

Summary
The Agrobacterium tumefaciens VirB4 ATPase functions with other VirB proteins to export T-DNA to susceptible plant cells and other DNA substrates to a variety of prokaryotic and eukaryotic cells. Previous studies have demonstrated that VirB4 mutants with defects in the Walker A nucleotide-binding motif are non-functional and exert a dominant negative phenotype when synthesized in wild-type cells. This study characterized the oligomeric structure of VirB4 and examined the effects of Walker A sequence mutations on complex formation and transporter activity. VirB4 directed dimer formation when fused to the amino-terminal portion of cI repressor protein, as shown by immunity of Escherichia coli cells to λ phage infection. VirB4 also dimerized in Agrobacterium tumefaciens, as demonstrated by the recovery of a detergent-resistant complex of native protein and a functional, histidine-tagged derivative by precipitation with anti-His6 antibodies and by Co2+ affinity chromatography. Walker A sequence mutants directed repressor dimerization in E. coli and interacted with His-VirB4 in A. tumefaciens, indicating that ATP binding is not required for self-association. A dimerization domain was localized to a proposed N-terminal membrane-spanning region of VirB4, as shown by the dominance of an allele coding for the N-terminal 312 residues and phage immunity of host cells expressing cI repressor fusions to alleles for the first 237 or 312 residues. A recent study reported that the synthesis of a subset of VirB proteins, including VirB4, in agrobacterial recipients has a pronounced stimulatory effect on the virB-dependent conjugal transfer of plasmid RSF1010 by agrobacterial donors. VirB4′312 suppressed the stimulatory effect of VirB proteins for DNA uptake when synthesized in recipient cells. In striking contrast, Walker A sequence mutants contributed to the stimulatory effect of VirB proteins to the same extent as native VirB4. These findings indicate that the oligomeric structure of VirB4, but not its capacity to bind ATP, is important for the assembly of VirB proteins as a DNA uptake system. The results of these studies support a model in which VirB4 dimers or homomultimers contribute structural information for the assembly of a transenvelope channel competent for bidirectional DNA transfer, whereas an ATP-dependent activity is required for configuring this channel as a dedicated export machine.
Introduction
Agrobacterium tumefaciens induces the formation of crown gall disease in plants by delivering T-DNA in the form of a nucleoprotein particle termed the T-complex to susceptible plant cells (for reviews, see Christie, 1997; Das, 1998; Zupan and Zambryski, 1995; Sheng and Citovsky, 1996). Recent studies have indicated that the transporter dedicated to the transmission of T-DNA across the A. tumefaciens cell envelope is evolutionarily related to (i) conjugation systems responsible for the movement of plasmid DNA between bacterial cells and (ii) toxin export systems identified in a number of bacterial pathogens of humans (Christie, 1997; Christie and Covacci, 1999). These transporters are designated here as the ‘adapted conjugation systems’ to distinguish this secretion family from other conserved protein-targeting mechanisms identified in bacteria, such as the ATP-binding cassette (ABC) transporter superfamily exemplified by Escherichia coli haemolysin export (Fath and Kolter, 1993), the terminal branch of the general secretory pathway exemplified by Klebsiella oxytoca pullulanase secretion (Pugsley, 1993) and contact-dependent transporters exemplified by Salmonella typhimurium Inv and Yersinia pestis Yop secretion (Hueck, 1998).
The adapted conjugation systems that function to transmit DNA between cells include the T-DNA exporter and transfer (tra) systems of the narrow-host-range F plasmid of E. coli and the broad-host-range plasmids of the IncN, IncW and IncP incompatibility groups (Winans et al., 1996; Christie, 1997). Systems dedicated to toxin export include the Bordetella pertussis Ptl system for exporting multisubunit pertussis toxin (Weiss et al., 1993; Winans et al., 1996), the Helicobacter pylori Cag system for exporting a toxin that induces interleukin-8 secretion by mammalian cells (Tummuru et al., 1995; Covacci et al., 1997; Akopyants et al., 1998) and the Legionella icm/dot system for exporting a toxin involved in phagosome trafficking and macrophage killing (Segal and Schuman, 1997; Vogel et al., 1998). Very recently, the Rickettsia prowazekii genome has been shown to carry a set of genes whose deduced products are homologues of several components of the adapted conjugation systems; these proteins may also assemble into a dedicated transport system for the export of virulence factors (Anderson et al., 1998). The Ptl system excretes functional pertussis toxin into the extracellular milieu when grown in culture, yet it is not known whether any of the toxin exporters of the adapted conjugation family function in a cell contact-dependent fashion or elaborate extracellular filaments in the mammalian host. For most of these systems, the toxin substrates have not been identified but, interestingly, the Icm/Dot system appears to have retained a functional vestige of the DNA transfer system from which it evolved in its ability to deliver the non-self-transmissible IncQ plasmid RSF1010 to bacterial recipient cells by a process requiring cell-to-cell contact (Vogel et al., 1998).
Numerous laboratories are attempting to generate structural information for the adapted conjugation systems. The T-DNA export system, composed minimally of products of the virB operon and the virD4 gene, is thought to consist of a transenvelope channel for mediating substrate transfer and an extracellular pilus for making contact with recipient cells (Winans et al., 1996; Christie, 1997). Although the pilus (T-pilus) has been visualized and its major subunit identified (Fullner et al., 1996; Lai and Kado, 1998), the architecture of the presumed transenvelope channel has not been defined. Progress towards the definition of steps in the T-complex transporter assembly pathway has been made by the construction of non-polar virB gene deletion mutants (Berger and Christie, 1994). Analyses of these mutants led to the discovery that some VirB proteins, including VirB7 and VirB9, provide important stabilizing functions for other VirB proteins (Berger and Christie, 1994; Fernandez et al., 1996a). VirB7, an outer membrane lipoprotein (Fernandez et al., 1996b), has been shown to stabilize itself as well as VirB9 through the assembly of a disulphide cross-linked heterodimer (Anderson et al., 1996; Fernandez et al., 1996b; Spudich et al., 1996; Baron et al., 1997). Synthesis of the heterodimer is critical for the elaboration of the T-pilus and for the stabilization of several putative channel components, including VirB10 and the two cytoplasmic membrane VirB ATPases, VirB4 and VirB11. A current model proposes that the VirB7–VirB9 heterodimer, once positioned at the outer membrane, functions as a nucleation centre for the recruitment and stabilization of other VirB channel and T-pilus components (Christie, 1997).
Recent genetic findings further support the notion that both of the VirB ATPases function as components of larger multimeric complexes. For example, codon substitutions in the Walker A sequence abolish VirB4 function and also exert dominant negative effects when the mutated alleles are expressed in wild-type cells (Berger and Christie, 1993; Fullner et al., 1994). The simplest model to explain these findings is that the Walker sequence mutant proteins perturb the ability of the native protein to interact with itself or with other VirB proteins. Interestingly, in contrast to VirB4 and most other traffic ATPases examined, mutations in the Walker A motif of VirB11 are recessive (Stephens et al., 1995; Rashkova et al., 1997). However, a collection of 11 dominant virB11 alleles was isolated, and genetic suppression studies showed that the dominance of each of these mutations could be suppressed by the overexpression of the virB9, virB10 and/or virB11 genes. Thus, VirB11, like VirB4, most probably also functions as a multimer (Zhou and Christie, 1997).
In this report, we have characterized the oligomeric state of the VirB4 ATPase further. We present several lines of evidence that VirB4 assembles minimally as a homodimer via an N-terminal dimerization domain, and we show that VirB4 homomultimers form independently of other VirB proteins as well as having the capacity to bind ATP. Our results support a model in which dimer formation is a prerequisite for the productive assembly of VirB4 with other VirB proteins during transporter biogenesis.
Results
VirB4-mediated dimerization of the λ cI repressor protein
VirB4 dimerization was assessed with the λ cI repressor fusion system. The cI repressor functions as a dimer to repress transcription at operator sites in λ Pr. Dimer formation is mediated by the C-terminus, whereas the N-terminal DNA-binding domain is only weakly interactive. Fusion of a self-associating peptide or protein to the N-terminus can significantly enhance repressor binding (Hu, 1995). We used a λ phage immunity assay to monitor the dimerization potential of a hybrid protein composed of the N-terminus of cI, designated cI′, and full-length VirB4. AG1688 cells expressing cI ′-virB4 were cross-streaked against phage λKH54, and immunity to phage was determined by visual inspection of the plates. As shown in Fig. 1, AG1688(pSR58) expressing cI ′ were sensitive to phage, while AG1688(pJH157) expressing full-length cI were immune to phage infection and killing. Similarly, AG1688(pTAD181) expressing cI ′-virB4 and AG1688(pTAD180) expressing cI ′ fused to full-length virB11 were also immune to phage. Phage dilution experiments showed that the levels of immunity for cells synthesizing the cI′-VirB4 and cI′-VirB11 fusion proteins approximated that for cells synthesizing full-length cI repressor protein (data not shown). These findings suggest that both VirB4 and VirB11 direct dimer formation of the DNA-binding domain of cI repressor. Elsewhere, we report additional evidence for VirB11 self-association (Zhou and Christie, 1997; S. Rashkova et al., submitted).
Fig. 1.
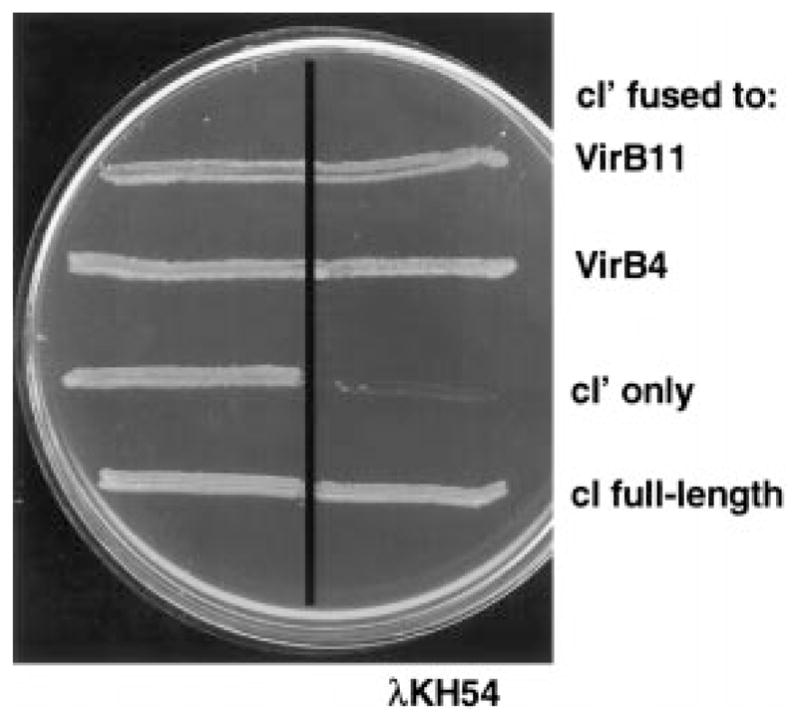
Immunity of E. coli AG1688 cells expressing cI chimeric genes. Strains were streaked across a line of λKH54 phage (vertical line) for the determination of phage-sensitivity phenotypes. Strains synthesizing cI′ fused to proteins indicated at the right carried the following expression plasmids: VirB11 (pTAD180), VirB4 (pTAD181), cI′ N-terminal 117 residues of cI (pSR58), cI full-length (pJH157).
We used a competitive disruption assay (Joung et al., 1995) to confirm that VirB4 directs the dimerization of the cI-VirB4 hybrid. The premise for this assay is that, if VirB4 dimerizes, its overproduction should disrupt the formation of cI-VirB4 dimers. AG1688 strains expressing full-length cI, cI ′ alone or cI ′ fused to virB4 or virB11 were transformed with the compatible IncP plasmid pPC40 expressing native virB4 from Plac. Preliminary experiments showed that strains carrying pPC40 accumulated native VirB4 at levels similar to those of the cI-VirB4 hybrid protein, establishing that both proteins are stable in E. coli (data not shown). As shown in Fig. 2, cells expressing full-length cI were immune to phage, and cells expressing cI ′ coding for the N-terminal fragment were sensitive. These findings show that native VirB4 had no effect on the dimerization of native repressor and also did not direct the dimerization of the DNA-binding domain when synthesized in a trans configuration. In contrast, synthesis of the native protein did interfere with cI-VirB4 dimer formation as shown by conversion of AG1688(pTAD181) to phage sensitivity (Fig. 2). These findings suggest that wild-type VirB4 was able to interact with and titrate the cI-VirB4 hybrid. In some experiments, overexpression of virB4 appeared to interfere with cI-VirB11 dimerization slightly (see Fig. 2), but these findings were not reproducible. Moreover, phage dilution experiments failed to show a reproducible effect of virB4 overexpression on VirB11 dimerization (data not shown). We also examined the possibility that other VirB proteins might influence the ability of VirB4 to dimerize. AG1688(pTAD181) cells were transformed with IncP plasmids expressing virB5, virB7 or virB10, and the resulting strains were assayed for phage sensitivity. None of these strains displayed a conversion to phage sensitivity, suggesting that overproduction of these VirB proteins does not interfere with VirB4 dimerization (data not shown).
Fig. 2.
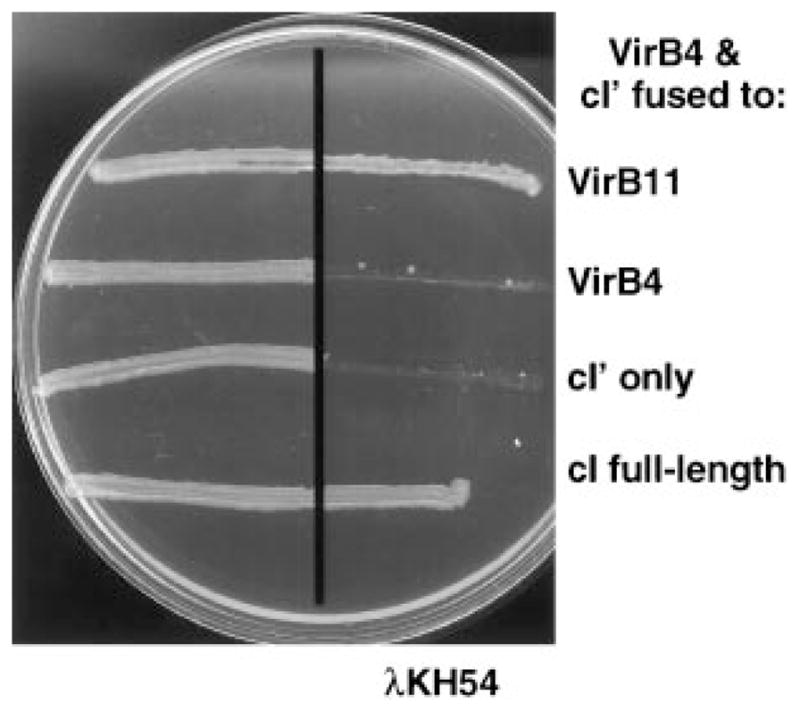
Immunity of E. coli AG1688 cells co-expressing wild-type virB4 and cI chimeric genes. VirB4 was synthesized from Plac carried on IncP plasmid pPC40. Proteins listed at the right were synthesized from the pBR322-based plasmids listed in Fig. 1.
Co-precipitation of His-VirB4 and VirB4
To test for VirB4 self-association in A. tumefaciens cells, we constructed a VirB4 derivative with an ≈2 kDa N-terminal polyhistidine tag and assayed for its interaction with native VirB4. his-virB4 exhibits wild-type function, as shown by its ability to complement the non-polar ΔvirB4 mutation of strain PC1004 (see below). Furthermore, the 89 kDa His-VirB4 protein migrates at a position slightly higher in SDS–polyacrylamide gels than the 87 kDa native protein (Fig. 3), and an anti-His6 monoclonal antibody reacted specifically with the His-tagged protein in immunoblots (data not shown). As shown in Fig. 3, the His6 antibody co-precipitated both forms of VirB4 from dodecyl-maltoside (DM)-solubilized extracts of PC1004(pTAD2141), which co-expresses virB4 and his-virB4. In contrast, the His6 antibody failed to precipitate native VirB4 from extracts of wild-type A348 or PC1004(pTAD214) expressing only virB4. These findings show that the native protein was precipitated only when it was co-synthesized with His-VirB4. Further, both proteins were co-precipitated from PC1000(pTAD2141) cells, showing that the production of other VirB proteins did not interfere with the formation of the putative His-VirB4/VirB4 interaction (Fig. 3). Between experiments, some variability was obtained in the relative amounts of His-VirB4 and VirB4 recovered in the precipitates. This variability could result from the fact that his-virB4 and virB4 were expressed from the constitutive Plac and the AS-inducible PvirB promoters, respectively, which could affect both the amount and the timing of HisVirB4 and VirB4 synthesis relative to other VirB proteins. Of interest, we were unable to detect other VirB proteins immunologically, including VirB5 or VirB7 to VirB11, in immunoprecipitates from DM-solubilized A348(pTADB150) extracts (data not shown). Although no direct interactions between VirB4 and other VirB proteins have been identified, the observation that several VirB proteins contribute to the stabilization of VirB4 is suggestive of complex formation (Fernandez et al., 1996b). It is possible that the precipitated VirB4 homomultimers correspond to an assembly intermediate. We also cannot exclude the possibility that solubilization with DM disrupted protein interactions other than those mediating VirB4 dimerization or homo-oligomerization.
Fig. 3.
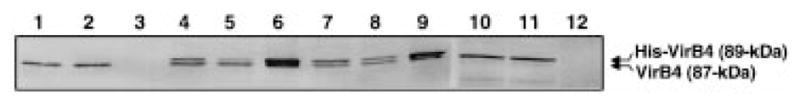
Immunoprecipitation (IP) of VirB4 complexes from detergent-solubilized A. tumefaciens extracts. Immunoprecipitates were analysed by SDS–PAGE and immunostaining with anti-VirB4 antibodies. Upper reactive species is the 89 kDa His-VirB4; lower species is the 87 kDa native VirB4 protein. Strains: wild-type A348 (lanes 1–3), PC1004(pTAD2141) (lanes 4–6), PC1000(pTAD2141) (lanes 7–9), PC1000(pTAD214) (lanes 10–12). Lanes 1, 4, 7 and 10 are cell extracts before IP; lanes 2, 5, 8 and 11 are supernatants after IP; lanes 3, 6, 9 and 12 are immunoprecipitates.
Co-retention of His-VirB4 and VirB4 on cobalt affinity columns
An interaction between VirB4 and His-VirB4 was also demonstrated by immobilized metal affinity chromatography (IMAC). Initial experiments using Ni2+ affinity resins resulted in a low background retention of VirB4 in the absence of His-VirB4. This background binding was completely eliminated using Co2+ affinity resins. As shown in Fig. 4A, native VirB4 was retained on the Co2+ affinity columns when material loaded onto the columns was derived from PC1004(pTAD2141) or PC1000(pTAD2141) cells co-synthesizing VirB4 and His-VirB4 in the presence or absence of other VirB proteins respectively (lanes 6 and 9). As with the immunoprecipitates described above, we were unable to detect VirB proteins other than His-VirB4 and VirB4 in eluates recovered from chromatography of PC1004(pTAD2141) extracts. We next tested whether His-VirB4 interacts with the Walker A sequence mutants. As shown in Fig. 4B and C, VirB4K439Q and VirB4Δ438–440 were retained on the column only when these proteins were co-synthesized with His-VirB4. The Walker A sequence mutants interacted with His-VirB4 in strains independently of the presence of other VirB proteins (Fig. 4B and C).
Fig. 4.
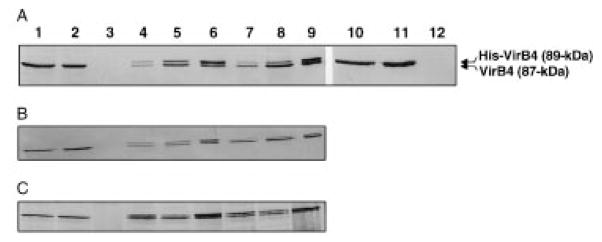
Cobalt affinity chromatography of DM-solubilized A. tumefaciens extracts. Column fractions were analysed by SDS–PAGE and immunostaining with anti-VirB4 antibodies. Upper reactive species is the 89 kDa His-VirB4; lower species is the 87 kDa native VirB4 protein.
A. Strains: wild-type A348 (lanes 1–3), PC1004(pTAD2141) (lanes 4–6), PC1000(pTAD2141) (lanes 7–9), PC1000(pTAD214) (lanes 10–12).
B. Strains: PC1004(pBB15) (lanes 1–3), PC1004(pPC43) (lanes 4–6), PC1000(pPC43) (lanes 7–9).
C. Strains: PC1004(pBB17) (lanes 1–3), PC1004(pPC44) (lanes 4–6), PC1000(pPC44) (lanes 7–9).
Lanes 1, 4, 7 and 10 are cell extracts before chromatography; lanes 2, 5, 8 and 11 are flowthrough fractions; lanes 3, 6, 9 and 12 are fractions eluted with 100 mM imidazole.
As controls for these studies, we examined the potential for these interactions to be generated in situ. In one set of experiments, we mixed cells expressing only his-virB4 or virB4 before cell lysis, DM solubilization and passage through Co2+ affinity columns. In a second set of experiments, we preloaded columns with His-VirB4, washed the columns, then applied solubilized extracts from cells expressing wild-type virB4. Although we found His-VirB4 in the column eluates from both sets of experiments, we were unable to detect the native protein (data not shown). These findings suggest that VirB4 assembles as dimers or higher order oligomers in vivo and does not undergo further complex formation upon cell lysis.
virB4 truncation derivatives are transdominant
Several VirB4 truncation derivatives were constructed with the aim of localizing a region of VirB4 with dominant negative effects (Fig. 5A). These alleles were tested for synthesis of detectable protein by immunoblot analysis, functionality by expression in a non-polar virB4 null mutant and dominance by expression in the wild-type A348 genetic background. Several of the truncation derivatives, including VirB4′780, VirB4′611 and VirB4′312, accumulated at levels slightly lower than that of native VirB4 expressed from the Ti plasmid. The remaining truncation derivatives, including VirB4′237, VirB4′101, VirB4Δ1–273 and VirB4Δ13–157, were undetectable even when gels were overloaded with protein. Efforts to visualize these derivatives by fusion to the green fluorescence protein (with antibodies kindly supplied by W. Margolin) were also unsuccessful, suggesting that these mutant proteins are intrinsically unstable in A. tumefaciens.
Fig. 5.

VirB4 derivatives and their phenotypes.
A. Native VirB4 (789 residues) at the top with positions noted for proposed transmembrane regions 1 and 2, with dark stipples denoting proposed periplasmic loops and light stipples denoting flanking transmembrane segments. The Walker A nucleotide-binding sequence is denoted by a black box. The His tag is denoted with a box filled with diagonal lines. Synthesis of detectable protein was assessed by immunostaining. Functionality was assessed by the ability to restore virulence to strain PC1004. Dominance was assessed by the virulence of A348 merodiploids.
B. Virulence assays showing the dominance of VirB4 truncation proteins. Strains synthesized native VirB4 (top left), His-VirB4 (top right) or both native VirB4 and the truncated proteins listed (two side-by-side inoculations per strain).
Figure 5B shows the results of virulence assays of mero-diploid strains co-expressing wild-type virB4 and alleles coding for the three truncated proteins visualized by immunostaining. As shown previously, overexpression of wild-type virB4 from an IncP replicon does not alter the virulence of host cells (see Berger and Christie, 1993; 1994). Interestingly, each of the truncated alleles displayed negative dominance, as shown by the attenuated virulence of the virB4*/virB4 merodiploids compared with the level of virulence of wild-type A348 or PC1004 strains engineered to express wild-type virB4 (data not shown) or his-virB4 from an IncP replicon. The virB4′312 allele synthesizing the N-terminal third of VirB4 reproducibly displayed the strongest dominance of the three alleles tested. To assess the dominance of the truncated alleles more quantitatively, we inoculated strains over a range of cell concentrations on plants. The virB4′312/virB4 merodiploid strain consistently failed to induce tumour formation with inoculum sizes of ≤ 105 cells, whereas wild-type A348 cells induced tumour formation with inoculum sizes as low as 103 cells. Results of these serial dilution experiments also showed that merodiploid strains co-expressing virB4 and either virB4′780 or virB4′611 exhibited only a slight reduction in virulence compared with wild-type A348 (data not shown). Taken together, our results suggest that VirB4 possesses a domain within the first 312 residues that interferes with the function of the native protein in intercellular DNA transmission. We have not been able to localize further the region of VirB4 conferring negative dominance using truncated proteins because of the apparent instabilities of N-terminal fragments of less than 312 residues in A. tumefaciens.
Dimerization of the N-terminus and of Walker A sequence mutants
To test whether the N-terminus of VirB4 self-interacts, we assayed for the capacity of VirB4′312 and two smaller N-terminal fragments, VirB4′101 and VirB4′237, to direct cI′ dimer formation (Fig. 6A). Cross-streak analysis of AG1688(pTAD182) expressing cI ′-virB4′312 showed clearly that the hybrid protein mediates immunity to phage infection. Interestingly, AG1688(pTAD183) cells expressing cI ′-virB4′237 were also immune to phage (Fig. 6A). Phage dilution experiments failed to reveal any difference in levels of immunity conferred by cI-virB4′312 and cI-virB′237 (data not shown). In contrast, AG1688(pTAD184) cells expressing cI ′-virB4′101 were sensitive to phage. We assayed for the synthesis of hybrid proteins in E. coli by immuno-staining. Both cI′-VirB4 and cI′-VirB4′312 accumulated at similar levels (Fig. 6B), but we were unable to detect either the cI′-VirB4′237 or cI′-VirB4′101 hybrid proteins. The VirB4′237 hybrid may be unstable in E. coli but may still accumulate to levels sufficient for conferring phage immunity. Alternatively, this hybrid protein might react poorly with our anti-VirB4 antibodies.
Fig. 6.
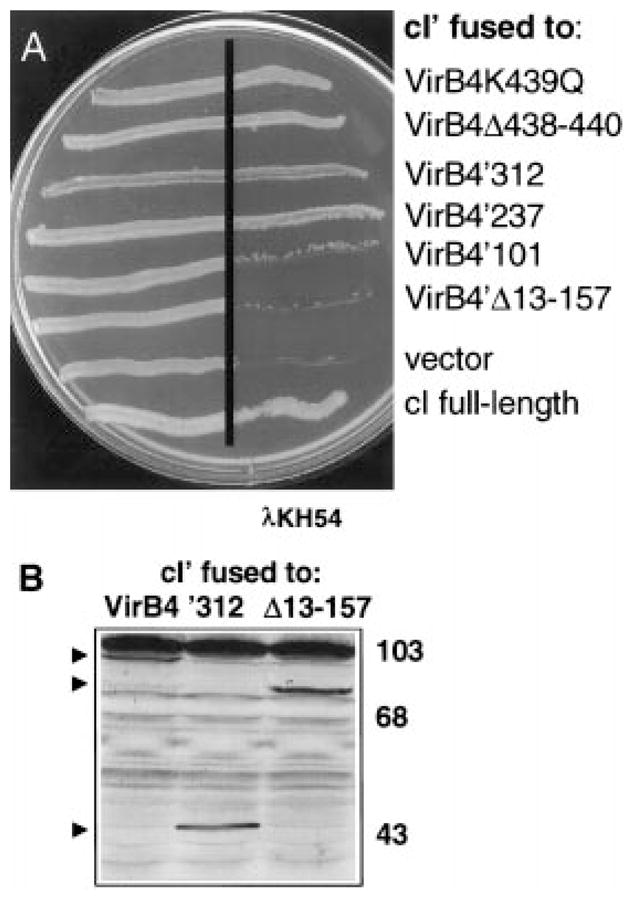
Immunity of E. coli AG1688 cells expressing cI′ fused to virB4 deletions.
A. Strains synthesizing cI′ fused to proteins indicated at the right carried the following expression plasmids: VirB4K439Q (pPC45), VirB4Δ438–440 (pPC46), VirB4′312 (pTAD182), VirB4′237 (pTAD183), VirB4′101 (pTAD184), VirB4′Δ13–157 (pTAD185), vector (pSR58), cI full-length (pJH157).
B. Immunoblot analysis of extracts from AG1688 strains synthesizing cI′ fused to the VirB4 derivatives listed. Blots developed with anti-VirB4 antisera show the presence of hybrid proteins (indicated by arrowheads) of the expected sizes. VirB4 antisera cross-reacted with a high-molecular-weight species (> 103 kDa) in the AG1688 cell extracts.
To establish further the importance of the N-terminus for VirB4 dimerization, we assessed the ability of a derivative with an N-terminal deletion to direct cI dimerization. As shown in Fig. 6A, AG1688(pTAD184) cells expressing cI-virB4Δ13–157 are sensitive to phage. Immunoblot analyses confirmed that the cI′-VirB4Δ13–157 hybrid protein accumulated at levels approximating the abundance of the cI′-VirB4 full-length and the cI′-VirB4′312 hybrid proteins (Fig. 6B). Therefore, residues 157 to the end of VirB4 do not direct dimerization. virB4Δ13–157 was recessive when expressed in A. tumefaciens merodiploid cells, and we were unable to detect the VirB4Δ13–157 mutant protein by immunostaining. Taken together, the phenotypic differences displayed by alleles coding minimally for the N-terminal third of VirB4 and alleles coding for derivatives lacking some or all of these residues suggest that the N-terminus contributes to both dimerization and protein stabilization.
The localization of a dimerization domain to the N-terminus of VirB4 suggested that dimerization occurs independently of any ATP-dependent function(s) mediated by the C-terminal region of the protein. To test this hypothesis, we assayed for dimerization potential of VirB4 derivatives with Walker A sequence mutations. Two alleles, one with a Gln codon substitution for Lys-439, and the second deleted of codons for residues Gly-438, Lys-439 and Thr-440, code for non-functional proteins, as shown by a failure to restore virulence to a virB4 null mutant. These alleles also exert negative dominance when expressed in wild-type cells (Fig. 5A). We fused these alleles to cI ′, and the resulting strains, AG1688(pPC45) expressing cI ′-virB4K439Q and AG1688(pPC46) expressing cI ′-virB4Δ438–440, were tested for resistance to phage. As shown in Fig. 6A, both strains grew in the presence of phage. Lys-439 corresponds to an invariant residue within the Walker A sequence that binds the β- and γ-phosphates of ATP (Hyde et al., 1990). Mutational analyses of numerous traffic ATPases have demonstrated that Lys substitution mutations abolish ATP hydrolysis and diminish or abolish ATP binding (see Schneider and Hunke, 1998). While it is formally possible that the VirB4K439Q mutant retains some ATP-binding activity, this is very unlikely for the more severe VirB4Δ438–440 deletion mutant. Therefore, VirB4 self-association most probably occurs independently of ATP.
Contribution of VirB4 to the stimulation of IncQ plasmid uptake during conjugation
The T-DNA transport system mobilizes the IncQ plasmid RSF1010 to plant and A. tumefaciens recipient cells (Buchanan-Wollaston et al., 1987; Beijersbergen et al., 1992). Recently, Bohne et al. (1998) reported the intriguing finding that a subset of the VirB proteins enhances RSF1010 transfer frequencies when synthesized in A. tumefaciens recipients. One model to account for these findings is that this subset of VirB proteins, which includes VirB3, VirB4 and VirB7 to VirB10, assembles as a transenvelope structure in recipient cells that can mediate DNA uptake (see Discussion).
The negative dominance of virB4′312 with respect to T-DNA export suggests that VirB4 dimer formation is important for transporter assembly. To examine the contribution of VirB4 dimerization for the assembly of a functional VirB protein complex in recipient cells, we assayed the effect of VirB4′312 synthesis on the acquisition of an RSF1010 derivative by conjugation. Consistent with the previous report (Bohne et al., 1998), A348(pML122G) donor cells transferred the IncQ plasmid at approximately three orders of magnitude higher frequency to A348 recipient cells expressing the virB genes than to isogenic PC1000 recipients deleted of the virB operon (Table 1). A348(pML122G) donors transferred the IncQ plasmid at a low frequency to the ΔvirB4 strain PC1004, demonstrating the importance of virB4 expression in this transfer process. A348(pML122G) donors also transferred the IncQ plasmid at a low frequency to A348(pTADB46) recipient cells expressing virB4′312. Thus, VirB4′312 impairs the assembly of VirB proteins both as a T-DNA exporter in donor cells and as an IncQ plasmid uptake system in recipients. The IncP plasmid pSW172 is non-self-transmissible, does not move through the VirB pore and does not influence IncQ plasmid transfer when carried in donor or recipient cells (Bohne et al., 1998).
Table 1.
Effects of expressing virB4 derivatives on IncQ plasmid uptake by A. tumefaciens recipientsa.
| Recipient strain | Transconjugants | Output donors (×106) | Tcs/donorb (± SD) | Percentage of A348(pXZ1000K)b |
|---|---|---|---|---|
| A348(pXZ1000K) | 74 132 | 871 | 8.78 (± 1.3) × 10−5 | 100 |
| PC1000(pXZ1000K) | 61 | 648 | 9.51 (± 0.9) × 10−8 | 0.11 |
| PC1004(pXZ1000K) | 54 | 591 | 9.36 (± 1.2) × 10−8 | 0.11 |
| PC1004(pZDH10) | 60 571 | 547 | 1.16 (± 0.3) × 10−4 | 132 |
| A348(pTADB46) | 642 | 414 | 1.61 (± 2.6) × 10−6 | 1.8 |
| A348(pBB15) | 75 331 | 790 | 9.48 (± 2.1) × 10−5 | 108 |
| A348(pBB17) | 60 382 | 665 | 9.21 (± 1.4) × 10−5 | 105 |
| PC1004(pBB15) | 50 443 | 496 | 9.96 (± 0.8) × 10−5 | 114 |
| PC1004(pBB17) | 78 140 | 883 | 8.94 (± 2.7) × 10−5 | 102 |
Donor strain was A348(pML122G) for all matings. Transconjugants (Tcs) and output donors were recovered after 3 days of incubation on induction media with 250 μM AS at 19°C. Donor and recipient cells were mixed in a 1:1 ratio.
Data shown are means of triplicates with standard deviations (± SD) from a single experiment. Experiments were repeated five times with similar results.
The finding that the VirB4 ATPase is important for enhanced DNA uptake in recipients, whereas the VirB11 ATPase is dispensable for this process, prompted the suggestion that VirB4 uses the energy of ATP hydrolysis to drive the assembly of the DNA transfer channel, whereas VirB11 drives substrate export (Bohne et al., 1998). If this model is correct, recipients expressing alleles for VirB4 Walker A sequence mutants should show a reduced capacity to stimulate IncQ plasmid uptake. To test this idea, we first assessed whether alleles for the Walker A sequence mutants exert negative dominance with respect to IncQ plasmid uptake. Unexpectedly, A348(pML122G) donors transferred the IncQ plasmid at similar frequencies to A348 recipients and the isogenic virB4*/virB4 merodiploid recipients (Table 1). This finding contrasts with previous demonstrations that alleles for the VirB4 Walker A sequence mutants exert dominant negative effects with respect to the export of T-DNA to plants (Berger and Christie, 1993) and IncQ plasmids to bacteria (Fullner et al., 1994). Thus, the Walker A sequence mutants appear to disrupt DNA export, but have no effect on the assembly of VirB proteins as a complex that stimulates IncQ plasmid uptake.
Next, we assessed the recipient competence of cells expressing alleles for the VirB4 Walker A sequence mutants in the absence of wild-type virB4. As shown in Table 1, A348(pML122G) donors also transferred the IncQ plasmid as efficiently to PC1004(pBB15) or PC1004(pBB17) cells synthesizing the Walker A sequence mutants as to wild-type A348 or PC1004(pZDH10) cells expressing wild-type virB4 from an IncP plasmid. The finding that the Walker A sequence mutants stimulate IncQ plasmid uptake to the same extent as native VirB4 suggests that VirB4 contributes structurally, not catalytically through ATP hydrolysis, to the assembly of the complex responsible for this DNA uptake process. The finding that Walker A sequence mutants display phenotypes specifically related to DNA export supports a model in which the ATPase activity of VirB4 contributes to the configuration of VirB proteins as a dedicated export machine.
Discussion
The T-DNA transporter displays extreme versatility in its ability to deliver DNA to a wide range of plants, bacteria, yeast and filamentous fungi and in its recognition of a diversity of substrates, including any T-DNA border-containing DNA, the mobilizable IncQ plasmid RSF1010 and several proteins in association with or independently of DNA (see Christie, 1997; de Groot et al., 1998). In addition, the recent discovery that the presence of this transport system in recipient cells greatly enhances IncQ plasmid uptake frequencies during conjugation has added a novel perspective that this export machine might also function as a DNA uptake system (Bohne et al., 1998).
How this DNA transfer system is configured architecturally and how it functions in a dynamic sense to achieve this level of promiscuity is a subject of great interest. Previous studies in this laboratory have shown that VirB4 levels are diminished by the mutation of genes for several presumed transporter components, most notably VirB6, VirB7 and VirB9. Furthermore, we have demonstrated that VirB4 levels can be modulated through the regulated synthesis of VirB6 (Dang, 1998) and the disulphide-cross-linked VirB7–VirB9 heterodimer (Fernandez et al., 1996b; X.-R. Zhou, unpublished data). These findings, coupled with the finding that alleles for VirB4 Walker A sequence mutants display negative dominance (Berger and Christie, 1993), indicate that VirB4 is a structural component of the T-DNA transport system rather than being an enzyme that, for example, catalyses transporter assembly and dissociates from the structure upon its completion.
In the present study, we have extended our characterization of the oligomeric state of VirB4. We have supplied evidence that VirB4 dimerizes in the heterologous E. coli host using the cI repressor system (Figs 1 and 2). We also demonstrated that VirB4 dimers or higher order homo-oligomers can be recovered from A. tumefaciens cell extracts by immunoprecipitation and IMAC (Figs 3 and 4). Finally, we have shown that a dimerization domain maps within the N-terminal third of VirB4 and that the allele coding for this region of VirB4 exerts dominant negative effects with respect to T-DNA export and IncQ plasmid uptake by recipient cells (Fig. 5, Table 1). Together, these findings indicate that the formation of VirB4 dimers or higher order homo-oligomers is critical for the assembly of this transport system.
The localization of a dimerization domain to the N-terminus is of interest in view of recent findings that VirB4 is configured as a polytopic membrane protein with two extra-cytoplasmic regions (Dang and Christie, 1997). One of the regions (region 1) resides within the N-terminus between residues 14 and 131, whereas the second (region 2) is in the middle of the protein immediately after the Walker A motif positioned at residues 432–440. The results of the present study suggest that region 1 contributes to VirB4 dimer formation. By analogy with other dimeric integral membrane proteins (Bormann, 1992), one or both of the TMs in region 1 might form the VirB4 dimerization interface. Examples of this family of proteins include the E. coli Tar chemoreceptor and related methyl-accepting chemotaxis proteins (Milligan and Koshland, 1988; Pakula and Simon, 1992), the E. coli EnvZ and A. tumefaciens VirA sensor kinases (Pan et al., 1993; Hidaka et al., 1997) and eukaryotic proteins such as human erythrocyte glycophorin A and various members of the tyrosine kinase receptor family (Bormann, 1992). It also is possible that the periplasmic loop of region 1 is necessary for VirB4 dimerization, as similar domains have been shown to contribute to the dimerization of integral membrane proteins including the E. coli Tar and EnvZ proteins (Gardina and Manson, 1996; Surette and Stock, 1996; Tatsuno et al., 1996; Hidaka et al., 1997) and Vibrio cholerae ToxR protein (Dziejman and Mekalanos, 1994). Our analyses of the VirB4 truncation mutants suggest that the region of VirB4 from residue 157 to the end of the protein does not possess sufficient sequence information for dimerization. However, it should be noted that this region might be required for the formation of higher order homo-oligomers or productive contacts with other VirB proteins.
Recent studies have indicated that dimerization is a dominant theme among ATPase subunits associated with a variety of membrane transport systems (Driessen, 1993; Bianchet et al., 1997). Three classes of traffic ATPases for which dimerization has been demonstrated include (i) ATPase subunits or domains of bacterial and eukaryotic members of the ABC superfamily of transporters (Davidson et al., 1996; Nikaido et al., 1997; Schneider and Hunke, 1998); (ii) Klebsiella oxytoca PulE for pullulanase secretion (Possot and Pugsley, 1994) and its homologue P. aeruginosa XcpR for exotoxin secretion via the terminal branch of the general secretory pathway (Turner et al., 1997); and (iii) E. coli SecA translocase for ATP-dependent translocation of proteins bearing signal peptides (Driessen, 1993). Precisely how these dimeric ATPases co-ordinate their enzymatic activities for achieving substrate translocation has not yet been clearly defined. For two of the best-characterized traffic ATPases, the HisP and MalK components of the histidine and maltose transport systems, it has been demonstrated recently that the ATPase subunits show co-operativity for ATP hydrolysis (Davidson et al., 1996; Nikaido et al., 1997). The co-ordinated catalytic activities of the ATPase subunits together with substrate binding are thought to induce conformational changes in the cognate integral membrane components necessary for effecting transport (Davidson et al., 1996). While it is not known how SecA monomers co-ordinate their activities, the net effect of ATP binding and hydrolysis along with preprotein binding is a cycle of SecA insertion and deinsertion in the cytoplasmic membrane needed for preprotein translocation (Economu and Wickner, 1994).
To explore the roles of VirB4 oligomerization and ATP binding/hydrolysis in substrate movement, we characterized the effects of various virB4 mutations on T-DNA export and IncQ plasmid uptake by recipient cells. This latter assay for VirB4 function was made possible by the report by Bohne et al. (1998) that the synthesis of VirB proteins in recipient cells greatly stimulates the acquisition of the IncQ plasmid RSF1010 during conjugation with wild-type donor cells. Intriguingly, these investigators also showed that the production of only a subset of VirB proteins – VirB4, VirB5 and VirB7 to VirB10 – was necessary for stimulating IncQ plasmid uptake (Bohne et al., 1998). The VirB4 requirement for this phenomenon led to the proposal that VirB4 contributes enzymatically to the assembly of a trans envelope structure through which the RSF1010 transfer intermediate can pass to gain entry into recipient cells. In contrast, VirB11, which is dispensable for stimulating plasmid uptake, was postulated to use the energy of ATP hydrolysis to drive substrate export.
Recent findings from subcellular localization and protein–protein interaction studies from several laboratories are compatible with the notion that a subset of VirB proteins can assemble as a trans envelope channel. For example, the VirB7–VirB9 heterodimer assembles at the outer membrane (Anderson et al., 1996; Spudich et al., 1996; Baron et al., 1997) and plays a critical role in stabilizing other VirB proteins (Fernandez et al., 1996b). VirB4 has been shown to be essential for the stabilization of VirB3 at the outer membrane (Jones et al., 1994). These outer membrane proteins might assemble as a structure permitting the passage of DNA substrates across the outer membrane. Topology studies indicate that VirB8 and VirB10 are bitopic cytoplasmic membrane proteins with the bulk of the proteins situated in the periplasm. Recent genetic and biochemical studies suggest that VirB10 extends through the periplasm to make direct contact with the VirB7–VirB9 heterodimer (Ward et al., 1990; Thorstenson and Zambryski, 1994; Beaupré et al., 1997; Banta et al., 1998). These proteins may therefore assemble as a channel for the movement of DNA substrates across the periplasm. VirB4, VirB8 and VirB10 are integral inner membrane proteins and, thus, might form a structure permitting substrate movement across the inner membrane.
Of further interest, homologues of the VirB proteins shown to be important for enhancing plasmid uptake by recipient cells correspond to the most highly conserved components of the adapted conjugation secretion family (see Christie, 1997). The recent discovery of genes in the R. prowazekii genome coding for homologues of VirB4, VirB8, VirB9, VirB10 and VirB11 potentially adds another member to the adapted conjugation family of toxin exporters and illustrates the importance of maintaining the functions ascribed to this subset of VirB proteins through evolutionary time (Anderson et al., 1998). Homologues of some of the VirB proteins also contribute to other DNA transfer processes. For example, two com genes, comB2 and comB3, which code for homologues of VirB9 and VirB10, respectively, are required for the development of competence by Helicobacter pylori (Hofreuter et al., 1998). Based on these evolutionary relationships, it is intriguing to speculate that the structure that mediates IncQ plasmid uptake by A. tumefaciens recipients corresponds to an ancestral channel that cells have adapted for specific purposes, such as the acquisition of exogenous DNA or the export of novel virulence factors.
Our studies of virB4 truncation derivatives have indicated that VirB4 oligomerization is important for the assembly of both the T-DNA exporter (Fig. 5) and the IncQ plasmid uptake system (Table 1). We propose that oligomerization occurs soon after VirB4 protein synthesis and independently of other VirB proteins, and that it may contribute to the stabilization of newly synthesized monomers. Whether the formation of VirB4 oligomers is an obligatory intermediate event in the channel assembly pathway or is, instead, required for VirB4 to interact productively with previously assembled channel components is currently not known. Our further studies of alleles coding for VirB4 Walker A sequence mutants showed that, whereas these alleles do not functionally substitute for the wild-type gene to mediate T-DNA export to plants (Berger and Christie, 1993), they fully substitute for the wild-type gene to stimulate IncQ plasmid uptake during conjugation (Table 1). Furthermore, these alleles exert negative dominance over the wild-type gene when merodiploids are assayed for T-DNA export, whereas they are phenotypically silent when merodiploids are assayed for IncQ plasmid uptake (Table 1). Thus, whereas VirB4 ATP binding and/or hydrolysis is important for the export of T-DNA during the infection of plants, an ATP-dependent function(s) is completely dispensable for IncQ plasmid acquisition during interbacterial conjugation.
In summary, our findings support a model in which VirB4 contributes in two ways to promote the movement of DNA across the cell envelope. First, VirB4 supplies structural information, most probably in the context of dimers or higher order homo-oligomers, for the assembly of a trans envelope channel. This structural information but not the capacity of VirB4 to bind or hydrolyse ATP promotes the assembly of a channel that can function to take up DNA and perhaps other macromolecules during conjugation. Secondly, in addition to the capacity to oligomerize, VirB4 must bind and/or hydrolyse ATP for the configuration of the VirB proteins as a dedicated export machine. Based on previous studies of traffic ATPases, we predict that VirB4 ATPase activity induces conformational changes in the transporter required for activating the export process. Such conformational changes might be required for substrate docking at the cytoplasmic face of this export machine. Alternatively, or in addition, ATP-induced conformational changes may be used to drive substrate export, possibly by a mechanism reminiscent of that described for SecA translocase (Economu and Wickner, 1994).
Experimental procedures
Enzymes and reagents
IPTG, Triton X-100, 5-bromo-4-chloro-3-indolyl-β-D-galacto-pyranoside (Xgal), nitroblue tetrazolium (NBT), phenylmethyl-sulphonyl fluoride (PMSF), carbenicillin, kanamycin and tetracycline were purchased from Sigma. n-Dodecyl-β-D-maltoside (DM) was from Anatrace. Alkaline phosphatase (AP)-conjugated goat anti-rabbit immunoglobulin G and a Bio-Rad protein assay kit was from Bio-Rad Laboratories. AS (3′,5′-dimethoxy-4′-hydroxyacetophenone) was from Aldrich. Talon (Co2+) resin and histidine monoclonal antibody were from Clontech. Protein A Sepharose CL-4B beads were from Pharmacia Biotech. Restriction endonucleases and Klenow fragment of DNA polymerase I were purchased from New England Biolabs, Promega or Gibco BRL. The four deoxyribonucleoside triphosphates were from Boehringer Mannheim.
Bacterial strains, plasmids and growth conditions
Conditions for the growth of E. coli and A. tumefaciens have been described previously (Zhou and Christie, 1997). For the analysis of Vir protein content, cells were grown to an OD600 of 0.5 in MG/L media (Fernandez et al., 1996b) with antibiotic selection. A 1 ml culture was pelleted and resuspended in induction media (IM; see Fernandez et al., 1996a) with 200 μM AS at an OD600 of 0.2. Cells were incubated with shaking at 23°C for 18 h to induce vir gene expression. Plasmids were maintained in E. coli and A. tumefaciens by the addition of carbenicillin (50 μg ml−1), kanamycin (50 μg ml−1) or tetracycline (5 μg ml−1) to the growth media.
Recombinant DNA techniques
DNA manipulations and DNA electrophoresis were performed as described by Sambrook et al. (1989). Polymerase chain reaction (PCR) amplification was performed with a Perkin-Elmer Cetus DNA thermocycler, using Taq polymerase from Promega. Oligonucleotides were synthesized by Bioserve Technologies. DNA sequencing was carried out at the DNA Core Facility of the Department of Microbiology and Molecular Genetics with an ABI 373A DNA sequencer (Perkin-Elmer, Applied Biosystems Division) using Taq polymerase.
Construction of virB4 derivatives
Table 2 lists plasmids used in these studies. Plasmids with ColE1 replication origins were ligated to the IncP broad-host-range plasmid pSW172 (Chen and Winans, 1991) for introduction into A. tumefaciens. Such plasmids are given the ColE1 plasmid name plus a ‘B’ to indicate ligation to the BHR plasmid (i.e. pTAD46 ligated to pSW172 is pTADB46). New plasmids were constructed as follows.
Table 2.
Bacterial strains and plasmidsa.
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| AG1688 | MC1061 F′128 lacI q lacZ::Tn5 | Gift from J. Hu |
| A. tumefaciens | ||
| A348 | Octopine-type Ti plasmid pTiA6NC | Garfinkel et al. (1981) |
| PC1004 | A348 deleted of virB4 from pTiA6NC | Berger and Christie (1994) |
| PC1000 | A348 deleted of virB operon from pTiA6NC | Fernandez et al. (1996a) |
| Plasmids | ||
| Vectors | ||
| pSW172 | Tet r, broad-host-range IncP plasmid with Plac and multiple cloning site | Chen and Winans (1991) |
| pSW213 | Tet r, pSW172 with lacI q | Chen and Winans (1991) |
| pXZ1000K | Kanr, Tetr, BHR IncP pSW172 ligated at Kpn I site to ColE1 pBSKSK+ | This study |
| pML122G | Crbr, IncQ RSF1010 plasmid derivative with gentamicin resistance | Fullner and Nester (1996) |
| pET15B | Crbr, cloning vector for construction of N-terminal His-tagged proteins | Novagen |
| pBIISK+.NdeI | Crbr, pBIISK+ with an NdeI site at the lacZ translational start site | Berger and Christie (1994) |
| pBIIKS+.NcoI | Crbr, pBIIKS+ with an NcoI site at the lacZ translational start site | This study |
| virB4 expression plasmids | ||
| pTAD130 | Crbr, pBIISK+.NdeI with Plac ::virB4 | This study |
| pTAD944 | Crbr, pPC914KS+ with PvirB ::virB4 | This study |
| pPC40 | Tet r, IncP pSW172 with Plac ::virB4 | This study |
| pZDH10 | Crbr, Tetr, BHR IncP–ColE1 co-integrate plasmid with Plac ::virB4 | Berger and Christie (1993) |
| pTAD214 | Tetr, IncP pSW172 with PvirB ::virB4 | This study |
| pTAD204 | Crbr, IncP pSW213 with PvirB ::virB4 | This study |
| pPC949 | Crbr, pET15b (Novagen) with PT7::his-virB4 | This study |
| pTAD150 | Crbr, pBIIKS+.NcoI with Plac ::his-virB4 | This study |
| pTAD2141 | Crbr, Tet r, IncP pSW172 with PvirB ::virB4 and Plac ::his-virB4 | This study |
| virB4 truncations | ||
| pTAD140 | Crbr, pBIISK+.NdeI with Plac ::virB4 ′780 | Dang and Christie (1997) |
| pTAD45 | Kanr, pBKSK+.NdeI with Plac ::virB4 ′611 | Dang and Christie (1997) |
| pTAD46 | Kanr, pBKSK+.NdeI with Plac ::virB4 ′312 | Dang and Christie (1997) |
| pTAD47 | Kanr, pBKSK+.NdeI with Plac ::virB4 ′237 | Dang and Christie (1997) |
| pTAD48 | Kanr, pBKSK+.NdeI with Plac ::virB4 ′101 | This study |
| pTAD190 | Crbr, pBSK+.NdeI with Plac ::virB4Δ13–157 | This study |
| pTAD110 | Crbr, pBKS+.NcoI with Plac ::virB4Δ1–273 | This study |
| virB4 ABC cassette mutants | ||
| pBB11 | Crbr, pBSIIKS+ with Plac ::virB4K439Q | Berger and Christie (1993) |
| pBB13 | Crbr, pBSIIKS+ with Plac ::virB4Δ438–440 | Berger and Christie (1993) |
| pBB15 | Crbr, Tet r, IncP pSW172 with Plac ::virB4K439Q | Berger and Christie (1993) |
| pBB17 | Crbr, Tet r, IncP pSW172 with Plac ::virB4Δ438–440 | Berger and Christie (1993) |
| pPC43 | Crbr, Tet r, IncP pSW172 with Plac ::virB4K439Q and Plac ::his-virB4 | This study |
| pPC44 | Crbr, Tet r, IncP pSW172 with Plac ::virB4Δ438–440 and Plac ::his-virB4 | This study |
| cI fusion plasmids | ||
| pJH157 | Crbr, pBR322-based vector with Plac ::cI | Hu et al. (1990) |
| pJH391 | Crbr, pBR322-based vector with Plac ::cI ′ coding for N-terminal 132 residues of cI for construction of cI-hybrid proteins | Gift from J. Hu |
| pSR58 | Crbr, pJH391 with a mutated NdeI site | S. Rashkova |
| pTAD180 | Crbr, pSR58 with Plac ::cI ′-virB11 | This study |
| pTAD181 | Crbr, pSR58 with Plac ::cI ′-virB4 | This study |
| pTAD182 | Crbr, pSR58 with Plac ::cI ′-virB4′312 | This study |
| pTAD183 | Crbr, pSR58 with Plac ::cI ′-virB4′237 | This study |
| pTAD184 | Crbr, pSR58 with Plac ::cI ′-virB4′101 | This study |
| pTAD185 | Crbr, pSR58 with Plac ::cI ′-virB4Δ13–157 | This study |
| pPC45 | Crbr, pSR58 with Plac ::cI ′-virB4K439Q | This study |
| pPC46 | Crbr, pSR58 with Plac ::cI ′-virB4Δ438–440 | This study |
Except where indicated in Table 1, when ColE1 plasmids expressing virB4 constructs were ligated to IncP plasmids pSW172 or pSW213 for introduction into A. tumefaciens; the co-integrate plasmid in A. tumefaciens is given the ColE1 plasmid name plus a B for broad host range.
Plasmids expressing
virB4. pTAD130, a 2.34 kb fragment generated by NdeI and a Pst I partial digestion of pPC945 was introduced into similarly digested pBSIISK+.NdeI; pTAD944, a 2.4 kb NdeI–XhoI fragment from pTAD130 was substituted for the 0.82 kb fragment carrying virB1 of pPC914KS+ (Berger and Christie, 1994); pPC40, a 2.8 kb PvuII fragment from pTAD130 was introduced into the StuI site of pSW172; pTAD214 and pTAD204, a 3.0 kb XbaI–KpnI fragment from pTAD944, was introduced into similarly digested pSW172 and pSW213 respectively.
Plasmids expressing his-virB4
pPC949, a 3.09 kb NdeI–XhoI restriction fragment from pPC945 (Dang and Christie, 1997) was introduced into similarly digested pET15b (Novagen); pTAD150, first an NcoI restriction site was engineered at the lacZ translational start site of pBSIIKS+ by oligo-nucleotide-directed mutagenesis (Kunkel et al., 1991) to make pBSIIKS+.NcoI, then an ≈3.1 kb NcoI–XhoI fragment from pPC949 was introduced into similarly digested pBSIIKS+.NcoI; pTAD2141, StuI-digested IncP pTAD214 was ligated with SmaI-digested ColE1 pTAD150.
Plasmids expressing virB4 deletions
pTAD48, a 0.3 kb NdeI–HindIII fragment from pPC945, was introduced into pBSKSK+.NdeI; pTAD190, an ≈2.0 kb NdeI–XhoI fragment from pTAD185 (see below) was introduced into pBSK+.NdeI; pTAD110, first an NcoI site was engineered at basepair 819 of virB4 in pPC957 (Berger and Christie, 1994) by oligonucleotide-directed mutagenesis (Kunkel et al., 1991), then a 1.5 kb NcoI–NdeI (made blunt ended with Klenow) was introduced into NcoI- and EcoRV-digested pBKS+.NcoI.
Plasmids expressing virB4 alleles coding for Walker A sequence mutations
pPC43 and pPC44, ≈2.8 kb PvuII fragments from pBB11 and pBB13, respectively, were introduced into StuI-digested pSW172. Then the resulting plasmids were digested with XhoI and ligated to similarly digested ColE1 plasmid pTAD150.
Construction of cI gene fusions
To construct a cI ′-virB11 chimeric gene, pSR58 was generated by the removal of an NdeI site in pJH157 by digestion, treatment with Klenow to create blunt ends and religation. Next, an ~ 1.0 kb fragment containing virB11 was generated by PCR amplification using upstream oligonucleotide 5′-CAG AAG CTT CAT ATG GAA GTG GAT CCG-3′ (underlined HindIII and NdeI restriction site located at the virB11 translational start site) and downstream oligonucleotide 5′-GAAC GTG TGT CGA CGG CTC ACC-3′ (Sal I site underlined). The amplified product was digested with HindIII and Sal I, and the gel-isolated fragment was introduced into plas-mid pSR58 to create pTAD180. The virB11 gene and the cI ′-virB11 junction site was sequenced to confirm that pTAD180 expresses the desired chimeric gene devoid of PCR-generated mutations. cI ′-virB4 chimeric genes were constructed by substituting the following fragments containing virB4 alleles for virB11 in pTAD180: pTAD181, a 2.4 kb NdeI–XhoI fragment from pTAD944; pTAD182, a 0.94 kb NdeI–XhoI fragment from pTAD45; pTAD183, a 0.7 kb NdeI–XhoI fragment from pTAD47; pTAD184, a 0.3 kb NdeI–XhoI fragment from pTAD48; pTAD185, by deletion of a 0.453 kb Bgl II fragment within virB4 on pTAD181, treatment with Klenow fragment to make blunt ends and religation; pPC45, by first substituting an ≈1.4 kb EcoRI fragment carrying the K439Q mutation from pBB11 for the corresponding fragment on pTAD130, then moving the ≈2.4 kb Nde1–XhoI fragment carrying the entire virB4K439Q gene into similarly digested pSR58; pPC46, by first substituting an ≈1.4 kb EcoRI fragment carrying the Δ438–440 mutation from pBB13 for the corresponding fragment on pTAD130, then moving the ≈2.4 kb Nde1–XhoI fragment carrying the entire virB4Δ438–440 gene into similarly digested pSR58. The Bgl II junction site of pTAD185 and the regions of pPC45 and pPC46 encoding the Walker A motif were sequenced to ensure that the plasmids expressed the desired chimeric genes.
Solubilization of membrane proteins
A. tumefaciens cultures (250 ml) induced with AS were centrifuged at 12 000 × g, and cell pellets were washed three times in 50 mM Tris (pH 8.0). Cells were resuspended in a solubilization buffer containing 20 mM Tris (pH 8.0), 0.1 M NaCl, 5 mM imidazole and 1 mM PMSF. Total protein extracts were prepared by cell lysis using a French pressure cell (14 000 psi). Unbroken cells were removed by centrifugation at 7000 × g for 15 min. The supernatant was transferred to a second tube, and the centrifugation step was repeated. The cleared cell lysate was diluted to a final protein concentration of 1 mg ml−1, and proteins were solubilized by the addition of dodecyl-maltoside (DM) to a final concentration of 2% and incubation overnight at 4°C with gentle inversion. The particulate fraction was removed by centrifugation for 30 min at 100 000 × g. Solubilized proteins served as the starting material for biochemical studies of the oligomeric state of VirB4.
Immunoprecipitation
We used an immunoprecipitation procedure described by Kerpolla et al. (1991) for the recovery of transport proteins in their native oligomeric state. Briefly, cell extracts were treated with 2% DM as described above to solubilize membrane proteins. The supernatant from centrifugation at 100 000 × g was diluted in 10 volumes of buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 5 mM sodium EDTA, 0.1% BSA, 0.01% DM and 1 mM PMSF. Monoclonal antiserum raised to a poly-6-histidine peptide (Clontech) was added at a titre of 1:250, and the resulting mixture was incubated at 4°C for 3 h with gentle inversion. Insoluble complexes were pelleted by brief centrifugation, followed by the addition of 100 μl of a 10% (w/v) solution of Protein A Sepharose (Pharmacia) suspended in 20 mM Tris (pH 8.0), 2% DM and 1 mM PMSF. The resulting mixture was incubated for 1.5 h at 4°C with gentle inversion. Sepharose beads were collected by centrifugation at 10 000 × g for 1 min at room temperature. Immunoprecipitates were washed several times sequentially, as described by Kerpolla et al. (1991). The pellets recovered from centrifugation of the final wash were resuspended in 50 μl of protein gel loading buffer, the samples were boiled for 5 min, and 15–20 μl were electrophoresed through SDS–polyacrylamide gels.
Immobilized ion affinity chromatography
Talon (Co2+) resin was equilibrated by washing in solubilization buffer containing 2% DM. A batch method was used to bind His-VirB4 to the Talon resin. Briefly, 100 μl of equilibrated resin was added to 1 ml of DM-solubilized proteins, and the resulting mixture was incubated overnight at 4°C with gentle inversion. The resin was pelleted by centrifugation at 14 000 × g for 1 min and washed three times in 500 μl of solubilization buffer containing 1% DM. Proteins bound to the resin were eluted with 500 μl of 100 mM imidazole in 20 mM Tris (pH 8.0). An equal volume of 2× protein sample buffer was added, and proteins were boiled for 5 min at 100°C and analysed by SDS–PAGE and immunoblotting.
Protein gel electrophoresis and immunoblot analysis
Proteins were resolved by sodium SDS–PAGE. For resolution of the 87 kDa native VirB4 and the 89 kDa His-tagged protein, protein samples were electrophoresed through a 9% (30:0.8 acrylamide:bisacrylamide) SDS–polyacrylamide gel. Proteins were transferred to nitrocellulose membranes, and blots were developed with anti-VirB4 antisera raised in rabbits to the overproduced native protein (T. A. Dang, unpublished data) or with monoclonal antibodies reactive with a 6-histidine oligopeptide (Clontech). Goat anti-rabbit antibodies conjugated to alkaline phosphatase were used for immunodetection, as described previously (Christie et al., 1988).
Lambda phage sensitivity assay
Lambda phage KH54 (≈109 pfu μl−1) was streaked down the centre of LB plates and allowed to air dry. E. coli AG1688 strains synthesizing cI repressor fusion proteins were then streaked across the lambda phage and incubated for 12, 18 or 24 h at 37°C. Resistance to phage was scored as the ability of the E. coli strain to grow on the far side of the streak (Hu, 1995; Turner et al., 1997).
Conjugation assays
Transfer of the IncQ plasmid pML122G to agrobacterial recipients was performed essentially as described by Fullner and Nester (1996). Overnight cultures of donors and recipients were suspended at an OD600 of 0.2 in induction media with or without AS and incubated with shaking for 6 h at 23°C. Five microlitres of the AS-induced donor and recipient cell cultures were spotted individually or together onto Whatman filter discs on induction media with 200 μM AS. Plates were incubated at 19°C for 3 days, and then cells were resuspended for the determination of donor, recipient and transconjugant cell numbers. Mating experiments were repeated five times, each time in triplicate.
Virulence assays
A. tumefaciens strains were tested for virulence on uniformly wounded K. daigremontiana leaves, as described previously (Berger and Christie, 1994). Controls for the tumorigenesis assays included co-inoculation of the same leaf with the wild-type strain A348 and avirulent strain A136. Assays were repeated at least five times for each strain on separate leaves. All strains were grown identically before the inoculation of plants, approximately the same numbers of cells were inoculated, and leaves used in the assays were of approximately the same age. Tumours were photographed 4–5 weeks after inoculation.
Acknowledgments
We thank Jim Hu for supplying us with advice, strains, plasmid constructs and phage for the cI repressor fusion assays. We also thank William Margolin for supplying us with anti-GFP antibodies and plasmid constructs, and Amy Davidson for suggesting that we try the Talon resin. We thank other members of the laboratory for helpful comments and suggestions. This work was supported by NIH grant GM48746.
References
- Akopyants NS, Clifton SW, Kersulyte D, Crabtree JE, Youree BE, Reece CA, et al. Analyses of the cag pathogenicity island of Helicobacter pylori. Mol Microbiol. 1998;28:37–53. doi: 10.1046/j.1365-2958.1998.00770.x. [DOI] [PubMed] [Google Scholar]
- Anderson LB, Hertzel AV, Das A. Agro-bacterium tumefaciens VirB7 and VirB9 form a disulfide-linked protein complex. Proc Natl Acad Sci USA. 1996;93:8889–8894. doi: 10.1073/pnas.93.17.8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SGE, Zomorodipour A, Andersson JO, Sicheritz-Ponten T, Alsmark UCM, Podowski RM, et al. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. doi: 10.1038/24094. [DOI] [PubMed] [Google Scholar]
- Banta LM, Bohne J, Lovejoy SD, Dostal K. Stability of the Agrobacterium tumefaciens VirB10 protein is modulated by growth temperature and periplasmic osmoadaptation. J Bacteriol. 1998;180:6597–6606. doi: 10.1128/jb.180.24.6597-6606.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron C, Thorstenson Y, Zambryski P. The lipoprotein VirB7 interacts with VirB9 in the membranes of Agrobacterium tumefaciens. J Bacteriol. 1997;179:1211–1218. doi: 10.1128/jb.179.4.1211-1218.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaupré C, Bohne J, Dale E, Binns A. Interactions between VirB9 and VirB10 membrane proteins involved in movement of DNA from Agrobacterium tumefaciens into plant cells. J Bacteriol. 1997;179:78–89. doi: 10.1128/jb.179.1.78-89.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beijersbergen A, Dulk-Ras AD, Schilperoort RA, Hooykaas PJJ. Conjugative transfer by the virulence system of Agrobacterium tumefaciens. Science. 1992;256:1324–1327. doi: 10.1126/science.256.5061.1324. [DOI] [PubMed] [Google Scholar]
- Berger BR, Christie PJ. The Agrobacterium tumefaciens virB4 gene product is an essential virulence protein requiring an intact nucleoside triphosphate-binding domain. J Bacteriol. 1993;175:1723–1734. doi: 10.1128/jb.175.6.1723-1734.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger BR, Christie PJ. Genetic complementation analysis of the Agrobacterium tumefaciens virB operon: virB2 through virB11 are essential virulence genes. J Bacteriol. 1994;176:3646–3660. doi: 10.1128/jb.176.12.3646-3660.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchet MA, Ko YH, Amzel LM, Pedersen PL. Modeling of nucleotide binding domains of ABC transporter proteins based on a F1-ATPase/RecA topology: structural model of the nucleotide binding domains of the cystic fibrosis transmembrane conductance regulator (CFTR) J Bioenerg Biomembr. 1997;29:503–524. doi: 10.1023/a:1022443209010. [DOI] [PubMed] [Google Scholar]
- Bohne J, Yim A, Binns AN. The Ti plasmid increases the efficiency of Agrobacterium tumefaciens as a recipient in virB-mediated conjugal transfer of an IncQ plasmid. Proc Natl Acad Sci USA. 1998;95:8057–7062. doi: 10.1073/pnas.95.12.7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bormann BJ. Intramembrane helix–helix association in oligomerization and transmembrane signaling. Annu Rev Biophys Biomol Struct. 1992;21:223–242. doi: 10.1146/annurev.bb.21.060192.001255. [DOI] [PubMed] [Google Scholar]
- Buchanan-Wollaston V, Passiatore JE, Cannon F. The mob and oriT mobilization functions of a bacterial plasmid promote its transfer to plants. Nature. 1987;328:172–175. [Google Scholar]
- Chen CY, Winans SC. Controlled expression of the transcriptional activator gene virG in Agrobacterium tumefaciens by using the Escherichia coli lac promoter. J Bacteriol. 1991;173:1139–1144. doi: 10.1128/jb.173.3.1139-1144.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie PJ. The Agrobacterium tumefaciens T-complex transport apparatus: a paradigm for a new family of multifunctional transporters in eubacteria. J Bacteriol. 1997;179:3085–3094. doi: 10.1128/jb.179.10.3085-3094.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie PJ, Covacci A. Bacterial type IV secretion systems: systems utilizing components of DNA conjugation machines for export of virulence factors. In: Cossart P, Boquet P, Normark S, Rappuoli R, editors. Cellular Microbiology. Washington DC: American Society for Microbiology Press; 1999. (in press) [Google Scholar]
- Christie PJ, Ward JE, Winans SC, Nester EW. The Agrobacterium tumefaciens virE2 gene product is a single-stranded-DNA-binding protein that associates with T-DNA. J Bacteriol. 1988;170:2659–2667. doi: 10.1128/jb.170.6.2659-2667.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covacci A, Falkow S, Berg D, Rappuoli R. Did the inheritance of a pathogenicity island modify the virulence of Helicobacter pylori? Trends Microbiol Sci. 1997;5:205–208. doi: 10.1016/S0966-842X(97)01035-4. [DOI] [PubMed] [Google Scholar]
- Dang TAT. PhD Thesis. The University of Texas Health Science Center at Houston; Houston, TX: 1998. Analysis of VirB4, a membrane-associated ATPase subunit essential for T-complex transport in Agrobacterium tumefaciens. [Google Scholar]
- Dang T, Christie P. The VirB4 ATPase of Agro-bacterium tumefaciens is a cytoplasmic membrane protein exposed at the periplasmic surface. J Bacteriol. 1997;179:453–462. doi: 10.1128/jb.179.2.453-462.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A. DNA transfer from Agrobacterium to plant cells in crown gall tumor disease. Subcellular Biochem. 1998;29:343–363. doi: 10.1007/978-1-4899-1707-2_11. [DOI] [PubMed] [Google Scholar]
- Davidson A, Laghaeian S, Mannering D. The maltose transport system of Escherichia coli displays positive cooperativity in ATP hydrolysis. J Biol Chem. 1996;271:4858–4863. [PubMed] [Google Scholar]
- Driessen AJ. SecA, the peripheral subunit of the Escherichia coli precursor protein translocase, is functional as a dimer. Biochemistry. 1993;32:13190–13197. doi: 10.1021/bi00211a030. [DOI] [PubMed] [Google Scholar]
- Dziejman M, Mekalanos JJ. Analysis of membrane protein interaction: ToxR can dimerize the amino terminus of phage lambda repressor. Mol Microbiol. 1994;13:485–494. doi: 10.1111/j.1365-2958.1994.tb00443.x. [DOI] [PubMed] [Google Scholar]
- Economu A, Wickner W. SecA promotes pre-protein translocation by undergoing ATP-driven cycles of membrane insertion and deinsertion. Cell. 1994;78:835–843. doi: 10.1016/s0092-8674(94)90582-7. [DOI] [PubMed] [Google Scholar]
- Fath MJ, Kolter R. ABC transporters: bacterial exporters. Microbiol Rev. 1993;57:995–1017. doi: 10.1128/mr.57.4.995-1017.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez D, Dang TAT, Spudich GM, Zhou XR, Berger BR, Christie PJ. The Agrobacterium tumefaciens virB7 gene product, a proposed component of the T-complex transport apparatus, is a membrane-associated lipoprotein exposed at the periplasmic surface. J Bacteriol. 1996a;178:3156–3167. doi: 10.1128/jb.178.11.3156-3167.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez D, Spudich GM, Zhou XR, Christie PJ. The Agrobacterium tumefaciens VirB7 lipoprotein is required for stabilization of VirB proteins during assembly of the T-complex transport apparatus. J Bacteriol. 1996b;178:3168–3176. doi: 10.1128/jb.178.11.3168-3176.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullner KJ, Nester EW. Temperature affects the T-DNA transfer machinery of Agrobacterium tumefaciens. J Bacteriol. 1996;178:1498–1504. doi: 10.1128/jb.178.6.1498-1504.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullner KJ, Stephens KM, Nester EW. An essential virulence protein of Agrobacterium tumefaciens, VirB4, requires an intact mononucleotide binding domain to function in transfer of T-DNA. Mol Gen Genet. 1994;245:704–715. doi: 10.1007/BF00297277. [DOI] [PubMed] [Google Scholar]
- Fullner KJ, Lara JC, Nester EW. Pilus assembly by Agrobacterium T-DNA transfer genes. Science. 1996;273:1107–1109. doi: 10.1126/science.273.5278.1107. [DOI] [PubMed] [Google Scholar]
- Gardina PJ, Manson MD. Attractant signaling by an aspartate chemoreceptor dimer with a single cytoplasmic domain. Science. 1996;274:425–426. doi: 10.1126/science.274.5286.425. [DOI] [PubMed] [Google Scholar]
- Garfinkel DJ, Simpson RB, Ream LW, White FF, Gordon MP, Nester EW. Genetic analysis of crown gall: fine structure map of the T-DNA by site-directed mutagenesis. Cell. 1981;27:143–153. doi: 10.1016/0092-8674(81)90368-8. [DOI] [PubMed] [Google Scholar]
- de Groot MJA, Bundock P, Hooykaas PJJ, Beijersbergen AGM. Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nature Biotechnol. 1998;16:839–842. doi: 10.1038/nbt0998-839. [DOI] [PubMed] [Google Scholar]
- Hidaka Y, Park H, Inouye M. Demonstration of dimer formation of the cytoplasmic domain of a transmembrane osmosensor, EnvZ, of Escherichia coli using Ni-histidine tag affinity chromatography. FEBS Lett. 1997;400:238–242. doi: 10.1016/s0014-5793(96)01396-8. [DOI] [PubMed] [Google Scholar]
- Hofreuter D, Odenbreit S, Henke G, Haas R. Natural competence for DNA transformation in Helicobacter pylori : identification and genetic characterization of the comB locus. Mol Microbiol. 1998;28:1027–1038. doi: 10.1046/j.1365-2958.1998.00879.x. [DOI] [PubMed] [Google Scholar]
- Hu J. Repressor fusions as a tool to study protein–protein interactions. Structure. 1995;3:431–433. doi: 10.1016/s0969-2126(01)00176-9. [DOI] [PubMed] [Google Scholar]
- Hu JC, O’Shea EK, Kim PS, Sauer RT. Sequence requirements for coiled-coils: analysis with lambda repressor-GCN4 leucine zipper fusions. Science. 1990;250:1814–1822. doi: 10.1126/science.2147779. [DOI] [PubMed] [Google Scholar]
- Hueck CJ. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev. 1998;62:379–433. doi: 10.1128/mmbr.62.2.379-433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde SC, Emsley P, Hartshorn MJ, Mimmack MM, Gileadi U, Pearce SR, et al. Structural model of ATP-binding proteins associated with cystic fibrosis, multidrug resistance, and bacterial transport. Nature. 1990;346:362–365. doi: 10.1038/346362a0. [DOI] [PubMed] [Google Scholar]
- Jones AL, Shirasu K, Kado CI. The product of virB4 gene of Agrobacterium tumefaciens promotes accumulation of VirB3 protein. J Bacteriol. 1994;176:5255–5261. doi: 10.1128/jb.176.17.5255-5261.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung JK, Chung EH, King G, Yu C, Hirsh AS, Hochschild A. Genetic strategy for analyzing specificity of dimer formation: Escherichia coli cyclic AMP receptor protein mutant altered in its dimerization specificity. Genes Dev. 1995;9:2986–2996. doi: 10.1101/gad.9.23.2986. [DOI] [PubMed] [Google Scholar]
- Kerpolla RE, Shyamala VK, Klebba P, Ames GF. The membrane-bound proteins of periplasmic permeases form a complex: Identification of the histidine permease HisQMP complex. J Biol Chem. 1991;266:9857–9865. [PubMed] [Google Scholar]
- Kunkel TA, Bebenek K, McClary J. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol. 1991;204:125–139. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- Lai EM, Kado CI. Processed VirB2 is the major subunit of the promiscuous pilus of Agrobacterium tumefaciens. J Bacteriol. 1998;180:2711–2717. doi: 10.1128/jb.180.10.2711-2717.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan DL, Koshland DE., Jr Site-directed cross-linking. Establishing the dimeric structure of the aspartate receptor of bacterial chemotaxis. J Biol Chem. 1988;263:6268–6275. [PubMed] [Google Scholar]
- Nikaido K, Liu PQ, Ames GF. Purification and characterization of HisP, the ATP-binding subunit of a traffic ATPase (ABC transporter), the histidine permease of Salmonella typhimurium. Solubility, dimerization, and ATPase activity. J Biol Chem. 1997;272:27745–27752. doi: 10.1074/jbc.272.44.27745. [DOI] [PubMed] [Google Scholar]
- Pakula AA, Simon MI. Determination of trans-membrane protein structure by disulfide cross-linking: the Escherichia coli Tar receptor. Proc Natl Acad Sci USA. 1992;89:4144–4148. doi: 10.1073/pnas.89.9.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan WQ, Charles T, Jin S, Wu ZL, Nester EW. Preformed dimeric state of the sensor protein VirA is involved in plant–Agrobacterium signal transduction. Proc Natl Acad Sci USA. 1993;90:9939–9943. doi: 10.1073/pnas.90.21.9939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possot O, Pugsley AP. Molecular characterization of PulE, a protein required for pullulanase secretion. Mol Microbiol. 1994;12:287–299. doi: 10.1111/j.1365-2958.1994.tb01017.x. [DOI] [PubMed] [Google Scholar]
- Pugsley AP. The complete general secretory pathway in Gram-negative bacteria. Microbiol Rev. 1993;57:50–108. doi: 10.1128/mr.57.1.50-108.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashkova S, Spudich GM, Christie PJ. Mutational analysis of the Agrobacterium tumefaciens VirB11 ATPase: identification of functional domains and evidence for multimerization. J Bacteriol. 1997;179:583–589. doi: 10.1128/jb.179.3.583-591.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: a Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schneider E, Hunke S. ATP-binding-cassette (ABC) transport systems: functional and structural aspects of the ATP-hydrolyzing subunits/domains. FEMS Microbiol Rev. 1998;22:1–20. doi: 10.1111/j.1574-6976.1998.tb00358.x. [DOI] [PubMed] [Google Scholar]
- Segal G, Schuman HA. Characterization of a new region required for macrophage killing by Legionella pneumophila. Infect Immun. 1997;65:5057–5066. doi: 10.1128/iai.65.12.5057-5066.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng J, Citovsky V. Agrobacterium–plant cell DNA transport: have virulence proteins, will travel. Plant Cell. 1996;8:1699–1710. doi: 10.1105/tpc.8.10.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich GM, Fernandez D, Zhou XR, Christie PJ. Intermolecular disulfide bonds stabilize VirB7 homodimers and VirB7/VirB9 heterodimers during biogenesis of the Agrobacterium tumefaciens T-complex transport apparatus. Proc Natl Acad Sci USA. 1996;93:7512–7517. doi: 10.1073/pnas.93.15.7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens KM, Roush C, Nester E. Agrobacterium tumefaciens VirB11 protein requires a consensus nucleotide-binding site for function in virulence. J Bacteriol. 1995;177:27–36. doi: 10.1128/jb.177.1.27-36.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surette MG, Stock JB. Role of alpha-helical coiled-coil interactions in receptor dimerization, signaling, and adaptation during bacterial chemotaxis. J Biol Chem. 1996;271:17966–17973. doi: 10.1074/jbc.271.30.17966. [DOI] [PubMed] [Google Scholar]
- Tatsuno I, Homma M, Oosawa K, Kawagishi I. Signaling by the Escherichia coli aspartate chemo-receptor Tar with a single cytoplasmic domain per dimer. J Biol Chem. 1996;274:423–425. doi: 10.1126/science.274.5286.423. [DOI] [PubMed] [Google Scholar]
- Thorstenson YR, Zambryski PC. The essential virulence protein VirB8 localizes to the inner membrane of Agrobacterium tumefaciens : Implications for the formation of a T-DNA transport structure. J Bacteriol. 1994;176:1711–1717. doi: 10.1128/jb.176.6.1711-1717.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummuru MKR, Sharma SA, Blaser MJ. Helicobacter pylori, picB, a homologue of the Bordetella pertussis toxin secretion protein, is required for induction of IL-8 in gastric epithelial cells. Mol Microbiol. 1995;18:867–876. doi: 10.1111/j.1365-2958.1995.18050867.x. [DOI] [PubMed] [Google Scholar]
- Turner LR, Olson JW, Lory S. The XcpR protein of Pseudomonas aeruginosa dimerizes via its N-terminus. Mol Microbiol. 1997;26:877–887. doi: 10.1046/j.1365-2958.1997.6201986.x. [DOI] [PubMed] [Google Scholar]
- Vogel JP, Andrews HL, Wong SK, Isberg RR. Conjugative transfer by the virulence system of Legionella pneumophila. Science. 1998;279:873–876. doi: 10.1126/science.279.5352.873. [DOI] [PubMed] [Google Scholar]
- Ward JE, Dale EM, Nester EW, Binns AN. Identification of a virB10 protein aggregate in the inner membrane of Agrobacterium tumefaciens. J Bacteriol. 1990;172:5200–5210. doi: 10.1128/jb.172.9.5200-5210.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss AA, Johnson FD, Burns DL. Molecular characterization of an operon required for pertussis toxin secretion. Proc Natl Acad Sci USA. 1993;90:2970–2974. doi: 10.1073/pnas.90.7.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winans SC, Burns DL, Christie PJ. Adaptation of a conjugal transfer system for the export of pathogenic macromolecules. Trends Microbiol Sci. 1996;4:64–68. doi: 10.1016/0966-842X(96)81513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XR, Christie PJ. Suppression of mutant phenotypes of the Agrobacterium tumefaciens VirB11 ATPase by overproduction of VirB proteins. J Bacteriol. 1997;179:5835–5842. doi: 10.1128/jb.179.18.5835-5842.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupan JR, Zambryski P. Transfer of T-DNA from Agrobacterium to the plant cell. Plant Physiol. 1995;107:1041–1047. doi: 10.1104/pp.107.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]