 133.Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, et al. Science. 2006;312:233–6. doi: 10.1126/science.1121435. [DOI] [PubMed] [Google Scholar]
- 134.Chen WD, Wang YD, Zhang L, Shiah S, Wang M, et al. Hepatology. 2010;51:953–62. doi: 10.1002/hep.23390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang X, Krupczak-Hollis K, Tan Y, Dennewitz MB, Adami GR, Costa RH. J Biol Chem. 2002;277:44310–6. doi: 10.1074/jbc.M207510200. [DOI] [PubMed] [Google Scholar]
- 136.Guo GL, Lambert G, Negishi M, Ward JM, Brewer HB, Jr, et al. J Biol Chem. 2003;278:45062–71. doi: 10.1074/jbc.M307145200. [DOI] [PubMed] [Google Scholar]
- 137.Yamamoto Y, Moore R, Goldsworthy TL, Negishi M, Maronpot RR. Cancer Res. 2004;64:7197–200. doi: 10.1158/0008-5472.CAN-04-1459. [DOI] [PubMed] [Google Scholar]
- 138.Huang W, Zhang J, Washington M, Liu J, Parant JM, et al. Mol Endocrinol. 2005;19:1646–53. doi: 10.1210/me.2004-0520. [DOI] [PubMed] [Google Scholar]
- 139.Staudinger J, Liu Y, Madan A, Habeebu S, Klaassen CD. Drug Metab Dispos. 2001;29:1467–72. [PubMed] [Google Scholar]
- 140.Dai G, He L, Bu P, Wan YJ. Hepatology. 2008;47:1277–87. doi: 10.1002/hep.22129. [DOI] [PubMed] [Google Scholar]
- 141.Ethier C, Kestekian R, Beaulieu C, Dube C, Havrankova J, Gascon-Barre M. Endocrinology. 1990;126:2947–59. doi: 10.1210/endo-126-6-2947. [DOI] [PubMed] [Google Scholar]
- 142.Koldamova R, Lefterov I. Curr Alzheimer Res. 2007;4:171–8. doi: 10.2174/156720507780362227. [DOI] [PubMed] [Google Scholar]
- 143.Fitz NF, Cronican A, Pham T, Fogg A, Fauq AH, et al. J Neurosci. 2010;30:6862–72. doi: 10.1523/JNEUROSCI.1051-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Adighibe O, Arepalli S, Duckworth J, Hardy J, Wavrant-De Vrieze F. Neurobiol Aging. 2006;27:1431–4. doi: 10.1016/j.neurobiolaging.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 145.Mooijaart SP, Kuningas M, Westendorp RG, Houwing-Duistermaat JJ, Slagboom PE, et al. J Gerontol A Biol Sci Med Sci. 2007;62:343–9. doi: 10.1093/gerona/62.4.343. [DOI] [PubMed] [Google Scholar]
- 146.Keisala T, Minasyan A, Lou YR, Zou J, Kalueff AV, et al. J Steroid Biochem Mol Biol. 2009;115:91–7. doi: 10.1016/j.jsbmb.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 147.Dardenne O, Prud’homme J, Arabian A, Glorieux FH, St-Arnaud R. Endocrinology. 2001;142:3135–41. doi: 10.1210/endo.142.7.8281. [DOI] [PubMed] [Google Scholar]
- 148.Lanske B, Razzaque MS. J Nutr Biochem. 2007;18:771–7. doi: 10.1016/j.jnutbio.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, et al. Science. 2005;309:1829–33. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.McElwee JJ, Schuster E, Blanc E, Thomas JH, Gems D. J Biol Chem. 2004;279:44533–43. doi: 10.1074/jbc.M406207200. [DOI] [PubMed] [Google Scholar]
- 151.Tsuchiya T, Dhahbi JM, Cui X, Mote PL, Bartke A, Spindler SR. Physiol Genomics. 2004;17:307–15. doi: 10.1152/physiolgenomics.00039.2004. [DOI] [PubMed] [Google Scholar]
- 152.Amador-Noguez D, Yagi K, Venable S, Darlington G. Aging Cell. 2004;3:423–41. doi: 10.1111/j.1474-9728.2004.00125.x. [DOI] [PubMed] [Google Scholar]
- 153.Amador-Noguez D, Dean A, Huang W, Setchell K, Moore D, Darlington G. Aging Cell. 2007;6:453–70. doi: 10.1111/j.1474-9726.2007.00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gerisch B, Rottiers V, Li D, Motola DL, Cummins CL, et al. Proc Natl Acad Sci U S A. 2007;104:5014–9. doi: 10.1073/pnas.0700847104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fisher AL, Lithgow GJ. Aging Cell. 2006;5:127–38. doi: 10.1111/j.1474-9726.2006.00203.x. [DOI] [PubMed] [Google Scholar]
- 156.Entchev EV, Kurzchalia TV. Semin Cell Dev Biol. 2005;16:175–82. doi: 10.1016/j.semcdb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 157.Cooke J, Sang JH. J Insect Physiol. 1970;16:801–12. doi: 10.1016/0022-1910(70)90214-3. [DOI] [PubMed] [Google Scholar]
- 158.Chitwood DJ. Crit Rev Biochem Mol Biol. 1999;34:273–84. doi: 10.1080/10409239991209309. [DOI] [PubMed] [Google Scholar]
- 159.Rewitz KF, Rybczynski R, Warren JT, Gilbert LI. Biochem Soc Trans. 2006;34:1256–60. doi: 10.1042/BST0341256. [DOI] [PubMed] [Google Scholar]
- 160.Gibbens YY, Warren JT, Gilbert LI, O’Connor MB. Development. 2011;138:2693–703. doi: 10.1242/dev.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Baker KD, Shewchuk LM, Kozlova T, Makishima M, Hassell A, et al. Cell. 2003;113:731–42. doi: 10.1016/s0092-8674(03)00420-3. [DOI] [PubMed] [Google Scholar]
- 162.Huang X, Warren JT, Gilbert LI. J Genet Genomics. 2008;35:1–10. doi: 10.1016/S1673-8527(08)60001-6. [DOI] [PubMed] [Google Scholar]
- 163.Wiese M, Antebi A, Zheng H. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yochem J, Tuck S, Greenwald I, Han M. Development. 1999;126:597–606. doi: 10.1242/dev.126.3.597. [DOI] [PubMed] [Google Scholar]
- 165.Li J, Brown G, Ailion M, Lee S, Thomas JH. Development. 2004;131:5741–52. doi: 10.1242/dev.01408. [DOI] [PubMed] [Google Scholar]
- 166.Kostrouchova M, Krause M, Kostrouch Z, Rall JE. Proc Natl Acad Sci U S A. 2001;98:7360–5. doi: 10.1073/pnas.131171898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gissendanner CR, Sluder AE. Dev Biol. 2000;221:259–72. doi: 10.1006/dbio.2000.9679. [DOI] [PubMed] [Google Scholar]
- 168.Tatar M, Yin C. Exp Gerontol. 2001;36:723–38. doi: 10.1016/s0531-5565(00)00238-2. [DOI] [PubMed] [Google Scholar]
- 169.Flatt T, Tu MP, Tatar M. Bioessays. 2005;27:999–1010. doi: 10.1002/bies.20290. [DOI] [PubMed] [Google Scholar]
- 170.Simon AF, Shih C, Mack A, Benzer S. Science. 2003;299:1407–10. doi: 10.1126/science.1080539. [DOI] [PubMed] [Google Scholar]
- 171.Maki A, Sawatsubashi S, Ito S, Shirode Y, Suzuki E, et al. Biochem Biophys Res Commun. 2004;320:262–7. doi: 10.1016/j.bbrc.2004.05.156. [DOI] [PubMed] [Google Scholar]
- 172.Angelo G, Van Gilst MR. Science. 2009;326:954–8. doi: 10.1126/science.1178343. [DOI] [PubMed] [Google Scholar]
- 173.Van Gilst MR, Hadjivassiliou H, Yamamoto KR. Proc Natl Acad Sci U S A. 2005;102:13496–501. doi: 10.1073/pnas.0506234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gerisch B, Weitzel C, Kober-Eisermann C, Rottiers V, Antebi A. Dev Cell. 2001;1:841–51. doi: 10.1016/s1534-5807(01)00085-5. [DOI] [PubMed] [Google Scholar]
- 175.Yabe T, Suzuki N, Furukawa T, Ishihara T, Katsura I. Development. 2005;132:3197–207. doi: 10.1242/dev.01909. [DOI] [PubMed] [Google Scholar]
- 176.Riddle DL, Swanson MM, Albert PS. Nature. 1981;290:668–71. doi: 10.1038/290668a0. [DOI] [PubMed] [Google Scholar]
- 177.Antebi A, Yeh WH, Tait D, Hedgecock EM, Riddle DL. Genes Dev. 2000;14:1512–27. [PMC free article] [PubMed] [Google Scholar]
- 178.Vowels JJ, Thomas JH. Genetics. 1992;130:105–23. doi: 10.1093/genetics/130.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Thomas JH, Birnby DA, Vowels JJ. Genetics. 1993;134:1105–17. doi: 10.1093/genetics/134.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jia K, Albert PS, Riddle DL. Development. 2002;129:221–31. doi: 10.1242/dev.129.1.221. [DOI] [PubMed] [Google Scholar]
- 181.Motola DL, Cummins CL, Rottiers V, Sharma KK, Li T, et al. Cell. 2006;124:1209–23. doi: 10.1016/j.cell.2006.01.037. [DOI] [PubMed] [Google Scholar]
- 182.Held JM, White MP, Fisher AL, Gibson BW, Lithgow GJ, Gill MS. Aging Cell. 2006;5:283–91. doi: 10.1111/j.1474-9726.2006.00218.x. [DOI] [PubMed] [Google Scholar]
- 183.Song C, Liao S. Endocrinology. 2000;141:4180–4. doi: 10.1210/endo.141.11.7772. [DOI] [PubMed] [Google Scholar]
- 184.Ludewig AH, Kober-Eisermann C, Weitzel C, Bethke A, Neubert K, et al. Genes Dev. 2004;18:2120–33. doi: 10.1101/gad.312604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Rottiers V, Motola DL, Gerisch B, Cummins CL, Nishiwaki K, et al. Dev Cell. 2006;10:473–82. doi: 10.1016/j.devcel.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 186.Yoshiyama T, Namiki T, Mita K, Kataoka H, Niwa R. Development. 2006;133:2565–74. doi: 10.1242/dev.02428. [DOI] [PubMed] [Google Scholar]
- 187.Dumas KJ, Guo C, Wang X, Burkhart KB, Adams EJ, et al. Dev Biol. 2010;340:605–12. doi: 10.1016/j.ydbio.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gerisch B, Antebi A. Development. 2004;131:1765–76. doi: 10.1242/dev.01068. [DOI] [PubMed] [Google Scholar]
- 189.Mak HY, Ruvkun G. Development. 2004;131:1777–86. doi: 10.1242/dev.01069. [DOI] [PubMed] [Google Scholar]
- 190.Wang Z, Zhou XE, Motola DL, Gao X, Suino-Powell K, et al. Proc Natl Acad Sci U S A. 2009;106:9138–43. doi: 10.1073/pnas.0904064106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ogawa A, Streit A, Antebi A, Sommer RJ. Curr Biol. 2009;19:67–71. doi: 10.1016/j.cub.2008.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Bento G, Ogawa A, Sommer RJ. Nature. 2010;466:494–7. doi: 10.1038/nature09164. [DOI] [PubMed] [Google Scholar]
- 193.Hannich JT, Entchev EV, Mende F, Boytchev H, Martin R, et al. Dev Cell. 2009;16:833–43. doi: 10.1016/j.devcel.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 194.Antebi A, Culotti JG, Hedgecock EM. Development. 1998;125:1191–205. doi: 10.1242/dev.125.7.1191. [DOI] [PubMed] [Google Scholar]
- 195.Bethke A, Fielenbach N, Wang Z, Mangelsdorf DJ, Antebi A. Science. 2009;324:95–8. doi: 10.1126/science.1164899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Abbott AL, Alvarez-Saavedra E, Miska EA, Lau NC, Bartel DP, et al. Dev Cell. 2005;9:403–14. doi: 10.1016/j.devcel.2005.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Mallanna SK, Rizzino A. Dev Biol. 2010;344:16–25. doi: 10.1016/j.ydbio.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Caygill EE, Johnston LA. Curr Biol. 2008;18:943–50. doi: 10.1016/j.cub.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Klass MR. Mech Ageing Dev. 1977;6:413–29. doi: 10.1016/0047-6374(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 200.Lee SJ, Kenyon C. Curr Biol. 2009;19:715–22. doi: 10.1016/j.cub.2009.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hsin H, Kenyon C. Nature. 1999;399:362–6. doi: 10.1038/20694. [DOI] [PubMed] [Google Scholar]
- 202.Yamawaki TM, Berman JR, Suchanek-Kavipurapu M, McCormick M, Maria Gaglia M, et al. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Berman JR, Kenyon C. Cell. 2006;124:1055–68. doi: 10.1016/j.cell.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 204.Lin K, Hsin H, Libina N, Kenyon C. Nat Genet. 2001;28:139–45. doi: 10.1038/88850. [DOI] [PubMed] [Google Scholar]
- 205.Wang MC, O’Rourke EJ, Ruvkun G. Science. 2008;322:957–60. doi: 10.1126/science.1162011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Flatt T, Min KJ, D’Alterio C, Villa-Cuesta E, Cumbers J, et al. Proc Natl Acad Sci U S A. 2008;105:6368–73. doi: 10.1073/pnas.0709128105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Cargill SL, Carey JR, Muller HG, Anderson G. Aging Cell. 2003;2:185–90. doi: 10.1046/j.1474-9728.2003.00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]