

Abstract
The proapoptotic BCL-2 family member BAD resides in a glucokinase-containing complex that regulates glucose-driven mitochondrial respiration. Here, we present genetic evidence of a physiologic role for BAD in glucose-stimulated insulin secretion by beta cells. This novel function of BAD is specifically dependent upon the phosphorylation of its BH3 sequence, previously defined as an essential death domain. We highlight the pharmacologic relevance of phosphorylated BAD BH3 by using cell-permeable, hydrocarbon-stapled BAD BH3 helices that target glucokinase, restore glucose-driven mitochondrial respiration and correct the insulin secretory response in Bad-deficient islets. Our studies uncover an alternative target and function for the BAD BH3 domain and emphasize the therapeutic potential of phosphorylated BAD BH3 mimetics in selectively restoring beta cell function. Furthermore, we show that BAD regulates the physiologic adaptation of beta cell mass during high-fat feeding. Our findings provide genetic proof of the bifunctional activities of BAD in both beta cell survival and insulin secretion.
The BCL-2 family of proteins constitutes a key control point in apoptosis by regulating the release of apoptogenic factors from mitochondria1. Recent studies have assigned new roles to certain BCL-2 family members in other physiologic pathways, such as metabolism2,3, Ca2+ homeostasis4 and mitochondrial morphology5. Whether such roles represent separate functions for these proteins, which are primarily defined as regulators of cell death, is a topic under active investigation. BAD belongs to a subset of BCL-2 family members, known as BH3-only proapoptotic proteins, that share sequence homology only within an α-helical BH3 motif, also known as the minimal death domain. The apoptotic activity of BAD is inhibited by phosphorylation6,7. We have previously reported that BAD nucleates a core complex at the mitochondrion containing glucokinase (also known as hexokinase IV), the product of the gene associated with maturity-onset diabetes of the young type 2 (MODY2)8. Studies in Bad−/− and phosphorylation knock-in (Bad3SA) mice suggested that BAD is required for full activation of the mitochondrial-tethered portion of glucokinase, regulation of glucose-driven respiration and maintenance of glucose homeostasis2.
How BAD influences glucose metabolism is unknown. Glucokinase is mainly expressed in hepatocytes, pancreatic beta cells and certain subgroups of hypothalamic neurons, and it constitutes a key component of the mammalian glucose-sensing machinery. In the liver, glucokinase controls glycogen synthesis and glucose output, whereas in the pancreas it regulates insulin secretion8–12. The role of BAD in these tissues and their individual contribution to the glucose homeostasis defect in Bad mouse models has not been determined. Because Bad −/− and Bad3SA mice represent loss- and gain-of-function models for the proapoptotic activity of this protein, respectively, the common metabolic abnormalities in these animals suggested that the metabolic role of BAD is distinct from its capacity to induce apoptosis. Thus, we undertook a detailed investigation of the molecular underpinnings of BAD’s metabolic activity. Here, we report the development and application of genetic and chemical approaches to elucidate and characterize the role of BAD in glucose-stimulated insulin secretion (GSIS) by beta cells. Furthermore, we tested the importance of BAD in the physiologic control of beta cell mass during high-fat feeding.
RESULTS
Beta cell dysfunction in Bad-deficient mice
To examine the role of BAD in beta cells in vivo, we assessed insulin secretion in cohorts of Bad +/+ and Bad −/− mice subjected to hyperglycemic clamp studies (Fig. 1a). Analysis of plasma insulin at multiple time points during the clamp period indicated significant impairment of insulin secretion in Bad −/− mice during both the acute (0–30 min) and late phases (30–120 min) of secretion (Fig. 1b,c).
Figure 1.
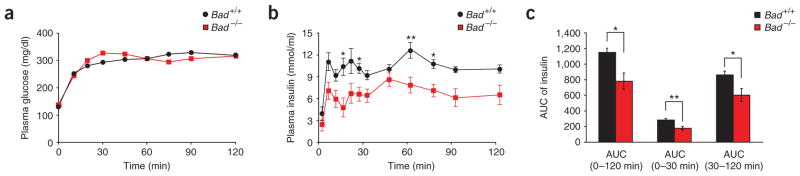
Impaired insulin secretion in Bad −/− mice. Plasma glucose (a) and insulin (b) levels and the area under the curve (AUC) for insulin secretion (c) during hyperglycemic clamp studies performed on overnight fasted Bad +/+ (n = 10) and Bad −/− (n = 12) mice. *P < 0.05, **P < 0.01, Bad +/+ versus Bad −/− mice, unpaired, two-tailed t-test.
To determine whether this insulin secretion defect likewise manifests in vitro, we performed perifusion assays on isolated islets (Fig. 2a). Bad −/− islets perifused with 25 mM glucose secreted significantly lower amounts of insulin (Fig. 2a,b). However, the total pool of insulin released by KCl was comparable in both genotypes (Fig. 2a).
Figure 2.

Characterization of the insulin secretion defect in Bad −/− islets. (a) Islet perifusion. Data are means ± s.e.m. of four independent experiments. DNA content per islet was 12.09 ± 0.65 ng and 13.17 ± 0.95 ng for Bad +/+ and Bad −/− mice, respectively. (b) AUC for insulin throughout the perifusion (min 0–40), first phase (min 8–15) and second phase (min 15–40) of release in a. (c) Glucose-induced changes in ATP/ADP ratio in Bad +/+ and Bad −/−islets. (d) Insulin release in response to 10 mM α-ketoisocaproic acid (KIC), 0.25 mM tolbutamide or 0.25 mM carbachol, as measured by static incubation method. Data are means ± s.e.m. of four separate experiments. Insulin content per islet was 115.1 ± 4.64 and 118.49 ± 4.09 ng, Bad +/+ and Bad −/−, respectively. (e) Glucokinase activity in homogenates of primary islets isolated from Bad +/+ and Bad −/− mice. Data represent means ± s.e.m. of four independent measurements. (f) Insulin secretion in Bad +/+ and Bad −/− islets perifused with increasing concentration of glucose. Data are means ± s.e.m. of four independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t-test.
Characterization of the secretory defect in Bad −/− beta cells
The glucose-induced insulin secretion pathway in beta cells consists of both mitochondrial proximal and distal events13. Mitochondria generate important metabolic coupling factors required for the induction of insulin release by glucose and other nutrients13,14. The increase in the intracellular ATP/ADP ratio leads to closure of ATP-sensitive K (KATP) channels at the plasma membrane, followed by membrane depolarization and opening of the voltage-sensitive Ca2+ channels. The increase in intracellular Ca2+ concentration ([Ca2+]i) in turn stimulates insulin release15. Bad −/− islets did not show a robust increase in ATP/ADP ratio (Fig. 2c), suggesting a lack of sufficient metabolic coupling.
We used several well-characterized secretagogues to examine the mitochondrial distal and proximal steps of the insulin secretion pathway in Bad −/− islets. KIC (α-ketoisocaproate), the deamination product of leucine, fuels the mitochondrial TCA cycle independent of glucokinase16. KIC-induced insulin secretion by Bad −/− islets was comparable to that seen in wild-type islets (Fig. 2d). We tested the mitochondrial distal steps, using the sulfonylurea tolbutamide and the muscarinic receptor agonist carbachol. Tolbutamide binds and closes the KATP channel independently of changes in the ATP/ADP ratio17, allowing examination of signaling downstream of this channel. We used carbachol to assess whether the IP3-mediated release of intra-cellular Ca2+ stores occurred properly. The response of Bad −/− islets to both tolbutamide and carbachol was comparable to the responses of control islets (Fig. 2d).
The glucose-selective aspect of the secretory defect in Bad −/− beta cells prompted us to closely examine glucokinase. We previously reported that BAD associates with the hepatic isoform of glucokinase at mitochondria2, a finding that was subsequently confirmed by other independent studies18. Because the hepatic and beta cell isoforms of glucokinase are regulated differently, we evaluated the potential regulatory effect of BAD on the beta cell isoform. The beta cell glucokinase indeed associates with BAD at the mitochondria in a complex similar to that described for hepatocytes2 (Supplementary Fig. 1 online). Furthermore, homogenates prepared from primary Bad −/− islets showed reduced glucokinase activity compared to control islets (Fig. 2e). A signature of beta cell dysfunction associated with impaired glucokinase activity is loss of glucose sensing8. Of note, Bad −/− islets required more glucose to release the same magnitude of insulin secreted by control islets, indicating perturbation of glucose dose responsiveness of insulin release (Fig. 2f).
The efficiency of glucose and other fuel secretagogues to stimulate insulin secretion correlates with their capacity to hyperpolarize the mitochondrial membrane potential (ΔΨm)19. In beta cells, the characteristic features of glucose-driven mitochondrial respiration correspond to that of glucose phosphorylation by glucokinase20. To examine the efficiency of respiration in Bad −/− beta cells, we recorded changes in Δψm in response to different fuels (Fig. 3a). Hyperpolarization of the mitochondrial membrane potential is evident by an increase in the fluorescence intensity of the mitochondrial potentiometric dye tetramethyl rhodamine ethyl ester (TMRE), which can be used as an index for respiration21. Glucose-induced changes in ΔΨm were significantly reduced in Bad −/− beta cells (Fig. 3a, P < 0.05). This reduction is not due to a global impairment of mitochondrial respiratory chains, as both genotypes showed comparable changes in ΔΨm in response to KIC (Fig. 3a).
Figure 3.

Glucose-induced changes in mitochondrial membrane potential and [Ca2+]i in Bad +/+ and Bad −/− beta cells. (a) Changes in mitochondrial membrane potential (ΔΨm) in response to stimulatory fuels (n = 10). Images on the top are color-coded for fluorescence intensity: blue (low) and red (high). Scale bar, 5 μm. (b–g) Representative Ca2+ traces obtained from individual Bad +/+ (n = 116) and Bad −/− (n = 115) islet cells in response to 11 mM glucose and 35 mM KCl. Cells representing at least four different mice from each genotype were analyzed. Quantitative summary of the [Ca2+]i response is provided in Supplementary Table 1 online.
The increase in [Ca2+]i and the rise in ATP/ADP ratio after mitochondrial metabolism of glucose trigger insulin secretion15. We monitored changes in [Ca2+]i in individual islet cells exposed to glucose (Fig. 3b–g) and found that basal [Ca2+]i at 3 mM glucose was comparable in both genotypes, indicating that the basic control mechanisms for Ca2+ handling are preserved in Bad −/− cells. However, the average [Ca2+]i response to 11 mM glucose was significantly lower in Bad −/− cells (Fig. 3b–g and Supplementary Table 1 online). Of note, depolarization of plasma membrane by KCl induced a similar [Ca2+]i response in control and Bad−/− cells (Fig. 3b–g). This is consistent with unaltered KCl-induced insulin secretion in Bad −/− islets (Fig. 2a).
The role of BAD BH3 domain in insulin secretion
Genetic reconstitution assays provided a stringent test for relating the secretory defect in Bad −/− islets directly to the intrinsic function of BAD in beta cells or to secondary changes that may have occurred in Bad −/− mice (Fig. 4). Prior to genetic correction, Bad −/− islets did not show a stepwise increase in insulin release when exposed to incremental increases in glucose concentration (Fig. 4a). Adenovirus-mediated reintroduction of BAD in these islets restored a robust dose-responsive secretory behavior, supporting a direct role for BAD in GSIS (Fig. 4a).
Figure 4.

Regulation of GSIS by the BAD BH3 domain and its phosphorylation status. (a) Genetic reconstitution of the GSIS defect in Bad −/− islets. Data are means ± s.e.m. of three separate experiments performed with two independent preparations of viral stocks. n.s., not significant. Asterisks compare release at 5.5 mM versus 12.5 or 25 mM glucose within each group of islets, ***P < 0.001. ‡P < 0.05, comparing Bad +/+ versus Bad −/− islets infected with control (GFP) viruses; †P < 0.001, comparing Bad −/− islets infected with control viruses versus viruses expressing wild-type BAD; ¶P < 0.05, comparing Bad −/− islets infected with viruses expressing wild-type BAD versus viruses expressing the L151A mutant. Insulin content per islet was 122.75 ± 12.34 and 127.13 ± 5.09 ng, Bad +/+ and Bad −/−, respectively. (b) GSIS in Bad +/+, Bad 3SA and Bad S155A islets. Data are means ± s.e.m. of three independent experiments. Asterisks compare release at 5.5 mM versus 12.5 or 25 mM glucose within each group of islets, *P < 0.05, ***P < 0.001. ‡P < 0.05, Bad +/+ versus Bad 3SA or Bad +/+ versus Bad S155A. Insulin content per islet was 118.74 ± 3.86, 96.46 ± 3.42 and 106.5 ± 6.24 ng, Bad +/+, Bad3SA and Bad S155A, respectively. (c,d) Blood insulin (c) and glucose (d) abundance after intraperitoneal glucose tolerance test performed on Bad +/+ (n = 10) and Bad S155A (n = 10) mice. *P < 0.05, **P < 0.01, Bad +/+ versus Bad S155A mice, Student’s t-test. (e) BAD phosphorylation status in islets isolated from fed or overnight fasted mice. The ratio of phospho-BAD to total BAD in fed versus fasted state was 0.72 versus 0.01 for Ser155 and 0.80 versus 0.03 for Ser136. WB, western blot.
As the established biochemical property of BAD is high-affinity binding to antiapoptotic BCL-2 family members, we investigated whether the effect of BAD on insulin secretion was mediated by its interaction with BCL-2 and BCL-XL. The BH3 domain of BAD is an amphipathic α-helix that binds BCL-2 and BCL-XL and neutralizes their antiapoptotic activity. Leu151 (mouse BADL enumeration), a highly conserved amino acid of the BH3 domain, is important for BCL-2 and BCL-XL binding22. Notably, the L151A mutant did not correct the GSIS defect in Bad −/− islets, suggesting a previously unforeseen role for this domain in GSIS (Fig. 4a).
The requirement of an intact BH3 domain in GSIS raised the possibility that the metabolic activity of BAD could be mediated by its binding to BCL-2 and BCL-XL. To explore this mechanism, we examined a BAD mutant in which the three serine phosphorylation sites (Ser112, Ser136 and Ser155) were converted to alanine (BAD 3SA)23. As the interaction between BAD and BCL-2 and BCL-XL is inhibited by phosphorylation24, BAD 3SA constitutes a nonrepressible mutant whose binding to BCL-2 and BCL-XL cannot be inhibited. Because adenoviruses carrying this mutant were toxic to islets in genetic reconstitution assays, we evaluated islets isolated from the Bad3SA knock-in mice. Of note, glucokinase activity is blunted in Bad3SA islets (data not shown) and, like Bad −/− islets, Bad3SA islets showed impaired GSIS (Fig. 4b). Whereas the BAD 3SA and L151A mutations share similar defects in insulin secretion, they show opposite capacities to engage BCL-2 and BCL-XL. Hence, the abilities of BAD to regulate insulin secretion and to interact with antiapoptotic partners do not cosegregate, highlighting a novel functionality for BAD that is distinct from its role in targeting BCL-2 or BCL-XL and regulating apoptosis.
Stimulation of GSIS by hydrocarbon-stapled BAD BH3 peptides
To determine whether the BAD BH3 domain itself is sufficient to correct the GSIS defect in Bad −/− beta cells, we synthesized a panel of cell-permeable stapled BAD BH3 peptides. We modified the BAD BH3 peptide by hydrocarbon stapling, a chemical synthesis strategy recently developed to generate compounds known as stabilized α-helices of BCL-2 domains (SAHBs) that show full BH3 bioactivity25 (Fig. 5a and Supplementary Fig. 2a,b online). Unlike the unmodified BAD BH3 peptide, BAD SAHB compounds maintain predominant α-helical structure (Supplementary Fig. 2b) and are readily cell permeable26. Of note, treatment with 3 μM BAD SAHBA restored the secretion defect in Bad −/− islets (Fig. 5b). In contrast, BID SAHBA (ref. 25), a distinct SAHB modeled after the BH3 domain of another BCL-2 family protein, BID, did not correct the secretory defect in Bad −/− islets (Fig. 5b), underscoring the sequence specificity of the observed BAD SAHB effect. Mutating the conserved leucine and aspartic acid residues of the BAD BH3 sequence (BAD SAHBA(L,D→A)) abrogated its effect on insulin release (Fig. 5b). Notably, the efficiency of BAD SAHBA in restoring GSIS accompanied its effect on glucose-induced changes in mitochondrial membrane potential (Fig. 5c). At 1 μM concentration, BAD SAHB conferred significant hyperpolarization of the mitochondrial membrane potential (ΔΨm) in response to glucose (P < 0.05), whereas the effect of BID SAHB on ΔΨm was statistically indistinguishable from vehicle-treated counterparts (Fig. 5c).
Figure 5.

Metabolic activity of SAHB compounds in beta cells. (a) Panel of human SAHBs generated for islet treatment. Conserved leucine and aspartic acid residues are highlighted in yellow and serine is marked in gray. Residues altered in different SAHB compounds are marked in green and purple. NL, norleucine. *The S5 non-natural amino acid (see Methods). (b) Effect of 3 μM SAHB on GSIS. Data are means ± s.e.m. of five independent experiments. Asterisks compare release at 5.5 mM versus 12.5 or 25 mM glucose within each group of islets, **P < 0.01, ***P < 0.001. ‡P < 0.05 or P < 0.001, Bad +/+ versus Bad −/− islets treated with vehicle. †P < 0.05 or P < 0.01, Bad −/− islets treated with vehicle versus BAD SAHBA. ¶P < 0.05 or P < 0.01, Bad −/− islets treated with BAD SAHBA versus phosphomimetic SAHBs (SAHBA(S→pS) or SAHBA(S→D)). Insulin content per islet was 114.61 ± 5.18 and 105.54 ± 4.63 ng, Bad +/+ and Bad −/−, respectively. (c) Effect of 1 μM SAHB compounds on glucose-induced changes in ΔΨm. (d) Binding affinities of SAHBs to recombinant BCL-XL ΔC protein. (e) Effect of 3 μM SAHBs on glucokinase activity in INS-1 cells. *P < 0.05 and ***P < 0.001. (f) Panel of derivatized mouse SAHBs containing a photoactivatable benzophenone moiety. (g) Identification of glucokinase as a direct BAD BH3 target. Formation of a covalent complex between SAHBA(S→pS) and glucokinase upon UV photoactivation. (h) Competition of SAHB compounds with full-length IVTT BAD for binding to IVTT glucokinase. IP, immunoprecipitation.
Phosphorylation of BAD on Ser155 is required for GSIS
The specificity of BAD BH3 compared to BID BH3 in stimulating GSIS raised the possibility that residues other than Leu151 and Asp156, which are conserved across the BH3 domains of BCL-2 family proteins, may also be required. Notably, Ser155 is a distinguishing feature of the BAD BH3 sequence whose phosphorylation is essential for dissociation of BAD from BCL-2 and BCL-XL27. To test the role of Ser155 in insulin secretion, we examined a previously unpublished Bad knock-in model in which only this serine residue was replaced with alanine (Supplementary Fig. 3 online). Bad S155A knock-in beta cells showed abnormalities in insulin secretion both in vitro, when purified islets were examined (Fig. 4b), and in vivo, when insulin levels were assessed by intraperitoneal glucose tolerance test (Fig. 4c,d).
To further investigate the significance of Ser155 phosphorylation, we assessed changes in BAD phosphorylation under physiologic settings of blunted insulin secretion. During fasting, islet glucokinase activity and insulin secretion in response to glucose are inhibited28. Notably, Ser155 phosphorylation is diminished in islets isolated from fasted wild-type mice compared to fed mice (Fig. 4e). Previous studies have indicated that, in the context of the full-length BAD protein, phosphorylation at Ser155 requires a priming phosphorylation at Ser136 upstream of the BH3 domain27. In agreement with this, Ser136 phosphorylation was also reduced in islets isolated from fasted mice (Fig. 4e). Thus, the diminution of BAD phosphorylation parallels the physiologic change in glucokinase activity and secretory behavior of islets during fasting.
To test the pharmacologic effect of Ser155 phosphorylation, we generated new phospho-BAD SAHBs (SAHBA(S→pS) and SAHBA(S→D); Fig. 5a and Supplementary Fig. 2b). SAHBA(S→pS) contains a phosphorylated serine, whereas SAHBA(S→D) contains a serine-to-asparagine replacement previously shown to simulate a constitutively phosphorylated BAD BH3 domain27. Both compounds showed robust activity in restoring the GSIS defect of Bad −/− islets (Fig. 5b). Of note, these SAHB compounds rescued the secretory defect despite their inability to bind BCL-XL (Fig. 5d and Supplementary Fig. 2c).
Glucokinase is a BAD BH3 target
To determine the mechanism by which the BAD BH3 domain restores GSIS, we examined the effect of BAD SAHBs on glucokinase activation. When added to INS-1 cells, BAD SAHB and its phosphomimetics stimulated glucokinase activity, whereas BAD SAHBA(L,D→A) had no effect (Fig. 5e). These findings prompted us to evaluate whether the BAD BH3 domain targets glucokinase directly. To test this mechanism, we derivatized BAD SAHBs to contain both FITC and a photoactivatable benzophenone moiety for covalent capture of protein targets29 (Fig. 5f). The photo-crosslinkable SAHBs were engineered so that benzophenone, an aromatic moiety, was substituted for a conserved native aromatic residue, such as the tyrosine of the BH3 sequence (Fig. 5f). Upon ultraviolet (UV) light exposure, the benzophenone photoprobe is excited to generate a radical intermediate that intercalates into covalent bonds of the target binding protein. As the reactive radius of the excited benzophenone intermediate is ~3.1 Å, only the protein that is in direct association with the BH3 domain is captured. If the interaction between BAD BH3 domain and glucokinase is direct, UV light exposure should produce a covalent complex between BAD SAHB and glucokinase, resulting in a shift in glucokinase mobility corresponding to the molecular mass of the SAHB compound. We further reasoned that the phosphomimetic SAHBs, which cannot bind BCL-XL (Fig. 5d) but show robust metabolic activity (Fig. 5b,e), would be more efficient at crosslinking glucokinase. Indeed, incubation of the SAHBA(S→pS) compound with INS-1 cell lysates followed by UV photoactivation caused a shift in glucokinase mobility, an effect not seen in vehicle-treated samples (Fig. 5g). The covalent complex contained both glucokinase and the SAHBA(S→pS) compound as shown by its immunoreactivity with antibodies to glucokinase and FITC (Fig. 5g). Similar results were obtained with the SAHBA(S→D) phosphomimetic compound (data not shown). Notably, a BAD BH3 mutant, in which the three residues required for the metabolic activity of BAD (Leu151, Ser155 and Asp156; Fig. 4a,b and Fig. 5b) were converted to alanine (SAHBA(L,S,D→A)), was severely compromised in glucokinase binding (Fig. 5g). The inactivity of the negative control mutant confirms the specificity of interaction between the BAD BH3 domain and glucokinase. The results of glucokinase capture assays using BAD SAHB and its mutants correlate with their metabolic activity, indicating that direct engagement of glucokinase enables their activation of glucokinase and regulation of GSIS.
As an independent approach to test the specificity of the BAD BH3-glucokinase interaction, we performed binding competition assays (Fig. 5h). Full-length BAD and glucokinase proteins were produced by an in vitro transcription-translation (IVTT) system. The ability of BAD and glucokinase to associate was examined in the presence or absence of the indicated BAD SAHB compounds. Notably, SAHBA(S→pS), which binds glucokinase, diminished the amount of IVTT BAD coimmunoprecipitated with glucokinase, whereas the negative control mutant SAHBA(L,S,D→A) did not effectively compete with IVTT BAD for glucokinase binding (Fig. 5h), highlighting the specificity of the BH3-dependent interaction of BAD and glucokinase.
The role of BAD in diet-induced beta cell mass compensation
Beta cells normally undergo compensatory changes to meet the secretory demand of the insulin-resistant state. These adaptive mechanisms include beta cell mass expansion. A lack of proper adaptation results in diabetes30–33. Furthermore, chronic exposure to high glucose and lipids can eventually lead to impairment of insulin secretion and beta cell apoptosis34.
In addition to the newly identified role for the BAD BH3 domain in glucokinase regulation and GSIS, this domain has been implicated in the effect of BAD on cell survival. For example, BAD is an important sentinel in beta cell death induced by streptozotocin and chronic exposure to high glucose concentrations (N.N.D., unpublished observations). To further evaluate how BAD’s metabolic and apoptotic functions in beta cells may be relevant to the pathophysiology of diabetes, we examined its role in susceptibility to high-fat diet (HFD)-induced obesity and diabetes. Of note, the relative Bad mRNA levels were significantly increased in islets isolated from wild-type mice upon high-fat feeding (Fig. 6a). In addition, a recent study reported increased amounts of phosphorylated BAD in islets of Zucker fatty rats, which coincided with increased beta cell survival and enhanced beta cell mass before development of obesity and hyperinsulinemia35. These observations prompted us to examine the genetic requirement of BAD in physiologic adaptation of beta cell mass using Bad-null and Bad3SA mice subjected to HFD. Notably, these two distinct mouse models allow genetic dissection of BAD’s apoptotic and metabolic roles. The Bad −/− and Bad3SA mutations are associated with a similar decrease in GSIS, yet they represent loss- and gain-of-function mutations in BAD’s apoptotic function, respectively. Bad −/− islets have a survival advantage despite their reduced insulin secretion, while Bad3SA islets are more sensitive to apoptosis in addition to their blunted insulin-secretory capacity.
Figure 6.

Sensitivity of Bad genetic models to HFD. (a) Relative islet Bad mRNA levels in wild-type mice on HFD for 16 weeks. *P < 0.05. (b) Weekly blood glucose (left) and body weights (right) of Bad +/+ and Bad −/−(n = 20) treated with HFD for 16 weeks. (c) Percentage islet area of cohorts in b. Representative pancreatic sections developed with an antibody to insulin are shown on the right. Scale bar, 250 μm. (d) Fed blood insulin levels of Bad +/+ and Bad −/− on control or HF diet for 8 weeks. *P < 0.05, Bad −/− versus Bad +/+. (e) Weekly blood glucose (left) and body weights (right) of a cohort of Bad +/+ and Bad 3SA (n = 8) treated with HFD for 16 weeks. (f) Percentage islet area of cohorts in e. Scale bar, 250 μm. (g) Fed blood insulin levels of Bad +/+ and Bad 3SA on control or HFD for 8 weeks. *P < 0.05, Bad 3SA versus Bad +/+ on HFD. (h) Proposed model for dual role of BAD in insulin secretion and beta cell survival. The BH3 domain endows BAD with bifunctional activities in GSIS and apoptosis. Ser155 phosphorylation in this domain instructs BAD to assume a metabolic role by targeting glucokinase and regulating insulin secretion. When dephosphorylated, BAD BH3 targets BCL-XL to control apoptosis. Apoptosis, together with other negative and positive regulators of beta cell growth, proliferation and survival contribute to physiologic control of beta cell mass.
Cohorts of Bad +/+ and Bad −/− mice were subjected to HFD for 16 weeks and compared to parallel cohorts placed on control diet. On control diet, the body weight and fed glucose levels in wild-type and Bad −/− mice were comparable throughout the study (data not shown). On HFD, however, Bad −/− mice were resistant to the development of hyperglycemia despite gaining weight at a similar rate as control animals (Fig. 6b). Given the metabolic abnormalities of Bad −/− mice, their resistance to HFD may seem surprising. However, this resistance was associated with a significantly larger increase in percentage islet area in Bad −/− mice compared to controls (Fig. 6c). This increase derived from markedly higher numbers of islets in pancreatic sections from Bad −/− mice compared to Bad +/+ mice on HFD (Supplementary Fig. 4a,b online). Concordant with these observations, blood insulin levels were higher in Bad −/− mice compared to Bad +/+ controls (Fig. 6d). Thus despite their decreased insulin secretory capacity, Bad −/− beta cells have an advantageous capacity to expand their mass, allowing sufficient production of insulin to counter the HFD challenge.
Based on the above observations, we predicted that gain of function in the proapoptotic activity of BAD may in turn negatively affect beta cell mass expansion, leading to enhanced sensitivity to HFD. Indeed, Bad3SA mice were more sensitive to the HFD challenge, having, on average, higher blood glucose levels (Fig. 6e). The body weights of Bad3SA mice were initially lower than those of control cohorts, but did not show any significant differences during the subsequent weeks. The percentage islet area in pancreatic sections was significantly lower in Bad3SA mice compared to littermate controls, suggesting that beta cell mass adaptation was inadequate (Fig. 6f and Supplementary Fig. 4c,d). Consistent with this observation, blood insulin levels were significantly lower in Bad3SA mice on HFD (Fig. 6g). Collectively, our findings suggest that the accompanying beta cell mass compensation and sensitivity to HFD is influenced by BAD phosphorylation status. Moreover, the opposite response of Bad −/− and Bad3SA to a HFD challenge in spite of the common beta cell secretory defect in these genetic models further highlights the dual role of BAD in beta cell apoptosis and insulin secretion, each having important physiologic consequences (Fig. 6h).
DISCUSSION
Targeting BCL-2 proteins for therapeutic manipulation of islets requires a careful assessment of the impact of distinct family members on both cellular metabolism and beta cell survival. Using BAD mutants and stapled BAD BH3 peptides that are either defective in binding to BCL-XL (BAD L151A, BAD SAHBA(L,D→A), SAHBA(S→pS) and SAHBA(S→D)) or whose binding to BCL-XL cannot be repressed (BAD 3SA and BAD S155A), we show that the BAD BH3 domain possesses the bifunctional capacity to differentially control GSIS and engage BCL-XL to regulate apoptosis. These findings are consistent with the observation that loss of BAD in beta cells does not entirely phenocopy BCL-XL overexpression. Transgenic islets expressing BCL-XL had a general secretory impairment, including a lack of response to KCl, tolbutamide and KIC36. This is consistent with the function of BCL-XL at the endoplasmic reticulum and probably reflects perturbations in Ca2+ homeostasis associated with BCL-XL overexpression4. In contrast to BCL-XL–overexpressing islets, the glucose-selective defect of Bad −/− islets is consistent with a functional and biochemical interaction between BAD and glucokinase, which can be pharmacologically manipulated by a BAD BH3 helix that has been chemically reinforced by the hydrocarbon-stapling synthetic strategy.
The finding that BAD, an apoptotic sentinel, also serves as a binding partner and regulator of glucokinase exemplifies the evolution of signaling molecules to integrate distinct homeostatic pathways. Other examples of proteins that serve as integral components of multiple signaling pathways include the γ isoform of phosphoinositide-3 kinase, which also binds and regulates phosphodiesterase-3B to modulate both the AKT and cAMP pathways37. Glyceraldehyde-3-phosphate dehydrogenase, an abundant glycolytic enzyme, can also interact with the E3-ubiquitin-ligase Siah and translocate to the nucleus to degrade selected proteins, inducing cell death38. Proapoptotic BAX and BAK, which are best known for their regulation of cytochrome c release during apoptosis, control mitochondrial dynamics in healthy cells5.
Our observations suggest that the BH3 domain of BAD toggles between apoptotic and metabolic functions in a phosphorylation-dependent manner (Fig. 6h). Ser155 phosphorylation inactivates the proapoptotic function of BAD and is required for its control of GSIS. Several kinases converge to coordinately regulate phosphorylation of Ser155. First, a priming phosphorylation of Ser136 upstream of the BH3 domain by AKT-p70S6 kinase7,27 is believed to induce a conformational change in BAD, which subsequently allows access of Ser155 kinases, including PKA and RSK1 (also known as p90 ribosomal S6 kinase)27,39. Ultimately, Ser155 phosphorylation disrupts the interaction between BAD and BCL-XL (ref. 27). Ser155 phosphorylation downstream of growth factors and nutrient signaling pathways may be stimulated through simultaneous activation of both the AKT-p70S6 kinase and the PKA-RSK1 kinases, or through sufficient activation of AKT-p70S6 kinases alone, which upon phosphorylation of Ser136 allow access of basally active PKA-RSK1 to Ser155 (ref. 27). Notably, several of the kinases that phosphorylate BAD integrate nutrient and hormonal signaling in different cell types. For example, in the beta cell, p70S6 kinase and cAMP-dependent protein kinase activities are stimulated by both glucose and glucagon-like peptide-1, an incretin hormone released from the gut upon feeding that stimulates both glucose-associated insulin secretion and beta cell survival40. Thus, both BAD phosphorylation and the upstream kinases in charge of this modification are nutrient sensitive. Moreover, our results implicate BAD and its phosphorylation status in the physiologic control of beta cell mass expansion upon nutrient challenge imposed by high-fat feeding. Notably, the upstream signaling events that target BAD phosphorylation, including AKT, p70S6 kinase and cAMP pathways, have been implicated in beta cell survival41–43. Hence, activation of BAD’s metabolic function and inactivation of its proapoptotic function seem to be physiologically coordinated on the basis of the metabolic demands of beta cells.
To date, the best-characterized function of BAD is its high-affinity interaction with BCL-2 and BCL-XL mediated by the essential proapoptotic BH3 domain. Applying genetic and chemical methods, we identify a novel physiologic function for BAD in GSIS that is dependent upon its BH3 domain but does not cosegregate with its capacity to engage BCL-2 and BCL-XL. Mutational analysis of the BAD BH3 sequence reveals that its ability to bind glucokinase correlates with its activation of glucokinase and stimulation of insulin secretion. Our studies mechanistically identify glucokinase as a novel and direct physiologic target of the BAD BH3 domain in beta cells. Moreover, genetic evidence combined with the pharmacologic activity of novel stapled BAD BH3 peptides indicate that phosphorylation within the BH3 domain drives the metabolic functionality of BAD and serves as a physiologic switch between its apoptotic and metabolic effects.
The molecular dissection and pharmacologic targeting of glucokinase activation holds therapeutic promise and has recently led to the development of several glucokinase activator compounds8. By uncovering an alternative binding partner and functionality for the BAD BH3 domain, we highlight the potential therapeutic application of BAD SAHBs and other BAD BH3 mimetics in restoring insulin secretion. Our findings also indicate that the distinct functions of BAD are amenable to selective pharmacologic targeting. For example, a phosphorylated BAD SAHB that activates GSIS but does not affect the survival function of BCL-XL may serve as a prototype therapeutic in diabetes and islet transplantation.
METHODS
Animal models
The Bad −/− and Bad 3SA knock-in genetic models have been described23,44. We generated the Bad S155A knock-in mice as described in the Supplementary Methods online. Mice received a standard chow or HFD (58% fat energy, D12331, Research Diets)45 as indicated. All animal procedures were approved by the Institutional Animal Care and Use Committee of Dana-Farber Cancer Institute and Yale University School of Medicine.
Metabolic studies
We performed hyperglycemic clamp studies as described in the Supplementary Methods. We carried out glucose tolerance tests as previously published2.
Islet isolation and insulin secretion studies
We isolated islets as previously described46. Experimental details for insulin secretion studies are available in the Supplementary Methods.
ATP/ADP ratio measurements
We preincubated batches of 50 size-matched islets in Krebs buffer containing 3 mM glucose for 30 min before a 1-h incubation in Krebs solution containing 5.5 mM or 25 mM glucose at 37 °C. We measured ATP and ADP contents as described previously47.
Glucokinase assays in primary islets and INS-1 cells
We used a previously published protocol for measurement of beta cell glucokinase activity48, as detailed in the Supplementary Methods.
Measurement of mitochondrial membrane potential
We plated dispersed islets on coverslips. After overnight incubation at 37 °C, we loaded the cells with mitochondrial dyes followed by glucose stimulation to calculate ΔΨm as described in Supplementary Methods.
Ca2+ measurement
We loaded single islet cells with fura-2 and measured Ca2+ traces as previously described21.
Islet infection using adenoviruses
We generated recombinant adenoviruses using the pAdEasy system49. We employed the services of the Vector Core of the Harvard Gene Therapy Initiative for virus amplification, purification, titration and verification. We infected batches of 150 islets with adenoviruses at 1 × 108 plaque-forming units (PFU) in RPMI medium containing 11 mM glucose and 10% serum for 90 min at 37 °C50. At this PFU, infections did not cause significant islet apoptosis. We then replaced the medium with fresh RPMI and cultured the islets for 24–48 h before insulin release assays. We hand picked the GFP-expressing islets in each group for insulin secretion studies.
SAHB synthesis, circular dichroism and fluorescence polarization binding assays
The amino acid sequence of the BAD SAHBs are based on the human (NLWAAQRYGRELRRMSDEFVDSFKK) and mouse (NLWAAQRYGRELRRM SDEFEGSFK) BAD BH3 domains, as indicated in Figure 5. We performed Fmoc-based peptide synthesis incorporating the S5 alkenyl non-natural amino acids, olefin metathesis, FITC derivatization, reverse-phase HPLC purification and microanalyses as previously reported25 (Supplementary Fig. 2a). We synthesized the benzophenone-containing BAD SAHBA derivatives by incorporating the corresponding non-natural amino acid, Fmoc-Bpa-OH (Advanced ChemTech), at the desired location (see Fig. 5f). We determined the percentage helicity of the SAHBs using circular dichroism and performed fluorescence polarization binding assays as detailed in the Supplementary Methods.
SAHB treatment
For insulin secretion studies (Fig. 5b), we incubated 150 islets in RPMI medium containing 11 mM glucose, 10% serum and 3 μM SAHBs or vehicle control (0.3% DMSO) overnight at 37 °C. We washed the islets and measured insulin secretion as described. For ΔΨm measurements (Fig. 5c), we incubated dispersed islets with 1 μM SAHBs or 0.1% DMSO for 4 h before imaging. For glucokinase activity (Fig. 5e), we treated INS-1 cells with 3 μM SAHB compounds for 4 h.
Examination of BAD phosphorylation status in islets
We solubilized islets isolated from fed and overnight-fasted wild-type mice in RIPA buffer (1.25% (wt/vol) NP-40, 1.25% (wt/vol) sodium deoxycholate, 0.0125 M sodium phosphate, pH 7.2, 2 mM EDTA) supplemented with protease and phosphatase inhibitors (Roche). We fractionated 80 μg total protein for western blotting with antibodies to phospho-Ser155 BAD (ab28825, Abcam), phospho-Ser136 or total BAD (#9295 and 9292, respectively, Cell Signaling Technology). We quantified the western blot signals using the ImageJ software.
BH3 domain target capture and binding competition assays
We incubated RIPA-solubilized whole-cell lysates from INS-1 cells with 20 μM SAHBs containing the photoactivatable benzophenone moiety for 30 min preincubation at 23 °C followed by exposure to 350 nm light for 2 h at 4 °C. We immunoprecipitated glucokinase using glucokinase affinity columns (Amino-Link Plus Coupling gel, Pierce).
For binding competitions, we cotranslated BAD and glucokinase using in vitro transcription-translation in rabbit reticulocyte lysates (TNT quick coupled transcription-translation systems, Promega) per the manufacturer’s instructions. We examined the amount of BAD coimmunoprecipitated with glucokinase in RIPA buffer in the presence of 30 μM SAHB compounds.
Real-time PCR
We prepared total RNA from islets using the RNeasy Plus Mini Kit (Qiagen) and performed real-time PCR as described in the Supplementary Methods.
Histological analysis and islet area measurements
We prepared serial 5-μm pancreatic sections from pancreata that were fixed in 10% formalin and embedded in paraffin. We developed the pancreatic sections using an antibody to insulin as described in the Supplementary Methods.
Statistical analysis
Data are presented as means ± s.e.m. Statistical analysis was carried out using Student’s t-test when comparing two groups and ANOVA when comparing multiple groups. Differences were considered significant at P < 0.05.
Supplementary Material
Acknowledgments
We thank M. Godes and C. Gramm for technical assistance, R. Pasquier and B. Szlyk for islet isolation and animal husbandry, G. Weir (Joslin Diabetes Center, Boston, MA) for antibody to glucokinase, B. Spiegelman, B. Malynn, M. Vander Heiden, A. Schinzel, J. Labelle, G. Verdine, F. Bernal, A. Saghatelian, A.-M. Richards and G. Yaney for helpful discussion and E. Smith for manuscript preparation. N.N.D. and L.D.W. are recipients of the Burroughs Wellcome Fund Career Award in Biomedical Sciences. This work was supported by US National Institute of Health grants K01CA10659 (N.N.D.), 5R01CA50239 and 5R01DK68781 (S.J.K.), 5K08HL074049 (L.D.W.) and by Charles E. Culpeper Scholarship in Medical Science (L.D.W.).
Footnotes
Note: Supplementary information is available on the Nature Medicine website.
AUTHOR CONTRIBUTIONS
N.N.D. planned and performed the in vivo and in vitro experiments, supervised the project and wrote the manuscript. J.K.F. and K.R. assisted with conducting experiments. L.D.W., K.L.P., G.H.B. and J.M. designed and synthesized the SAHB compounds and characterized their chemical and biophysical properties. C.-Y.Z. made conceptual contributions and helped with data interpretation. C.S.C. performed the hyperglycemic clamp studies with help from A.K., S.N., S.K. and supervision from G.I.S. A.J.A.M. and J.D.W. performed the measurements of mitochondrial membrane potential with supervision from O.S.S. S.R.D. generated the BadS155A knock-in model with supervision from M.E.G. J.T.D. helped with islet perifusion studies. B.E.C. provided critical guidance and expertise. B.B.L. provided advice. The late S.J.K. provided the mentorship and laboratory resources that catalyzed the initial studies.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 2.Danial NN, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424:952–956. doi: 10.1038/nature01825. [DOI] [PubMed] [Google Scholar]
- 3.Plas DR, Thompson CB. Cell metabolism in the regulation of programmed cell death. Trends Endocrinol Metab. 2002;13:75–78. doi: 10.1016/s1043-2760(01)00528-8. [DOI] [PubMed] [Google Scholar]
- 4.Pinton P, Rizzuto R. Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ. 2006;13:1409–1418. doi: 10.1038/sj.cdd.4401960. [DOI] [PubMed] [Google Scholar]
- 5.Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- 6.Harada H, et al. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol Cell. 1999;3:413–422. doi: 10.1016/s1097-2765(00)80469-4. [DOI] [PubMed] [Google Scholar]
- 7.Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. p70S6 kinase signals cell survival as well as growth, inactivating the proapoptotic molecule BAD. Proc Natl Acad Sci USA. 2001;98:9666–9670. doi: 10.1073/pnas.171301998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magnuson MA, Matschinsky FM. Glucokinase and Glycemic Disease: From Basics to Novel Therapeutics. Karger; Basel, Switzerland: 2004. pp. 1–17.pp. 42–64.pp. 360–397. [Google Scholar]
- 9.Bali D, et al. Animal model for maturity-onset diabetes of the young generated by disruption of the mouse glucokinase gene. J Biol Chem. 1995;270:21464–21467. doi: 10.1074/jbc.270.37.21464. [DOI] [PubMed] [Google Scholar]
- 10.Grupe A, et al. Transgenic knockouts reveal a critical requirement for pancreatic beta cell glucokinase in maintaining glucose homeostasis. Cell. 1995;83:69–78. doi: 10.1016/0092-8674(95)90235-x. [DOI] [PubMed] [Google Scholar]
- 11.Postic C, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell–specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 12.Terauchi Y, et al. Pancreatic beta cell–specific targeted disruption of glucokinase gene. Diabetes mellitus due to defective insulin secretion to glucose. J Biol Chem. 1995;270:30253–30256. doi: 10.1074/jbc.270.51.30253. [DOI] [PubMed] [Google Scholar]
- 13.Wiederkehr A, Wollheim CB. Minireview: implication of mitochondria in insulin secretion and action. Endocrinology. 2006;147:2643–2649. doi: 10.1210/en.2006-0057. [DOI] [PubMed] [Google Scholar]
- 14.Newgard CB, McGarry JD. Metabolic coupling factors in pancreatic beta cell signal transduction. Annu Rev Biochem. 1995;64:689–719. doi: 10.1146/annurev.bi.64.070195.003353. [DOI] [PubMed] [Google Scholar]
- 15.Berggren PO, Larsson O. Ca2+ and pancreatic B-cell function. Biochem Soc Trans. 1994;22:12–18. doi: 10.1042/bst0220012. [DOI] [PubMed] [Google Scholar]
- 16.Gao Z, et al. Distinguishing features of leucine and α-ketoisocaproate sensing in pancreatic beta cells. Endocrinology. 2003;144:1949–1957. doi: 10.1210/en.2002-0072. [DOI] [PubMed] [Google Scholar]
- 17.Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Sulfonylurea stimulation of insulin secretion. Diabetes. 2002;51 (Suppl 3):S368–S376. doi: 10.2337/diabetes.51.2007.s368. [DOI] [PubMed] [Google Scholar]
- 18.Arden C, Baltrusch S, Agius L. Glucokinase regulatory protein is associated with mitochondria in hepatocytes. FEBS Lett. 2006;580:2065–2070. doi: 10.1016/j.febslet.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 19.Antinozzi PA, Ishihara H, Newgard CB, Wollheim CB. Mitochondrial metabolism sets the maximal limit of fuel-stimulated insulin secretion in a model pancreatic beta cell: a survey of four fuel secretagogues. J Biol Chem. 2002;277:11746–11755. doi: 10.1074/jbc.M108462200. [DOI] [PubMed] [Google Scholar]
- 20.Liang Y, et al. Glucose metabolism and insulin release in mouse beta HC9 cells, as model for wild-type pancreatic beta cells. Am J Physiol. 1996;270:E846–E857. doi: 10.1152/ajpendo.1996.270.5.E846. [DOI] [PubMed] [Google Scholar]
- 21.Heart E, Corkey RF, Wikstrom JD, Shirihai OS, Corkey BE. Glucose-dependent increase in mitochondrial membrane potential, but not cytoplasmic calcium, correlates with insulin secretion in single islet cells. Am J Physiol Endocrinol Metab. 2006;290:E143–E148. doi: 10.1152/ajpendo.00216.2005. [DOI] [PubMed] [Google Scholar]
- 22.Zha J, et al. BH3 domain of BAD is required for heterodimerization with BCL-XL and proapoptotic activity. J Biol Chem. 1997;272:24101–24104. doi: 10.1074/jbc.272.39.24101. [DOI] [PubMed] [Google Scholar]
- 23.Datta SR, et al. Survival factor–mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev Cell. 2002;3:631–643. doi: 10.1016/s1534-5807(02)00326-x. [DOI] [PubMed] [Google Scholar]
- 24.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14–3-3 not BCL-XL. Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 25.Walensky LD, et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walensky LD, et al. A stapled BID BH3 helix directly binds and activates BAX. Mol Cell. 2006;24:199–210. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 27.Datta SR, et al. 14–3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell. 2000;6:41–51. [PubMed] [Google Scholar]
- 28.Burch PT, et al. Adaptation of glycolytic enzymes: glucose use and insulin release in rat pancreatic islets during fasting and refeeding. Diabetes. 1981;30:923–928. doi: 10.2337/diab.30.11.923. [DOI] [PubMed] [Google Scholar]
- 29.Saghatelian A, Jessani N, Joseph A, Humphrey M, Cravatt BF. Activity-based probes for the proteomic profiling of metalloproteases. Proc Natl Acad Sci USA. 2004;101:10000–10005. doi: 10.1073/pnas.0402784101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Accili D, Kido Y, Nakae J, Lauro D, Park BC. Genetics of type 2 diabetes: insight from targeted mouse mutants. Curr Mol Med. 2001;1:9–23. doi: 10.2174/1566524013364040. [DOI] [PubMed] [Google Scholar]
- 31.Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in beta cell function. Nature. 2001;414:788–791. doi: 10.1038/414788a. [DOI] [PubMed] [Google Scholar]
- 32.Dickson LM, Rhodes CJ. Pancreatic beta cell growth and survival in the onset of type 2 diabetes: a role for protein kinase B in the Akt? Am J Physiol Endocrinol Metab. 2004;287:E192–E198. doi: 10.1152/ajpendo.00031.2004. [DOI] [PubMed] [Google Scholar]
- 33.Weir GC, Laybutt DR, Kaneto H, Bonner-Weir S, Sharma A. Beta cell adaptation and decompensation during the progression of diabetes. Diabetes. 2001;50 (Suppl 1):S154–S159. doi: 10.2337/diabetes.50.2007.s154. [DOI] [PubMed] [Google Scholar]
- 34.Prentki M, Joly E, El-Assaad W, Roduit R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: role in beta cell adaptation and failure in the etiology of diabetes. Diabetes. 2002;51 (Suppl 3):S405–S413. doi: 10.2337/diabetes.51.2007.s405. [DOI] [PubMed] [Google Scholar]
- 35.Jetton TL, et al. Mechanisms of compensatory beta cell growth in insulin-resistant rats: roles of Akt kinase. Diabetes. 2005;54:2294–2304. doi: 10.2337/diabetes.54.8.2294. [DOI] [PubMed] [Google Scholar]
- 36.Zhou YP, et al. Overexpression of Bcl-XL in beta cells prevents cell death but impairs mitochondrial signal for insulin secretion. Am J Physiol Endocrinol Metab. 2000;278:E340–E351. doi: 10.1152/ajpendo.2000.278.2.E340. [DOI] [PubMed] [Google Scholar]
- 37.Patrucco E, et al. PI3K-γ modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell. 2004;118:375–387. doi: 10.1016/j.cell.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 38.Hara MR, Snyder SH. Nitric oxide-GAPDH-Siah: a novel cell death cascade. Cell Mol Neurobiol. 2006;26:525–536. doi: 10.1007/s10571-006-9011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan Y, Demeter MR, Ruan H, Comb MJ. BAD Ser-155 phosphorylation regulates BAD/Bcl-XL interaction and cell survival. J Biol Chem. 2000;275:25865–25869. doi: 10.1074/jbc.M004199200. [DOI] [PubMed] [Google Scholar]
- 40.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 41.Jhala US, et al. cAMP promotes pancreatic beta cell survival via CREB-mediated induction of IRS2. Genes Dev. 2003;17:1575–1580. doi: 10.1101/gad.1097103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pende M, et al. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature. 2000;408:994–997. doi: 10.1038/35050135. [DOI] [PubMed] [Google Scholar]
- 43.Tuttle RL, et al. Regulation of pancreatic beta cell growth and survival by the serine/threonine protein kinase Akt1/PKBα. Nat Med. 2001;7:1133–1137. doi: 10.1038/nm1001-1133. [DOI] [PubMed] [Google Scholar]
- 44.Ranger AM, et al. Bad-deficient mice develop diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2003;100:9324–9329. doi: 10.1073/pnas.1533446100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winzell MS, Ahren B. The high-fat diet–fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes. 2004;53 (Suppl 3):S215–S219. doi: 10.2337/diabetes.53.suppl_3.s215. [DOI] [PubMed] [Google Scholar]
- 46.Larsson O, Deeney JT, Branstrom R, Berggren PO, Corkey BE. Activation of the ATP-sensitive K+ channel by long chain acyl-CoA. A role in modulation of pancreatic beta cell glucose sensitivity. J Biol Chem. 1996;271:10623–10626. doi: 10.1074/jbc.271.18.10623. [DOI] [PubMed] [Google Scholar]
- 47.Schultz V, Sussman I, Bokvist K, Tornheim K. Bioluminometric assay of ADP and ATP at high ATP/ADP ratios: assay of ADP after enzymatic removal of ATP. Anal Biochem. 1993;215:302–304. doi: 10.1006/abio.1993.1591. [DOI] [PubMed] [Google Scholar]
- 48.Trus MD, et al. Regulation of glucose metabolism in pancreatic islets. Diabetes. 1981;30:911–922. doi: 10.2337/diab.30.11.911. [DOI] [PubMed] [Google Scholar]
- 49.He TC, et al. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou YP, et al. Overexpression of repressive cAMP response element modulators in high glucose and fatty acid-treated rat islets. A common mechanism for glucose toxicity and lipotoxicity? J Biol Chem. 2003;278:51316–51323. doi: 10.1074/jbc.M307972200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.