

Abstract
Hypothesis
Intracranial vestibular schwannoma xenografts can be successfully established and followed with bioluminescent imaging (BLI).
Background
Transgenic and xenograft mouse models of vestibular schwannomas have been previously reported in the literature. However, none of these models replicate the intracranial location of these tumors to reflect the human disease. Additionally, traditional imaging methods (magnetic resonance imaging, computed tomography) for following tumor engraftment and growth are expensive and time consuming. BLI has been successfully used to longitudinally follow tumor treatment responses in a noninvasive manner. BLI’s lower cost and labor demands make this a more feasible approach for tumor monitoring in studies involving large numbers of mice.
Methods
Patient excised vestibular schwannomas were cultured and transduced with firefly luciferase expressing lentivirus. One million cells were stereotactically injected into the right caudate nucleus of 21 nonobese diabetic/severe combined immunodeficient mice. Schwannoma engraftment and growth was prospectively followed for 30 weeks after injection with BLI. After animal sacrifice, the presence of human tumor cells was confirmed with fluorescent in situ hybridization.
Results
Eight (38%) of 21 mice successfully engrafted the schwannoma cells. All of these mice were generated from 4 (67%) of the 6 patient excised tumors. These 8 mice could be differentiated from the nonengrafted mice at 21 weeks. The engrafted group emitted BLI of greater than 100,000 photons/s (range, 142,478–3,106,300 photons/s; average, 618,740 photons/s), whereas the nonengrafted group were all under 100,000 photons/s (range, 0–76,010 photons/s; average, 10,737 photons/s) (p < 0.001). Fluorescent in situ hybridization analysis confirmed the presence of viable human schwannoma cells in much greater numbers in those mice with stable or growing tumors compared with those whose tumors regressed.
Conclusion
We have successfully established an intracranial schwannoma xenograft model that can be followed with noninvasive BLI. We hope to use this model for in vivo testing of schwannoma tumor therapies.
Keywords: Acoustic neuroma, Bioluminescence, NOD/SCID mice, Vestibular schwannoma, Xenograft
Useful animal models for translational research and therapeutic testing before human studies are important for the study of any disease process. Schwannomas have been studied through 2 main mouse modeling strategies: xenograft tumors in immunocompromised mice and transgenic mouse models. PoCre conditional Nf2 knockout transgenic mice have been created that demonstrate peripheral nerve Schwann cell hyperplasia and a few peripheral schwannomas such as uterine schwannomas in the female mice. However, these transgenic models do not accurately replicate the intracranial location of schwannomas observed in human NF2-associated and sporadic schwannomas (1,2). Additionally, time and expense of creating these mouse models may limit studies that require large animal numbers to evaluate tumor therapies in vivo. Although transgenic models are extremely valuable in studying schwannoma tumorigenesis, xenograft models may enable the evaluation of a wider array of treatment responses in patient tumors with different underlying genetic abnormalities.
Xenografted tumors have shown some success in the ability to grow human schwannoma tumors, but the growth rates are not consistent, and the tumor location of most models (flank, thigh) do not mimic the human disease (intracranial tumors) (3–6). Orthotopic xeno-transplants have the advantage that they derive from human tumors; therefore, they most closely resemble the pathologic process, and the tumor or cells are implanted in the area of interest such as the cerebrospinal fluid space. Studies of other malignant central nervous system tumor types, such as gliomas, have used intracranial mouse xenografts as an experimental model. In one model, primary glioblastoma tumors were implanted and maintained as heterotopic flank tumors, and cells derived from these flank xenografts could be injected into the brains of athymic mice to establish tumors that mimic the tumor growth and histology of the human disease (7). A mouse xenograft model of benign intracranial meningiomas has also been developed in which 85% of mice developed tumors after subdural meningioma cell injection (8). The use of an intracranial schwannoma xenograft would be important to properly test targeted therapeutics and the potential barriers that they may face (i.e., poor cerebrospinal fluid penetration). An intracranial xenograft created from human vestibular schwannoma cells has not been previously reported.
Bioluminescent imaging (BLI), similar to other imaging modalities such as magnetic resonance imaging (MRI) and positron emission tomography, can provide a means to noninvasively follow tumor growth in animal models without sacrificing the animal until study completion. Bioluminescence is a naturally occurring reaction catalyzed by the enzyme luciferase, which facilitates the oxidation of luciferin. This reaction converts chemical energy into photon energy or light. Using a lentivirus encoding the firefly luciferase gene that will integrate into the genome, luciferase can be stably expressed in tumor cells for multiple generations. When a source of exogenous luciferin is introduced, only viable tumor cells will emit light. BLI has been used in many cancer mouse models to longitudinally follow tumor responses to treatment (9–11). Many of these studies showed that tumor growth and regression correlated to increases and decreases of bioluminescent intensity over time. This imaging modality is much cheaper and less labor intensive for imaging large numbers of mice as compared with other modalities, such as gadolinium-enhanced MRI scans that can also be used to follow schwannoma growth in vivo.
METHODS
Permission for this project was obtained through the institutional review board (07-001276) and the Institutional Animal Care and Use Committee (A8007).
Transduction Schwannoma Cells With Firefly Luciferase
Lentiviruses were generated by cotransfection of 293T cells with 3 plasmids: gag-pol, env, and pHR-SIN-CSGW dlNotI-luciferase (Fig. 1) (provided by Galanis Lab). Forty-eight hours after transfection, virus containing supernatant was collected and passed through a 22-μm filter. The filtered supernatant was used to infect the target cells.
FIG. 1.

Lentivirus construct used to transduce schwannoma cells with luciferase gene. Essential viral cis elements for vector infection: LTRs indicate long terminal repeats; RRE, rev-responsive element; ψ (packaging signal); SIN, self-inactivated vector; cPPT, HIV central polypurine tract or central flap; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element; SFFV promoter, spleen focus forming virus—has strong promoter activity in most human or mouse cells. BamHI and NotI are restriction endonuclease sites.
Six patient excised sporadic vestibular schwannomas were used to culture tumor cells that were injected into 1 to 5 mice, depending on the number of cells available. This gave a total of 21 mice for the study. Individual tumor samples were minced into small pieces (<1 mm) and then cultured in Dulbecco modified Eagle medium (Fisher Scientific, Pittsburgh, PA, USA) with 10% fetal clone III calf serum (HyClone; ThermoFischer Scientific, Waltham, MA, USA). After 1 to 7 days, the cells were further dissociated via digestion for 24 hours with 160 U/ml collagenase (Sigma, St. Louis, MO, USA) and 1.25 U/ml dispase (Boehringer Mannheim, Mannheim, Germany) in media, followed by repetitive pipetting to create a cell suspension. After short-term culture of the cells (7–14 d) in Dulbecco modified Eagle medium, 10% fetal clone III calf serum, 0.5 μmol/L forskolin (Sigma), 10 nmol/L B1-heregulin (R & D Systems, Minneapolis, MN, USA), 0.5 mmol/L 3-isobutyl-1-methylxanthine (Sigma), 2.5 μg/ml Insulin (Sigma), and 500 U/ml 1% penicillin/streptomycin (Fischer Scientific), cells were exposed to the nonreplicating lentivirus from the prepared supernatant. This effectively inserted the luciferase gene into the tumor cell deoxyribonucleic acid (DNA). A separate culture was transduced with the same conditions using a similar lentivirus encoding the gene for DS Red fluorescent protein instead of luciferase, and this served as a visual positive control for viral gene delivery (Fig. 2). The cells were trypsinized and washed 2 times in 2X phosphate-buffered saline. Cells were resuspended in phosphate-buffered saline at 105 cells/μl in preparation for injection with a Hamilton syringe.
FIG. 2.
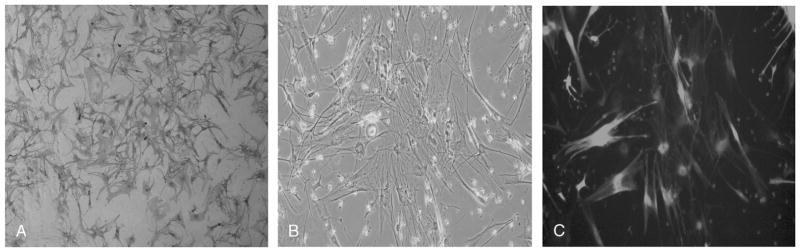
A, Positive immunohistochemical staining of schwannoma cells with S100 (Dako, Galanis Lab, Mayo Clinic, Rochester, NY, USA) after passage 2. B, Lentiviral (vector pHR-SIN-CSGW dlNotI-DS Red) transduction of schwannoma cells. Light microscope image (original magnification, ×10) of transduced schwannoma cells in culture. C, Same image with fluorescent light source. DS red fluorescence indicates transduction of these cells with lentivirus in greater than 90% of cells.
Creation of Intracranial Schwannoma Xenografts
Female nonobese diabetic/severe combined immunodeficient (SCID) mice (Jackson Laboratories, Bar Harbor, MN, USA), 6 to 8 weeks of age, were used for schwannoma xenograft experiments. After anesthesia with ketamine (90–120 mg/kg) and xylazine (10 mg/kg), a small midline scalp incision was used to expose the calvaria. A small burr hole was drilled 2.5 mm lateral (right) and 0.5 mm anterior to the bregma. With the aide of a small animal stereotactic frame (ASI Instruments, Houston, TX, USA), cells were injected at a rate of 5 μl/min for 2 minutes (total of 106 cells per mouse) at a depth of 3 mm. The injection syringe was left in place for 7 minutes after the completion of tumor cell injection. Injections were done at a depth of 3 mm below the outer table of the cranium, which introduced the cells into the right caudate nucleus.
Bioluminescent Imaging
The mice were anesthetized with an intraperitoneal injection of ketamine and xylazine (dosage above), which was combined with D-luciferin potassium (150 mg/kg; Caliper Life Sciences, Xenogen Corp., Hopkinton, MA, USA). Ten minutes after luciferin injection, mice were imaged with an IVIS 2.50 cooled charged-coupled device camera (Xenogen 200 series). For BLI analysis, an intracranial area of signal was defined using Living Image software, and the total number of photons per second per steradian centimeter was recorded. Because of the increased use of BLI at our institution, a step-down barrier room has been created in which only immunocompromised mice can be housed and handled because these mice cannot be returned to the formal barrier area after their initial imaging. The IVIS charged-coupled device camera is contained within this area because it is not currently a portable system. Mice were imaged every 3 to 4 weeks after implantation of schwannoma cells, and at the end of 30 weeks, surviving mice were killed. All of the study mice had their entire brain sectioned at 5-μm slices at 100-μm intervals.
Fluorescent in Situ Hybridization
The fluorescent in situ hybridization (FISH) protocol for paraffin-embedded tissues initially required deparaffinization in sequential room temperature baths of Citrosolv (Fischer Scientific) for 15 minutes X 2, and 100% ethanol for 5 minutes X 2. Slides were placed in Coplin jars filled with 10 mmol/L citric acid (pH 6.08) (3.84 g citric acid/2 L H2O) and boiled in a microwave water bath. Tissue digestion was done by placing slides in pepsin (4 mg pepsin/L 0.9% NaCl) for 15 minutes in a 37°C water bath. The slides were put in sequential alcohol baths for 2 minutes: 70% ethyl alcohol (ETOH), 80% ETOH, and 100% ETOH. Slides were codenatured and hybridized with a probe cocktail in the hybridization oven at 80°C for 5 minutes and then 37°C overnight. Slides were washed with 2× standard saline citrate/0.1% Nonidet P-40 for 2 minutes at 73°C, and 1 drop of 1× 4c6-diamidino-2-phenylindole/antifade was applied to the hybridized area. A fluorescent microscope was used to count red fluorescent human cells.
Mouse and human whole genomic probe cocktail was created as follows. Male mouse whole genomic DNA was extracted from several mouse spleens using the Qiagen DNeasy Blood & Tissue Kit. The extracted DNA was quantified and then labeled using the Nick Translation Kit and Spectrum Green deoxyuridine 5-triphosphate (Abbott Molecular, Abbott Park, IL, USA). After nick translation, the labeled DNA was precipitated and reconstituted to a final concentration of 50 ng. The commercially available human whole genomic probe (from Abbott Molecular, Abbott Park, IL, USA) labeled with SpectrumRed was used as the human probe in this research. One microliter of each probe was added to 8 μl of locus-specific/whole chromosome paint hybridization buffer (Abbott Molecular, Abbott Park, IL, USA) to create a working solution. After the paraffin pretreatment method, 10 μl of probe was applied to each specimen and hybridized according to the paraffin method (see above).
Statistical Analysis
A 2-sided Wilcoxon rank-sum test was used to compare the bioluminescent image intensity at 21 weeks between the 8 mice that showed stable or increasing tumors over the study time period and the 10 mice that failed to engraft their tumor. We excluded the 3 mice that died as a result of anesthetic complications from this analysis. This test compared the median BLI value between the 2 groups.
RESULTS
BLI was used to serially image 21 intracranial schwannoma xenograft nonobese diabetic/SCID mice over an extended 30-week period. Overall, 8 (38%) of 21 mice showed engraftment demonstrated by stable or slightly increasing bioluminescent emissions up to 30 weeks after injection (Fig. 3). All of these mice were generated from 4 (67%) of the 6 patient excised tumors. Patient Tumors 1 to 4 resulted in engraftment rates in the mice of 100% (1 of 1), 80% (4 of 5), 67% (2 of 3), and 20% (1 of 5), respectively. The remaining Patient Tumors 5 and 6 did not result in any successful engrafted tumors in the mice. We purposefully tried to select larger patient tumors from younger patients to generate cells for tumor injection: age range of 23 to 53 years and tumor size (largest cerebellopontine angle dimension) at presentation of 2.6, 2.2, 2.0, 3.9 (Tumors 1–4), 2.4, and 1.0 cm (Tumors 5 and 6). The tumors that led to mouse engraftment were Tumors 1 to 4, and there was no correlation between patient age and whether they showed engraftment. There was a trend toward larger tumors leading to engraftment; however, the sample size was too small to statistically analyze this. Figure 4 demonstrates consecutive images obtained from Mouse 6 (Patient Tumor 2, 2.2 cm), which had the most robust tumor emissions, and Mouse 19 (Patient Tumor 5, 1.0 cm) in which initial viable tumor cells failed to engraft. Of the remaining 13 mice, 3 mice died as a result of anesthetic complications, and either the remainder failed to show any tumor engraftment or the initial tumors declined to a reading of very low or unrecordable photon emission levels over a varying amount of time (most at <20 wk). In these mice, we saw a definite trend that the group of 8 stable or growing tumors had a bioluminescent emission greater than 100,000 photons/s (range, 142,478–3,106,300 photons/s; average, 618,740 photons/s) at 21 weeks, whereas the remaining mice had much lower intensities (range, 0–76,010 photons/s; average, 10,737 photons/s). All of these latter mice had their tumors either completely or nearly completely regressed by the study end point at 30 weeks. There was a statistically significant difference between the BLI at 21 weeks in the engrafting and nonengrafting group when compared by a 2-sided Wilcoxon rank-sum test (p < 0.001). Therefore, at 21 weeks after implantation, mice that are going to engraft their tumors can be differentiated from those mice that will not sustain or grow their tumors.
FIG. 3.

Graphs showing that 8 of 21 SCID mice injected intracranially with human schwannoma cells showed stable engraftment at 21 weeks of follow-up. The remainder of mice had little or no measurable recordings after a varying amount of time (most at <20 wk).
FIG. 4.
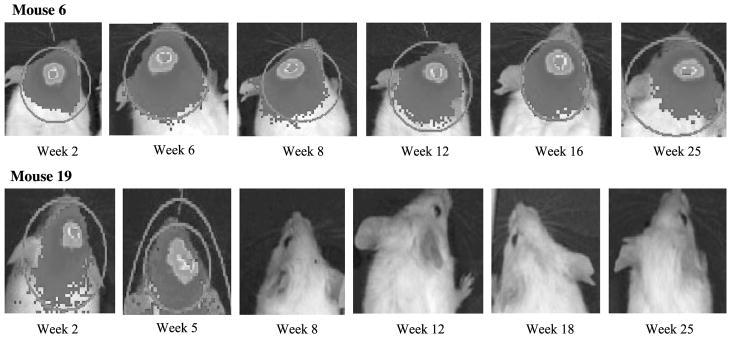
Imaging of bioluminescent emissions from Mice 6 (top; Patient Tumor 2, 2.2 cm) and 19 (bottom; Patient Tumor 5, 1.0 cm) over a 25-week period after tumor cell injection. Mouse 6 demonstrated significant early tumor growth and then persistence until the mouse was killed because of inanition at 25 weeks. Mouse 19 showed decline of emissions and tumor burden with failed engraftment.
Light microscopy was performed on hematoxylin and eosin–stained slides, and gross schwannoma tumor pathologic findings were difficult to observe with certainty in any of the mice. We think that this is due to injecting cell suspensions in which the tumor architecture and capsule are not maintained. As an alternative to preserve tumor architecture, implanting intracranial pieces of tumor is possible; however, we performed minicraniotomies on 10 mice and implanted 1 mm3 pieces of human schwannoma tumor. None of these mice demonstrated tumor on light microscopy after 16 weeks (data not shown). To aid in identifying viable human schwannoma cells, FISH was performed using a human whole genomic probe labeled with SpectrumRed and a mouse whole genomic probe labeled with SpectrumGreen (Abbott Molecular). All mice with bioluminescence demonstrated robust viable human staining cells ranging from 244 to 1,854 cells observed on imaged slices. In contrast, mice without emissions showed very few tumor cells on FISH analysis ranging from 9 to 20 cells observed on imaged sections (Fig. 5).
FIG. 5.

FISH analysis of mouse brain sections under fluorescent microscope (original magnification, ×5). Human schwannoma cells show red fluorescence in the background of green mouse brain cells. Large arrows, tumor islet; small arrows, sheets of tumor cells along the ventricle.
DISCUSSION
Recent reports of BLI in other xenograft models strongly support its use to effectively follow tumor therapy responses in longitudinal studies (12). The key advantages of BLI over more conventional imaging such as MRI or positron emission tomography is the number of mice that can be imaged in a given period with a corresponding significant savings in cost. BLI allows for relatively high throughput with imaging of 150 to 200 mice per day, which is approximately 15 times the number that could be done per day with MRI (13). Additionally, at our institution, the imaging cost per mouse is conservatively 10 times as much for MRI compared with BLI.
Strong correlations between BLI photon quantification and tumor size and growth as assessed by traditional methods or other animal imaging modalities have been demonstrated in several solid tumor models (14,15). Additionally, researchers have recently used a similar stereotactic injection to the methods used in our study to evaluate intracranial glioblastoma multiforme response to treatment. They found a high correlation (R2 = 0.94) between histopathologic volume of tumor and bioluminescent photon emissions (12). Likely because of the benign and noninvasive characteristics of human schwannoma cells, we were not able to identify large areas of tumor architecture by light microscopy. However, we were able to show abundant FISH-labeled human schwannoma cells in those mice with high bioluminescence recordings compared with very few or no cells in those with little or no photon emissions.
The overall engraftment rate (38%) in this study does not reach engraftment rates observed in xenografted malignancies such as acute myelogenous leukemia and multiple myeloma (70–80%); however, it is very similar to the xenograft take-rates in the Mayo glioblastoma multiforme xenograft model (unpublished data) and in other recent publications of xenografting with vestibular schwannomas (3,4,16,17). Attempts are currently being made to improve engraftment rates by selecting rapidly growing human tumors for mouse xenografting. Additionally, improved intracranial engraftment may be attained by passaging tumors in groups of mice (flank position) to select for more aggressive tumors before intracranial injection. This may also allow establishment of a larger cohort of mice derived from a single patient tumor to allow for better therapeutic testing. A conditional transgenic mouse model expressing luciferase in small cell lung carcinoma has been reported (18). This may be a future option for schwannoma modeling if transgenic mice can be designed to exhibit intracranial tumors.
A potential criticism of this model is that tumors showed either maintenance or very slow increases of photon emissions between Weeks 13 to 29, which is contrary to the experience of rapid sustained growth in malignant xenograft studies. Consequently, therapies will have to produce significant cytotoxic effects (decreased tumor size) to see a treatment outcome difference compared with a control group consisting of several slowly growing or stable tumors. However, this study does accurately model this benign tumor’s behavior in humans in which the average human schwannoma grows approximately 1 to 2 mm/yr (19). Thus, we think that this accurately reflects human tumor growth and that any potential human therapies will also have to show a cytotoxic effect in future human trials. A second criticism is that, although this model reflects the intracranial location of the tumor, the caudate nucleus location is artificial when considering that these tumors are Cranial Nerve VIII, extra-axial–derived tumors. Better image-guided xenograft injection techniques or transgenic models that initiate extra-axial tumor growth are needed to overcome this obstacle. A final criticism is that injection of tumor cells requires culturing of the cells that may alter the cell characteristics before implantation. However, this model used freshly created cell lines that were not subjected to multiple passages, and again, we were unable to establish any engrafted tumors in 10 mice in which we implanted tumor pieces directly into the intracranial space.
CONCLUSION
We report the first successful intracranial schwannoma xenograft with the goal of providing a model for studying schwannoma therapies in an anatomically appropriate context. As a secondary goal, we wanted to develop an inexpensive and readily available method of following tumor growth or regression, which will be necessary to initiate future animal therapy trials. BLI is such a method to allow rather inexpensive and noninvasive follow-up of mouse tumors.
Acknowledgments
This study was funded by a Mayo Institutional grant (Knowlton grant).
The authors thank Heather Flynn Gilmer, for assistance with the FISH protocol, and Amy L. Weaver, for statistical analysis. The authors also thank Bernd Scheithauer, M.D. (neuropathology), for expert review of the mouse brain histology.
References
- 1.Giovaninni M, Robanus-Maandag E, Niwa-Kawakita M, et al. Schwann cell hyperplasia and tumors in transgenic mice expressing a naturally occurring mutant NF2 protein. Genes Dev. 1999;13:978–86. doi: 10.1101/gad.13.8.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giovannini M, Robanus-Maandag E, van der Valk M, et al. Conditional biallelic Nf2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Genes Dev. 2000;14:1617–30. [PMC free article] [PubMed] [Google Scholar]
- 3.Messerli SM, Prabhakar S, Tang Y, et al. Treatment of schwannomas with an oncolytic recombinant herpes simplex virus in murine models of neurofibromatosis type 2. Hum Gene Ther. 2006;17:20–30. doi: 10.1089/hum.2006.17.20. [DOI] [PubMed] [Google Scholar]
- 4.Chang LS, Jacob A, Rock J, et al. Growth of benign and malignant schwannoma xenografts in severe combined immunodeficiency mice. Laryngoscope. 2006;116:2018–26. doi: 10.1097/01.mlg.0000240185.14224.7d. [DOI] [PubMed] [Google Scholar]
- 5.Stidham KR, Roberson JB. Human vestibular schwannoma growth in the nude mouse: evaluation of a modified subcutaneous implantation model. Am J Otol. 1997;18:622–6. [PubMed] [Google Scholar]
- 6.Kumazawa H, Kyoumito R, Matsumato A, et al. A severe combined immunodeficient mouse model for the in vivo study of acoustic schwannoma. Am J Otolaryngol. 1996;17:228–32. doi: 10.1016/s0196-0709(96)90086-1. [DOI] [PubMed] [Google Scholar]
- 7.Giannini C, Sarkaria JN, Saito A, et al. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol. 2005;7:164–76. doi: 10.1215/S1152851704000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCutcheon IE, Friend KE, Gerdes TM, et al. Intracranial injection of human meningioma cells in athymic mice: an orthotopic model for meningioma growth. J Neurosurg. 2000;92:306–14. doi: 10.3171/jns.2000.92.2.0306. [DOI] [PubMed] [Google Scholar]
- 9.Shu ST, Nadella MV, Fernandez SA, et al. A novel bioluminescent mouse model and effective therapy for adult T-cell leukemia/ lymphoma. Cancer Res. 2007;67:11859–66. doi: 10.1158/0008-5472.CAN-07-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Sun Z, Peng J, et al. Bioluminescent imaging of hepatocellular carcinoma in live mice. Biotechnol Lett. 2007;29:1665–70. doi: 10.1007/s10529-007-9452-0. [DOI] [PubMed] [Google Scholar]
- 11.Cowey S, Szafran AA, Kappes J, et al. Breast cancer metastasis to bone: evaluation of bioluminescence imaging and microSPECT/CT for detecting bone metastasis in immunodeficient mice. Clin Exp Metastasis. 2007;24:389–401. doi: 10.1007/s10585-007-9076-8. [DOI] [PubMed] [Google Scholar]
- 12.Dinca EB, Sarkaria JN, Schroeder MA, et al. Bioluminescence monitoring of intracranial glioblastoma xenograft: response to primary and salvage temozolomide therapy. J Neurosurg. 2007;107:610–16. doi: 10.3171/JNS-07/09/0610. [DOI] [PubMed] [Google Scholar]
- 13.Zinn KR, Chaudhuri TR, Szafran AA, et al. Noninvasive bioluminescence imaging in small animals. ILAR J. 2008;49:103–15. doi: 10.1093/ilar.49.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nogawa M, Yuasa T, Kimura S, et al. Monitoring luciferase-labeled cancer cell growth and metastasis in different in vivo models. Cancer Lett. 2005;217:243–53. doi: 10.1016/j.canlet.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 15.Scatena CD, Hepner MA, Oei YA, et al. Imaging of bioluminescent LNCaP-luc-M6 tumors: a new animal model for the study of metastatic human prostate cancer. Prostate. 2004;59:292–303. doi: 10.1002/pros.20003. [DOI] [PubMed] [Google Scholar]
- 16.Yaccoby S, Barlogie B, Epstein J. Primary myeloma cells growing in SCID-hu mice: a model for studying the biology and treatment of myeloma and its manifestations. Blood. 1998;92:2908–13. [PubMed] [Google Scholar]
- 17.Lee EM, Bachman PS, Lock RB. Xenograft models for the pre-clinical evaluation of new therapies in acute myelogenous leukemia. Leuk Lymphoma. 2007;48:659–68. doi: 10.1080/10428190601113584. [DOI] [PubMed] [Google Scholar]
- 18.Lyons SK, Meuwissen R, Krimpenfort P, et al. The generation of a conditional reporter that enables bioluminescence imaging of Cre/loxP-dependent tumorigenesis in mice. Cancer Res. 2003;63:7042–6. [PubMed] [Google Scholar]
- 19.Stangerup SE, Caye-Thomasen P, Tos M, et al. The natural history of vestibular schwannoma. Otol Neurotol. 2006;27:547–52. doi: 10.1097/01.mao.0000217356.73463.e7. [DOI] [PubMed] [Google Scholar]