

Abstract
Cerebral microvessel endothelial cells that form the bloodbrain barrier (BBB) have tight junctions (TJ) that are critical for maintaining brain homeostasis and low permeability. Both integral (claudin-1 and occludin) and membrane-associated zonula occluden-1 and -2 (ZO-1 and ZO-2) proteins combine to form these TJ complexes that are anchored to the cytoskeletal architecture (actin). Disruptions of the BBB have been attributed to hypoxic conditions that occur with ischemic stroke, pathologies of decreased perfusion, and high-altitude exposure. The effects of hypoxia and posthypoxic reoxygenation in cerebral microvasculature and corresponding cellular mechanisms involved in disrupting the BBB remain unclear. This study examined hypoxia and posthypoxic reoxygenation effects on paracellular permeability and changes in actin and TJ proteins using primary bovine brain microvessel endothelial cells (BBMEC). Hypoxia induced a 2.6-fold increase in [14C]sucrose, a marker of paracellular permeability. This effect was significantly reduced (∼58%) with posthypoxic reoxygenation. After hypoxia and posthypoxic reoxygenation, actin expression was increased (1.4- and 2.3-fold, respectively). Whereas little change was observed in TJ protein expression immediately after hypoxia, a twofold increase in expression was seen with posthypoxic reoxygenation. Furthermore, immunofluorescence studies showed alterations in occludin, ZO-1, and ZO-2 protein localization during hypoxia and posthypoxic reoxygenation that correlate with the observed changes in BBMEC permeability. The results of this study show hypoxia-induced changes in paracellular permeability may be due to perturbation of TJ complexes and that posthypoxic reoxygenation reverses these effects.
Keywords: endothelial cells, cytoarchitecture, blood-brain barrier, immunofluorescence
Whereas Research Involving hypoxic stress has traditionally focused on identifying and treating risk factors associated with ischemic stroke (17, 18, 43), hypoxia as a result of high-altitude exposure has also been associated with impairment in neurological function (26, 37). Early intervention after ischemic stroke has been shown to reduce tissue damage associated with increased cerebrovascular permeability (7, 39). Recently, research has expanded into examining cellular effects of cerebral ischemia-reperfusion. Ischemic stroke is a reduction or cessation of blood flow to the brain, which deprives the cerebral tissue of important nutrients and oxygen as well as removal of metabolic waste products. Whereas this decrease in oxygen supply to the brain (i.e., hypoxia) has been shown to increase cerebrovascular permeability (1, 19), there are also reports (42, 66) of increased permeability in peripheral and cerebral endothelial cells during posthypoxic reperfusion (i.e., reoxygenation). Little is known about cellular response of cerebral endothelial cells during high-altitude hypoxia. It remains unclear whether the hypoxic insult or the posthypoxic reoxygenation induces functional changes in the blood-brain barrier (BBB). Furthermore, the molecular alterations that occur during these changes in paracellular permeability require examination.
Brain microvessel endothelial cells (BMEC) form a metabolic and physical barrier separating the periphery from the brain to maintain cerebral homeostasis (5, 28). The lack of fenestrations and presence of tight junctional (TJ) complexes differentiate BMECs from peripheral microvascular endothelium. Whereas adherens junctions and other junctional proteins contribute to cell-to-cell contacts in the paracellular cleft, TJ complexes are critical for restricting paracellular diffusion in the cerebral microvasculature (4, 13, 55).
TJs are complexes of plasma membrane proteins that connect to the cytoarchitecture via membrane-associated accessory proteins (49). Claudins and occludin are integral transmembrane proteins, which interact with plasma membranes of adjacent cells forming the TJ barrier (63, 65). Cytoplasmic TJ accessory proteins [e.g., zonula occluden (ZO)-1, ZO-3, and cingulin] connect TJs to the cytoskeleton (i.e., actin) (8, 33). ZO-1 and ZO-2 are membrane-associated guanylate kinase proteins critical in forming and stabilizing TJs by binding occludin to the cytoarchitecture (33, 36). Figure 1 is a schematic of BMEC TJ complexes. Although TJ complexes have been identified, little is known about alterations in these proteins under pathological insult, i.e., hypoxia and reoxygenation conditions.
Fig. 1.
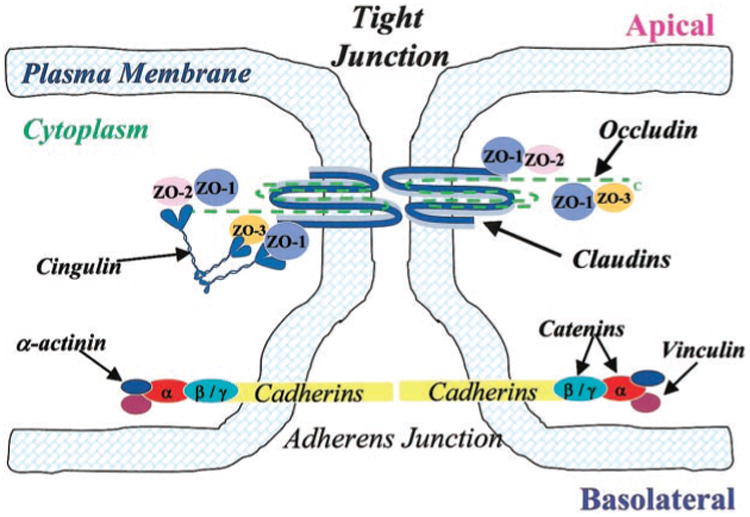
Schematic drawing of proposed junctional architecture of cerebromicrovessel endothelial cells. Tight junctions (TJ) consisting of the integral membrane proteins occludin and claudin are located towards the apical side of endothelial cells with their corresponding cytoplasmic accessory proteins [i.e., zonula occluden (ZO)-1, ZO-2, ZO-3, and cingulin] connecting TJs to the cytoskeleton (i.e., actin; not shown). Adherens junctions (AJ), located toward the basolateral side of endothelial cells, are composed of the integral membrane bound cadherins and cytoplasmic accessory proteins (i.e., α-and β-catenin).
Hypoxia and reoxygenation alter actin distribution (12). Cellular mechanisms induced by these changes in oxygen levels leading to increased permeability, changes in TJ complexes, and cytoarchitecture need to be investigated. During ischemic stroke with reduced perfusion, hypoglycemia and hypoxia result in energy depletion (62), ion imbalance, and release of cellular mediators (38, 53) that may lead to cell injury or death (38). Recent attention has been given to investigating cellular mechanisms affected by ischemia (1, 44). Whereas hypoxia-aglycemia has been shown to induce increased BMEC permeability via calcium-dependent pathways (1), the effects on TJ complexes are not well defined. Additionally, reoxygenation effects on intra-cellular mechanisms involved in altering BMEC permeability remain largely unknown.
This study used a well-characterized model of the BBB, bovine BMEC (BBMEC), to examine the relationship between functional and molecular alterations in cerebral microvasculature induced by hypoxia and posthypoxic reoxygenation.
Materials and Methods
Materials
Transwell polyester membrane inserts, minimal essential medium, Ham's F-12 medium, and Permanox tissue culture slide chambers were purchased from Fisher (St. Louis, MO). Bovine fibronectin, bovine serum albumin, 3-[4,5-dimethylthiazol-2-yl]2,5-diphenyltetrazolium bromide (MTT), and equine serum were purchased from Sigma (St. Louis, MO). Rat tail collagen (type I) was purchased from Collaborative Biomedical Products (Medford, MA). Bicinchoninic acid protein assay kit was purchased from Pierce (Indianapolis, IN). All other nutrients, salts, and antibiotics used in culture media or assay buffers were of cell culture quality from Sigma. Epithelial voltohmmeter and STX-2 “chopstick” electrodes were purchased from World Precision Instruments (Sarasota, FL).
Antibodies
Rabbit polyclonal anti-ZO-1, anti-ZO-2, anti-occludin, and anti-claudin-1 antibodies documented to react with human, mouse, rat, and dog were purchased from Zymed Laboratories (San Francisco, CA). Anti-ZO-1 is directed against amino acids 463–1109 of human ZO-1 cDNA. Anti-ZO-2 recognizes the central portion of the protein and has some cross-reactivity with unidentified proteins in the 50- to 85-kDa region. Anti-occludin is directed against the COOH-terminal 150 amino acids of human occludin. The anti-claudin-1 antibody recognizes the COOH-terminus region of the protein. Mouse monoclonal antibody (Sigma) directed against the COOH-terminus of actin (clone AC-40) reacts with human, bovine, chicken, dog, rat, and mouse species. Anti-mouse IgG and anti-rabbit IgG horseradish peroxidase-conjugated secondary antibodies were purchased from Amersham (Buckinghamshire, UK).
Isolation and culturing of BBMECs
Fresh bovine brains were obtained from the University of Arizona Animal Services for use as an in vitro BBB model. Primary BBMECs were collected from gray matter of bovine cerebral cortexes with the use of enzymatic digestion and centrifugal separation methods previously described (48). BBMECs were seeded (50,000 cells/cm2) on collagen-coated, fibronectintreated 75-cm2 tissue culture flasks or 12-well Transwell polyester membrane inserts (0.4 μm pore/12 mm diameter). Culture media was composed of 45% minimum essential medium, 45% Ham's F-12 nutrient mix, 10 mM HEPES, 13 mM NaHCO3, 50 μg/ml gentamicin, 10% equine serum, 2.5 μg/ml amphotericin B, and 100 μg/ml heparin sodium. Cell cultures were grown in a humidified 37°C incubator with 95% room air-5% CO2, and culture medium was replaced every other day until the BBMEC monolayers reached con-fluency (11–13 days).
Hypoxia-reoxygenation treatment
Several studies (19, 61) that examined the effects of hypoxia have used 95% N2-5% CO2 mixtures to reduce atmospheric O2, whereas others have used 100% N2 (12, 31). In this study, a hypoxic workstation (Coy Laboratories; Grass Lake, MI) was infused with 1% O2-99% N2 to treat confluent BBMEC monolayers for 24 h at 37°C. After 24-h hypoxia treatment, monolayers were used in permeability experiments or immunofluorescence studies, or protein was harvested for Western blot analyses. Alternatively, after exposure to hypoxic conditions, BBMEC monolayers were reoxygenated in 95% room air-5% CO2 at 37°C for 2 h, followed by permeability, Western blot, or immunofluorescent microscopy experiments. Changes in permeability, protein expression, or localization patterns after these treatment protocols were compared with control monolayers incubated for 24 h at 37°C in 95% room air-5% CO2 (normoxic conditions).
Gas analysis of cell culture medium
Gas values [pH, Pco2 (mmHg), Po2 (mmHg), and bicarbonate (mmol/l)] were measured in cell culture medium after exposure of BBMEC monolayers to normoxic (control), hypoxic, or posthypoxic reoxygenation conditions using a blood gas analyzer (model ABL-505 Radiometer; Copenhagen, Denmark).
Cytotoxicity study of BBMECs after hypoxia-reoxygenation
Viability of BBMECs after exposure to normoxia, hypoxia, or hypoxia with reoxygenation was assessed using the MTT cytotoxicity assay (27). Confluent BBMEC monolayers were exposed to normoxic, hypoxic, or hypoxic with reoxygenation conditions. The culture medium was removed and cells were rinsed with phosphate-buffered saline (PBS). The cells were incubated with 5 mg/ml of MTT for 2 h at 37°C. Excess MTT was removed and cells were rinsed with PBS before solubilizing in a 50:50 mixture of dimethyl formamide and 20% (wt/vol) sodium dodecyl sulfate (pH 4.7). Absorbance readings were taken at 550 nm using a Labsystems Microplate Reader (Fisher; Tustin, CA). Viability was expressed as a percentage of control cells exposed to normoxic conditions.
BBMEC monolayer permeability experiments
BBMEC monolayer integrity was assessed by transendothelial electrical resistance (TEER) measurements. Those monolayers with resistance values >120 Ω·cm2 were used to assess the permeability effects of hypoxia and reoxygenation on BBMEC monolayers. The passage of [14C]sucrose (a paracellular marker) across the BBMEC monolayer was employed because this method is more sensitive for detecting small changes in permeability compared with TEER measurements (20).
After treatment, confluent BBMEC monolayers were incubated with assay buffer composed of (in mM) 122 NaCl, 3 KCl, 1.4 CaCl2, 1.2 MgSO4, 25 NaHCO3, 10 HEPES, 10 glucose, and 0.5 K2HPO4 for 30 min at 37°C under the same gas conditions. Permeability across BBMEC monolayers was determined by adding [14C]sucrose (0.5 μCi) to the lumenal side (upper compartment of Transwell). Samples (50 μl) were removed from the ablumenal side (lower chamber of Trans-well) at 0, 60, and 120 min and replaced with fresh assay buffer. The concentration of [14C]sucrose applied to the lumenal side was determined by removing samples (50 μl) at time zero and replacing with fresh assay buffer and [14C]sucrose. The amount of radioactivity in the samples from permeability studies was determined by using a Beckman LS5000 TD beta counter. Permeability coefficients (PC) for [14C]sucrose were expressed in the following equation as previously described (15)
where V is volume in receiver chamber (1.5 cm3), SA is surface area of cell monolayer (1 cm2), Cd is the concentration of marker in the donor chamber at time 0, and Cr is the concentration of marker in the receiver at sample time T.
Western blot protein analysis
After being treated with hypoxia, hypoxia-reoxygenation, or normoxic (control) conditions, protein was isolated from confluent BBMEC monolayers using the TRI reagent protocol (Sigma). Briefly, protein was separated from RNA and DNA by chloroform and ethanol extraction and then precipitated using isopropanol, followed by washing with guanidinium chloride-95% ethanol before dissolving in 1% sodium dodecyl sulfate. Protein was quantified using the bicinchoninic acid method.
Protein samples (20 μg) were separated using an electrophoretic field on Novex 4–12% Tris-glycine gels at 125 V for 75–90 min. The proteins were transferred to polyvinylidene fluoride membranes with 240 mA at 4°C for 30 min. The membranes were blocked using 5% nonfat milk-Tris-buffered saline (20 mM Tris base-137 mM NaCl, pH 7.6) with 0.1% Tween 20 and incubated overnight at 4°C with primary antibodies (1:1,000–1:2,000 dilution) in PBS-0.5% BSA. The membranes were washed with 5% nonfat milk-Tris-buffered saline buffer before incubation with the respective secondary antibody at a 1:2,000 dilution (in PBS-0.5% BSA) for 30 min at room temperature. Membranes were developed using the enhanced chemiluminescence method (ECL+; Amersham) and protein bands were visualized on X-ray film. Semiquantitation of the protein was done with the use of imaging software (Scion) and the results are reported as a percentage of control.
Immunofluorescence of BBMECs
Confluent BBMECs monolayers grown on Permanox chamber slides (Fisher) were exposed to 24-h hypoxia, hypoxia with 2-h reoxygenation, or normoxic conditions before immunofluorescence staining. After treatment, culture medium was removed and monolayers were rinsed with prewarmed PBS. Cells were fixed with 3.7% formaldehyde-PBS for 10 min at room temperature (RT) and permeabilized using 1% Triton-X-PBS at RT for 10 min. After fixing and permeabilization, the monolayers were blocked with 1% BSA-PBS for 1 h at RT.
Confluent BBMEC monolayers from each treatment group (normoxia, hypoxia, and posthypoxic reoxygenation) were incubated with anti-occludin (10 μg/ml), anti-claudin-1 (10 μg/ml), anti-ZO-2 (5 μg/ml), or anti-ZO-1 (5 μg/ml) primary antibody at RT for 30 min. The cells were rinsed with 1% BSA/PBS, followed by incubation with a fluorescein-conjugated secondary antibody (4 μg/ml; Alexafluor 488 in 1% BSA-PBS; Molecular Probes) for 30 min at RT in the dark.
Confluent BBMEC monolayers from each treatment group were incubated with Alexafluor 488-conjugated phalloidin, which binds specifically to actin filaments. The phalloidin stain was reconstituted with 1% BSA-PBS (5 U/ml) applied to the luminal side of each monolayer and incubated for 30 min at RT in the dark. The fluorescent-stained cells were rinsed three times with PBS before being mounted on a coverslip with 50% glycerol-PBS and sealed.
Photographs were taken with the use of a ×60 oil immersion objective on a Nikon TE300 fluorescent microscope with a fluorescein filter.
Data analysis
Statistical analysis of the data was done with the use of one-way analysis of variance with Newman-Keuls multirange post hoc comparison of the means (10).
Results
Gas analysis of treated BBMECs
Table 1 shows the changes in pH, Pco2 (mmHg), Po2 (mmHg), and bicar-bonate ( ; mmol/l) levels in the BBMEC cell culture medium. As expected, Po2 levels dropped significantly (P value < 0.01) during the hypoxic (1% O2-99% N2) phase with a rapid return (within 5 min) to control levels during reoxygenation. Similarly, Pco2 levels decreased during hypoxia with a subsequent increase when O2 was reintroduced to the system. In concert with the declining Pco2, the pH increased significantly in the culture medium of BBMECs treated to 24-h hypoxia. There was no change in bicarbonate ( ) levels in any of the treatment groups.
Table 1. Gas analysis of cell culture medium.
| Treatment Group | pH | Pco2, mmHg | Po2, mmHg | , mmol |
|---|---|---|---|---|
| Normoxic | 7.24 ± 0.08 | 25.1±7.0 | 139.3 ±2.3 | 8.4 ± 1.5 |
| Hypoxic | 8.06 ± 0.09† | 1.7±0.1† | 46.5 ±7.3† | 6.5 ± 1.6 |
| Posthypoxic reoxygenation | 7.37 ± 0.03 | 10.8 ± 1.7* | 129.2 ±3.6 | 6.1 ± 0.8 |
Data are means ± SE; n = 5. Measurements were performed in culture medium.
P < 0.05 and
P < 0.01 compared with normoxic conditions using one-way ANOVA and Newman-Keuls post hoc analysis.
Cytotoxicity of hypoxia-treated BBMECs
There was no significant difference in cell viability of the BBMECs exposed to 24-h hypoxia (1% O2-99% N2) or hypoxia with reoxygenation compared with normoxic controls using the MTT assay (112% and 100%, respectively, compared with control; Table 2). This indicates that the decreased Po2 or increased pH levels were insufficient to cause cell death.
Table 2. BBMEC permeability and cell viability.
| Treatment Group | Permeability Coefficient of [14C]sucrose (cm/min ×10−4) | TEER, Ω·cm2 | Cell Viability, %Control |
|---|---|---|---|
| Normoxic | 4.80 ± 0.46 | 123 ± 2.3 | 100.0 ±5.6 |
| Hypoxic | 12.57 ± 1.31† | 112 ± 2.0 | 112.4 ±2.2 |
| Posthypoxic reoxygenation | 8.04 ± 1.10*‡ | 109 ± 1.0 | 100.3 ±6.1 |
| n | 9 | 6 | 6 |
Data are means ± SE; n, no. of cell cultures. BBMEC, bovine brain microvessel endothelial cells; TEER, transendothelial electrial resistance.
P < 0.05 and
P < 0.01 compared with normoxic conditions;
P < 0.01 compared with hypoxic conditions using one-way ANOVA and Newman-Keuls post hoc analysis.
Permeability of [14C]sucrose in BBMECs
Table 2 shows that, whereas there was no difference in BBMEC monolayer TEER measurements in any of the treatments (109–123 Ω·cm2), there was a significant increase (P < 0.01) in permeability of [14C] sucrose in the hypoxia-treated group compared with control BBMEC monolayers (12.57 vs. 4.80 cm/min ×10−4, respectively). Reoxygenation for 2 h attenuated the hypoxia-induced permeability increase by 58% (8.04 cm/min ×10−4).
Western blot analysis of cytoskeletal and TJ proteins
Alterations in expression of proteins that form TJs and cytoskeletal architecture were examined after hypoxia (24 h), hypoxia-reoxygenation (24 h/2 h), or normoxic (control) conditions. Table 3 summarizes the Western blot analyses of hypoxia and posthypoxic reoxygenated-treated BBMECs compared with controls. An increased expression (1.4-fold) of the major cytoskeletal protein (actin) occurred after hypoxia and continued to increase (2.3-fold) on reoxygenation (Fig. 2). In contrast, hypoxia alone had little effect on the expression of TJ proteins (i.e., claudin-1, occludin, ZO-1, or ZO-2; Fig. 2). Whereas there was little change in claudin-1 expression, a significant increase was observed in expression of occludin, ZO-1, and ZO-2 (1.4-, 1.8-, and 1.9-fold; respectively) with posthypoxia reoxygenation.
Table 3. Effects of treatment on BBMEC protein expression.
| Treatment Group | Actin, % | Claudin-1, % | Occludin, % | ZO-1, % | ZO-2, % |
|---|---|---|---|---|---|
| Normoxic | 100 ± 2 | 100 ± 1 | 100 ±2 | 100 ± 3 | 100 ± 1 |
| Hypoxic | 141±11* | 99 ±5 | 97 ±5 | 102 ±4 | 97 ±6 |
| Posthypoxic reoxygenation | 232 ±16† | 103 ±5 | 138 ±11† | 171±16† | 178±22† |
Data are percentage of control with means ± SE. ZO, zona occludens.
P < 0.05 and
P < 0.01 compared with normoxic conditions using one-way ANOVA and Newman-Keuls post hoc analysis.
Fig. 2.
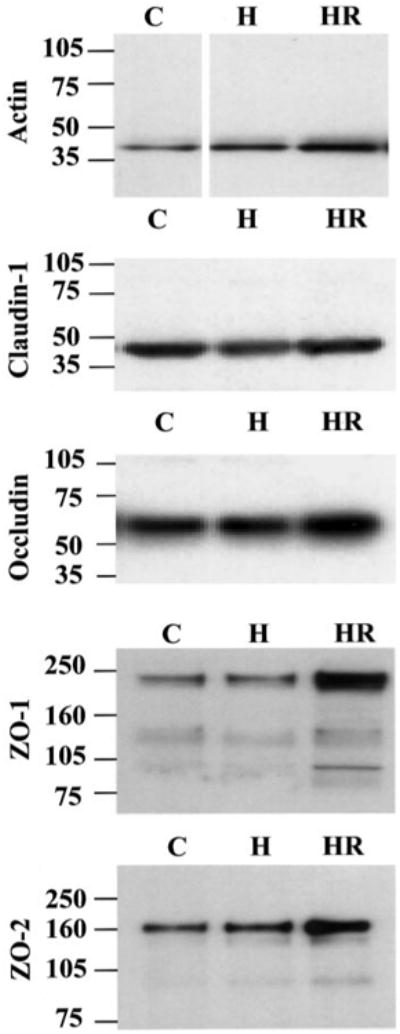
Hypoxia-reoxygenation effects on protein expression of bovine brain microvessel endothelial cells (BBMECs). Confluent monolayers were exposed to normoxia (C), 24-h hypoxia (H) or hypoxia with 2-h reoxygenation (HR). Representative Western blots for actin (42 kDa), claudin-1 (44 kDa dimer), occludin (65 kDa), ZO-1 (220 kDa), and ZO-2 (160 kDa) are shown (n = 4).
Immunofluorescence of hypoxia-treated BBMEC monolayers
BBMEC monolayers exposed to hypoxia for 24 h showed changes in actin localization, with increased staining of the filaments and development of stress tangles (arrowheads in Fig. 3). More of these actin stress tangles became apparent after reoxygenation. As with the protein expression of claudin-1, little change in the distribution pattern of this protein was seen after hypoxia or posthypoxic reoxygenation. In contrast, significant disruptions were seen in localization of occludin and ZO-2 after hypoxia (arrows in Fig. 3). These changes were less apparent in posthypoxic reoxygenation correlating to the increased protein expression (Fig. 2). The localization pattern of ZO-1 appears more diffuse throughout the cells after 24 h hypoxia. The distribution of ZO-1 and ZO-2 in posthypoxic reoxygenation BBMECs resembles that of control monolayers (Fig. 3); however, there is more intense staining at cell-to-cell contact points.
Fig. 3.
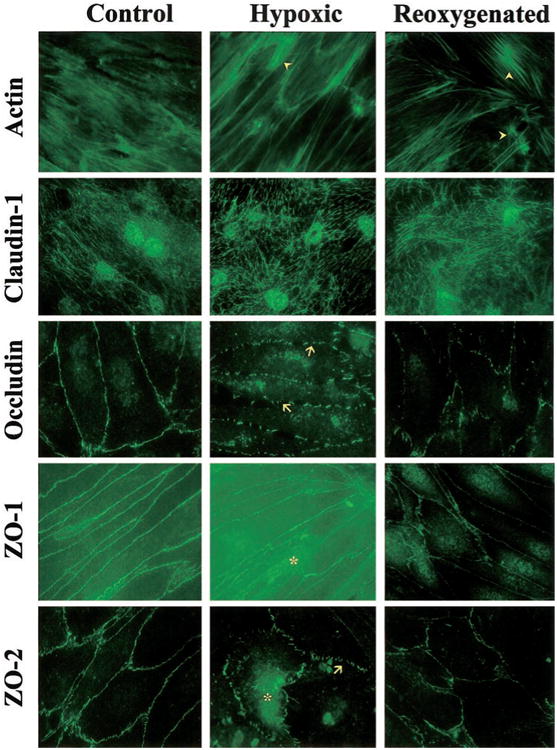
Immunofluorescence showing protein localization in BBMECs. Confluent monolayers were exposed to nor-moxic (Control), 24-h hypoxia (Hypoxic), or hypoxia with 2-h reoxygenation (Re-oxygenated) conditions. After treatment, the monolayers were incubated with primary antibodies directed against actin, claudin-1, occludin, ZO-1, and ZO-2, followed by Alexafluor 488-conjugated secondary antibodies. Representative pictures for each treatment group and protein are shown (n = 3). Arrowheads, actin stress tangles; arrows, disruptions in the expression of occludin and ZO-2. *Increased cytosolic staining of ZO-1 and ZO-2.
Discussion
Endothelial cells of the cerebral microvasculature serve as a frontline defense, protecting neurons and glial cells from harmful insult. This study and others focus on understanding how BMECs respond to changes in O2 content of blood seen in ischemic or high-altitude hypoxia. Under normal conditions of room air (21% O2), arterial Po2 (Pao2) levels range from 80–100 mmHg and interstitial Po2 (Pio2) values range from 15–25 mmHg or ∼25% of Pao2 (24). Because of the diffusion of O2 across membranes, decreases in Pao2 will result in similar reductions in Pio2. With high-altitude exposure or a narrowing of cerebral capillaries such as in ischemic hypoxia, the available O2 decreases with a subsequent reduction in Pao2 that varies depending on the severity of exposure. In fact, the severity and length of decreased O2 available to the cerebral microvessel endothelial cells will impact the degree of brain tissue damage.
Hypoxia as an element of ischemic stroke or high-altitude exposure has been examined using both in vivo and in vitro models. In vivo models for ischemic stroke have shown decreased Po2 values and increased Pco2 with corresponding decrease in pH (41, 47), whereas models for high-altitude hypoxia have shown decreased Po2 and Pco2 with a rise in pH (6, 51). Typically, O2 concentrations in the range of 6–12% have been used to study hypoxic effects in vivo with Po2 values of 35–47 mmHg (16, 25, 50). In Wistar rats, a combination of middle cerebral artery occlusion and hypoxia (Pao2 of 47 mmHg) resulted in increased brain tissue damage compared with controls (50).
Although middle cerebral artery occlusion provides a model for examining tissue damage (primarily neuronal) that occurs during focal ischemia, there is no documentation of Po2 values for the injury site. Furthermore, it does not allow for examination of paracellular permeability changes across the capillary endothelium nor protein localization changes that may occur during a hypoxic insult. Such changes are best examined with in vitro cell culture models. A variety of approaches for inducing a hypoxic environment have been used in peripheral and cerebral cell culture models, including O2 chelating agents (gaspaks) and anaerobic chambers infused with various gas mixtures (100% N2, 95% N2-5% CO2, or a mix of 85% N2-10% H2-5% CO2) to maintain an anoxic or hypoxic environment for periods ranging from 20 min to 72 h. To date, few studies have reported Po2, Pco2, and pH values from cell culture models of hypoxic insult (3, 54, 56, 60). In rat mesangial cells, Archer et al. (3) examined the effects of graded hypoxia on inducible nitric oxide (NO) synthase function using atmospheric O2 levels of 0, 2.5, 10, and 21%, resulting in culture medium Po2 levels of 32, 46, 85, and 140 mmHg, respectively (3).
The current study used primary BBMECs as an in vitro BBB model to investigate paracellular permeability changes induced by hypoxia and posthypoxic reoxygenation. Changes in expression and localization of cytoskeletal and TJ proteins were also examined under the same conditions. Whereas the benefits of astrocyte coculture conditions (astrocyte, glial cells, or conditioned media) have been debated, BBMECs alone were used in this study for three reasons. First, this model allows direct assessment of hypoxia-reoxygenation effects in cerebral microvascular endothelial cells without confounding their response by variations in astrocyte culturing techniques. Second, this allows a consistent model for evaluating the functional and molecular changes in brain microvessel endothelial cells (permeability, protein expression, and protein localization). Third, immunocytochemistry observations are best when the cells are grown on slides, which do not allow ablumenal access where astrocytes would exert their influence.
The current study used a decreased O2 level to simulate the reduced oxygen available to cerebral microvessel endothelial cells during hypoxic insults such as ischemic stroke or high-altitude exposure. The moderate hypoxic conditions (1% ± 0.5% O2 throughout the experiment) resulted in cell culture medium Po2 measurements of 47 ± 7 mmHg compared with normoxic controls of 139 ± 2 mmHg. These Po2 values are in agreement with the Po2 values used in both in vivo and in vitro studies of hypoxic exposure (2, 3, 50). Although the Pco2 declined with a subsequent increase in pH, there was no change in levels (Table 1). In a related study, BBMECs exposed to 1% O2-94% N2-5% CO2 had a mean pH value of 7.12, Pco2 of 33 mmHg, and Po2 value of 55 mmHg with similar permeability results (data not shown). Changes in pH, Pco2, and Po2 did not affect cell viability as demonstrated by the MTT cytotoxicity assay.
Whereas TEER measurements showed little change under the conditions in this study, radiolabeled sucrose (346 mol wt) proved to be a more sensitive marker for measuring changes in paracellular permeability (Table 2). Differences in permeability of [14C]sucrose were observed in hypoxia-treated BBMEC monolayers compared with normoxic (controls) and posthypoxic reoxygenated monolayers. BBMEC monolayers exposed to hypoxia for 24 h showed a significant (2.6-fold) increase in [14C]sucrose permeability compared with control monolayers. These findings are consistent with previous reports (1, 19, 23) showing increased permeability in brain capillary endothelial cells treated with hypoxia for periods ranging from 2 to 48 h.
Permeability changes shown in the current study correlate with alterations in TJ proteins and actin, as shown in Fig. 1. Increased expression of actin and the emergence of stress fiber tangles seen in Fig. 3 correlate with increased paracellular permeability suggesting hypoxia-induced alterations in cell morphology. This is supported by previous studies showing alterations in the cytoskeleton of 4 h hypoxia-treated bovine aortic endothelial cells (12) and actin filament rearrangement in brain endothelial cells after hypoxia for 24–48 h (53). This change in cell morphology and increased paracellular permeability suggests disruptions at TJ protein complexes.
Although no significant changes in protein expression of TJ proteins (claudin, occludin, ZO-1, and ZO-2) were observed in BBMECs after a 24-h hypoxic insult, there were changes in protein localization that correlate with functional changes (i.e., increased permeability) seen in this study. Claudin-1, which is considered the critical protein in forming TJ complexes, is embedded in the plasma membrane with short cytoplasmic sections that bind to occludin. Whereas the protein is important to the TJ complex, previous studies (11, 22, 67) examining claudin-1 have shown variations in im-munofluorescence patterns of claudin-1 protein including staining at cell boundaries and diffuse punctate distribution. In this study, little change in the protein expression or localization pattern of claudin-1 was observed with hypoxia or posthypoxic reoxygenation compared with normoxic controls (Figs. 2 and 3).
Occludin, another integral TJ protein, has two extracellular loops and long intracellular NH2-terminal and COOH-terminal regions suggesting that it connects claudins with TJ accessory proteins (i.e, ZO-1 and ZO-2) and the plasma membranes of adjacent cells. Figure 3 shows that under normoxic conditions, occludin has a continuous distribution pattern at the cell boundaries. Similarly, staining of the TJ accessory proteins ZO-1 and ZO-2 were localized along the plasma membrane at cell-to-cell contacts under control conditions. Exposure to hypoxia resulted in perturbations of the plasma membrane pattern of occludin and ZO-2 proteins compared with controls. In addition, both ZO-1 and ZO-2 show a diffuse pattern throughout hypoxic-treated BBMECs with discontinuous staining at the cell borders compared with control monolayers (Fig. 3). This is supported by other studies examining the hypoxic effects on localization of ZO-1 in BMECs (20). These disruptions in TJ proteins at cell-to-cell contact sites correlate with increased paracellular permeability and changes seen in actin distribution patterns (i.e., stress tangles). Together, these results indicate that hypoxic insult to brain endothelial cells induces restructuring of the TJ and cytoarchitecture (i.e., changes in protein localization) that correlate with observed functional changes (i.e., increased paracellular permeability) with little change in protein expression. This exposure to a hypoxic environment may trigger relocation of some TJ proteins from the plasma membrane to the cytoplasm. It is well known that protein phosphorylation regulation is a means of intra-cellular signaling, as cells use phosphorylation of enzymes and proteins to regulate cellular functions (protein degradation and enzyme activation) (58, 63). The possibility of TJ proteins relocating to the cytoplasm may be due to changes in phosphorylation states that occur with changes in phosphatase or kinase activity. The examination of the phosphorylation states of these TJ and cytoskeletal proteins during hypoxic insult will clarify whether this cellular mechanism is involved in protein localization and BBB permeability changes.
Whereas this study and others have shown perturbations of BBB permeability in various ischemia-hypoxia models (1, 19, 23, 52), others (12, 32, 42) have also reported increased paracellular permeability and disruptions in actin during posthypoxic reoxygenation. The present study showed that reoxygenation attenuated the hypoxia-induced increase in permeability by 58% (Table 2). This recovery with reoxygenation correlates with changes in protein expression and localization of actin and TJ proteins. Increasing TJ protein synthesis to enhance weakened cell-to-cell contacts may be an endothelial cell response to cytoarchitectural alterations and increased paracellular permeability. Whereas a modest rise (1.4-fold) in expression of actin protein was seen after hypoxic insult, these cells showed a 2.3-fold increase in protein expression with 2-h posthypoxic reoxygenation compared with normoxic monolayers. This increase in protein expression coincides with an increased number of stress fiber tangles (Fig. 3) and a reduction in permeability. Although no change in actin expression was observed with hypoxic exposure of bovine aortic endothelial cells, a 50% increase in expression was seen with posthypoxic reoxygenation (12). Because actin is the primary protein forming the cytoskeletal structure, these increases in actin protein and stress tangles may provide additional scaffolding for TJ proteins to reinforce cell-to-cell contacts that become weakened by hypoxic insult. Whereas studies (21, 29, 49) have demonstrated that claudin is vital to forming the tight junction seal, this study shows little change in protein expression or localization of claudin-1 after hypoxia or posthypoxic reoxygenation. This suggests that claudin is not involved in the hypoxia-induced paracellular permeability changes. In contrast, the increased expression of occludin, ZO-1, and ZO-2 after reoxygenation indicates alterations of TJ complexes and their connection to the cytoarchitecture during the reoxygenation stage of an ischemic event. This is further supported by the distribution patterns seen in these TJ proteins after a hypoxic insult when gaps are apparent and during posthypoxic reoxygenation when smaller and fewer gaps are seen. In addition, increased expression of the TJ proteins and actin with posthypoxic reoxygenation correlates well with a reduction in paracellular permeability of BBMEC monolayers compared with monolayers treated to hypoxia alone (Table 2). This suggests that O2 reintroduced to the system stimulates cellular mechanisms to reinforce cell-to-cell contact (TJ complexes), resulting in attenuation of paracellular permeability induced during hypoxic insult. Whether a sufficient reoxygenation period can induce cellular changes to reduce paracellular permeability to control levels requires further investigation.
The results from this study demonstrate that cerebral microvessel endothelial cells undergo molecular and functional changes during times of hypoxic-reperfusion stress, but the cellular signaling systems that connect the stress to these changes are not well characterized. In recent reviews by del Zoppo et al. (14) and Stanimirovic et al. (59), various mediators, including the proinflammatory cytokines tumor necrosis factor (TNF)-α and interleukin (IL)-1β, NO, and prostaglandins, are discussed in relation to their involvement with disruptions in BBB integrity during ischemic stroke. Sharkey et al. (56) showed an increased expression of the angiogenic cytokine vascular endothelial growth factor in endometrial cells exposed to decreased O2. In addition, hypoxia-induced increases in permeability of porcine brain endothelial cells have been correlated to such a rise in vascular endothelial growth factor (19). Temporal expression of cytokines (TNF-α, IL-6, and transforming growth factor-β) has been reported in focal ischemic model of stroke (30). In addition to release of these cytokines, the loss of O2 has also been shown to increase intracellular calcium levels, decrease levels of ATP, and the release of excitatory amino acids and NO (35, 38, 46). Abbruscato et al. (1) showed that calcium channel inhibitors (nifedipine and SKF-96365) prevented increased permeability induced by hypoxia-aglycemia (1). Posthypoxic reoxygenation has been shown to decrease the influx of calcium in bovine brain endothelial cells (34). This suggests that the recovery in permeability seen in the present study may be due to calcium mobilization by the endothelial cells. NO has been shown to increase permeability in cerebral microvasculature (45). Because NO is produced constitutively by calcium-dependent endothelial NO synthase and by calcium-independent inducible NO synthase, alterations in calcium levels may impact NO formation and other calcium-dependent cellular mechanisms (i.e., kinase activation). Decreased levels of ATP during hypoxic exposure have also been associated with increased permeability and actin rearrangement in brain endothelial cells (53). Changes in ATP levels are likely to impact phosphorylation states of cellular proteins. Whereas some proteins such as occludin are phosphorylated and tagged for degradation, phosphorylation is also a form of intracellular signaling (9, 57, 64). Recent studies (9, 57, 63, 64) have shown a correlation between increased phosphorylation of TJ proteins (ZO-1, ZO-2, and occludin) and increased permeability in both kidney and brain cell culture models. Indeed, it has been suggested that changes in phosphorylation of these cellular proteins is responsible for perturbations in cell-to-cell adhesion (40, 63). Whereas a 2.3-fold increase in tyrosine phosphorylation was seen in aortic endothelial cells exposed to 4-h hypoxia with 30-s posthypoxic reoxygenation (12), it is not clear which proteins are phosphorylated. Therefore, further work is required to determine which of these intracellular signaling systems connect the hypoxic insult and subsequent reoxygenation with the molecular and functional alterations demonstrated in the present study.
In summary, we report that hypoxia induces increased paracellular permeability that is significantly reduced on reoxygenation and these functional changes are associated with alterations in actin and TJ protein distribution. Whereas hypoxia-induced alterations in ZO-1 have been demonstrated previously (20), this is the first report examining the effects of hypoxia and posthypoxic reoxygenation on protein expression and localization of actin and TJ proteins in the cerebral microvasculature. Understanding cellular mechanisms induced by hypoxia and posthypoxic reoxygenation that alter BBB permeability will contribute to developing pharmacotherapies for treatment or prevention of decreased O2 conditions as seen with high-altitude exposure and ischemic stroke.
Acknowledgments
This study was supported by National Institute of Neurological Disorders and Stroke Grant R01-NS-39592 and University of Arizona Foundation Office of the Vice President for Research and Graduate Studies Grant 210777.
References
- 1.Abbruscato TJ, Davis TP. Combination of hypoxia/aglycemia compromises in vitro blood-brain barrier integrity. J Pharmacol Exp Ther. 1999;289:668–675. [PubMed] [Google Scholar]
- 2.Al-Haboubi HA, Ward BJ. Microvascular permeability of the isolated rat heart to various solutes in well-oxygenated and hypoxic conditions. Int J Microcirc Clin Exp. 1996;16:291–301. doi: 10.1159/000179188. [DOI] [PubMed] [Google Scholar]
- 3.Archer SL, Freude KA, Shultz PJ. Effect of graded hypoxia on the induction and function of inducible nitric oxide synthase in rat mesangial cells. Circ Res. 1995;77:21–28. doi: 10.1161/01.res.77.1.21. [DOI] [PubMed] [Google Scholar]
- 4.Audus KL, Borchardt RT. Bovine brain microvessel endothelial cell monolayers as a model for the blood-brain barrier. Ann NY Acad Sci. 1987;507:9–18. doi: 10.1111/j.1749-6632.1987.tb45787.x. [DOI] [PubMed] [Google Scholar]
- 5.Audus KL, Borchardt RT. Transport of macromolecules across the capillary endothelium. In: Juliano RL, editor. Targeted Drug Delivery. New York: Springer-Verlag; 1991. pp. 43–70. [Google Scholar]
- 6.Beck T, Krieglstein J. Cerebral circulation, metabolism, and blood-brain barrier of rats in hypocapnic hypoxia. Am J Physiol Heart Circ Physiol. 1987;252:H504–H512. doi: 10.1152/ajpheart.1987.252.3.H504. [DOI] [PubMed] [Google Scholar]
- 7.Betz AL, Yang GY, Kawai N, Fujisawa M, Abdelkarim GE, Ennis SR, Keep RF. Reperfusion-induced injury of the blood-brain barrier. In: Paulson OB, Knudsen GM, Moos T, editors. Brain Barrier Systems. Copenhagen, Denmark: Munksgaard; 1999. pp. 430–440. [Google Scholar]
- 8.Blum MS, Toninelli E, Anderson JM, Balda MS, Zhou J, O'Donnell L, Pardi R, Bender JR. Cytoskeletal rearrangement mediates human microvascular endothelial tight junction modulation by cytokines. Am J Physiol Heart Circ Physiol. 1997;273:H286–H294. doi: 10.1152/ajpheart.1997.273.1.H286. [DOI] [PubMed] [Google Scholar]
- 9.Braunton JL, Wong V, Wang W, Salter MW, Roder J, Liu M, Wang YT. Reduction of tyrosine kinase activity and protein tyrosine dephosphorylation by anoxic stimulation in vitro. Neuroscience. 1998;82:161–170. doi: 10.1016/s0306-4522(97)00286-8. [DOI] [PubMed] [Google Scholar]
- 10.Bruning JL, Kintz BL. Computational Handbook of Statistics. Glenview, IL: Scott, Foresman; 1977. pp. 112–131. [Google Scholar]
- 11.Chen Y, Lu Q, Schneeberger EE, Goodenough DA. Restoration of tight junction structure and barrier function by downregulation of the mitogen-activated protein kinase pathway in ras-transformed Madin-Darby canine kidney cells. Mol Biol Cell. 2000;11:849–862. doi: 10.1091/mbc.11.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crawford LE, Milliken EE, Irani K, Zweier JL, Becker LC, Johnson TM, Eissa NT, Crystal RG, Finkel T, Goldschmidt-Clermont PJ. Superoxide-mediated actin response in posthypoxic endothelial cells. J Biol Chem. 1996;271:26863–26867. doi: 10.1074/jbc.271.43.26863. [DOI] [PubMed] [Google Scholar]
- 13.Dejana E, Corada M, Lampugnani MG. Endothelial cell-to-cell junctions. FASEB J. 1995;9:910–918. [PubMed] [Google Scholar]
- 14.del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000;10:95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deli MA, Descamps L, Dehouck MP, Cecchelli R, Joo F, Abraham CS, Torpier G. Exposure of tumor necrosis factor-α to luminal membrane of bovine brain capillary endothelial cells co-cultured with astrocytes induces a delayed increase of permeability and cytoplasmic stress fiber formation of actin. J Neurosci Res. 1995;41:717–726. doi: 10.1002/jnr.490410602. [DOI] [PubMed] [Google Scholar]
- 16.Dijkhuizen RM, Knollema S, van der Worp HB, Ter Horst GJ, De Wildt DJ, Berkelbach van der Sprenkel JW, Tulle-ken KA, Nicolay K. Dynamics of cerebral tissue injury and perfusion after temporary hypoxia-ischemia in the rat: evidence for region-specific sensitivity and delayed damage. Stroke. 1998;29:695–704. doi: 10.1161/01.str.29.3.695. [DOI] [PubMed] [Google Scholar]
- 17.Dunbabin D. Cost-effective intervention in stroke. Pharmaco-economics. 1992;2:468–499. doi: 10.2165/00019053-199202060-00007. [DOI] [PubMed] [Google Scholar]
- 18.Easton JD. Current advances in the management of stroke. Neurology. 1998;51:S1–S2. doi: 10.1212/wnl.51.3_suppl_3.s1. [DOI] [PubMed] [Google Scholar]
- 19.Fischer S, Clauss M, Wiesnet M, Renz D, Schaper W, Karliczek GF. Hypoxia induces permeability in brain microvessel endothelial cells via VEGF and NO. Am J Physiol Cell Physiol. 1999;276:C812–C820. doi: 10.1152/ajpcell.1999.276.4.C812. [DOI] [PubMed] [Google Scholar]
- 20.Fischer S, Wobben M, Kleinstuck J, Renz D, Schaper W. Effect of astroglial cells on hypoxia-induced permeability in PBMEC cells. Am J Physiol Cell Physiol. 2000;279:C935–C944. doi: 10.1152/ajpcell.2000.279.4.C935. [DOI] [PubMed] [Google Scholar]
- 21.Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141:1539–1550. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furuse M, Sasaki H, Fujimoto K, Tsukita S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol. 1998;143:391–401. doi: 10.1083/jcb.143.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giese H, Mertsch K, Blasig IE. Effect of MK-801 and U83836E on a porcine brain capillary endothelial cell barrier during hypoxia. Neurosci Lett. 1995;191:169–172. doi: 10.1016/0304-3940(95)11584-j. [DOI] [PubMed] [Google Scholar]
- 24.Golanov EV, Reis DJ. Oxygen and cerebral blood flow. In: Welch KMA, Caplan LR, Reis DJ, Siesjo BK, Weir B, editors. Primer on Cerebrovascular Disease. San Diego, CA: Academic; 1997. pp. 58–60. [Google Scholar]
- 25.Guo Y, Ward ME, Beasjours S, Mori M, Hussain SN. Regulation of cerebellar nitric oxide production in response to prolonged in vivo hypoxia. J Neurosci Res. 1997;49:89–97. [PubMed] [Google Scholar]
- 26.Hackett PH. High altitude cerebral edema and acute mountain sickness. A pathophysiology update. Adv Exp Med Biol. 1999;474:23–45. doi: 10.1007/978-1-4615-4711-2_2. [DOI] [PubMed] [Google Scholar]
- 27.Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 28.Hawkins RA. Transport of essential nutrients across the blood-brain barrier of individual structures. Fed Proc. 1986;45:2055–2059. [PubMed] [Google Scholar]
- 29.Heiskala M, Peterson PA, Yang Y. The roles of claudin superfamily proteins in paracellular transport. Traffic. 2001;2:93–98. doi: 10.1034/j.1600-0854.2001.020203.x. [DOI] [PubMed] [Google Scholar]
- 30.Hill JK, Gunion-Rinker L, Kulhanek D, Lessov N, Kim S, Clark WM, Dixon MP, Nishi R, Stenzel-Poore MP, Eckenstein FP. Temporal modulation of cytokine expression following focal cerebral ischemia in mice. Brain Res. 1999;820:45–54. doi: 10.1016/s0006-8993(98)01140-8. [DOI] [PubMed] [Google Scholar]
- 31.Howard EF, Chen Q, Cheng C, Carroll JE, Hess D. NF-kappa B is activated and ICAM-1 gene expression is upregu-lated during reoxygenation of human brain endothelial cells. Neurosci Lett. 1998;248:199–203. doi: 10.1016/s0304-3940(98)00239-0. [DOI] [PubMed] [Google Scholar]
- 32.Ikeda K, Nagashima T, Wu S, Yamaguchi M, Tamaki N. The role of calcium ion in anoxia/reoxygenation damage of cultured brain capillary endothelial cells. Acta Neurochir Suppl (Wien) 1997;70:4–7. doi: 10.1007/978-3-7091-6837-0_2. [DOI] [PubMed] [Google Scholar]
- 33.Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol. 1999;147:1351–1363. doi: 10.1083/jcb.147.6.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimura C, Oike M, Ito Y. Hypoxia-induced alterations in Ca2+ mobilization in brain microvascular endothelial cells. Am J Physiol Heart Circ Physiol. 2000;279:H2310–H2318. doi: 10.1152/ajpheart.2000.279.5.H2310. [DOI] [PubMed] [Google Scholar]
- 35.Kish PE, Ueda T. Calcium-dependent release of accumulated glutamate from synaptic vesicles within permeabilized nerve terminals. Neurosci Lett. 1991;122:179–182. doi: 10.1016/0304-3940(91)90852-k. [DOI] [PubMed] [Google Scholar]
- 36.Kniesel U, Wolburg H. Tight junctions of the blood-brain barrier. Cell Mol Neurobiol. 2000;20:57–76. doi: 10.1023/a:1006995910836. [DOI] [PubMed] [Google Scholar]
- 37.Krasney JA. A neurogenic basis for acute altitude illness. Med Sci Sports Exerc. 1994;26:195–208. doi: 10.1249/00005768-199402000-00010. [DOI] [PubMed] [Google Scholar]
- 38.Kristian T, Siesjo BK. Calcium in ischemic cell death. Stroke. 1998;29:705–718. doi: 10.1161/01.str.29.3.705. [DOI] [PubMed] [Google Scholar]
- 39.Lassen NA. Pathophysiology of brain ischemia as it relates to the therapy of acute ischemic stroke. Clin Neuropharmacol. 1990;13:S1–S8. doi: 10.1097/00002826-199013003-00001. [DOI] [PubMed] [Google Scholar]
- 40.Lee TYJ, Gotlieb AI. Early stages of endothelial wound repair: conversion of quiescent to migrating endothelial cells involves tyrosine phosphorylation and actin microfilament reorganization. Cell Tissue Res. 1999;297:435–450. doi: 10.1007/s004410051370. [DOI] [PubMed] [Google Scholar]
- 41.Leniger-Follert E, Lubbers DW, Wrabetz W. Regulation of local tissue Po2 of the brain cortex at different arterial O2 pressures. Pflügers Arch. 1975;359:81–95. doi: 10.1007/BF00581279. [DOI] [PubMed] [Google Scholar]
- 42.Lum H, Barr DA, Shaffer JR, Gordon RJ, Ezrin AM, Malik AB. Reoxygenation of endothelial cells increases permeability by oxidant-dependent mechanisms. Circ Res. 1992;70:991–998. doi: 10.1161/01.res.70.5.991. [DOI] [PubMed] [Google Scholar]
- 43.Mark KS, Davis TP. Stroke: developement, prevention and treatment with pepitase inhibitors. Peptides. 2000;21:1750–1758. doi: 10.1016/s0196-9781(00)00346-6. [DOI] [PubMed] [Google Scholar]
- 44.Mason RB, Pluta RM, Walbridge S, Wink DA, Oldfield EH, Boock RJ. Production of reactive oxygen species after reperfusion in vitro and in vivo: protective effect of nitric oxide. J Neurosurg. 2000;93:99–107. doi: 10.3171/jns.2000.93.1.0099. [DOI] [PubMed] [Google Scholar]
- 45.Mayhan WG. Role of nitric oxide in disruption of the blood-brain barrier during acute hypertension. Brain Res. 1995;686:99–103. doi: 10.1016/0006-8993(95)00460-8. [DOI] [PubMed] [Google Scholar]
- 46.Mayhan WG, Didion SP. Glutamate-induced disruption of the blood-brain barrier in rats. Role of nitric oxide. Stroke. 1996;27:965–970. doi: 10.1161/01.str.27.5.965. [DOI] [PubMed] [Google Scholar]
- 47.McKinley BA, Morris WP, Parmley CL, Butler BD. Brain parenchyma Po2, Pco2, and pH during and after hypoxic, ischemic brain insult in dogs. Crit Care Med. 1996;24:1858–1868. doi: 10.1097/00003246-199611000-00016. [DOI] [PubMed] [Google Scholar]
- 48.Miller DW, Audus KL, Borchardt RT. Application of cultured endothelial cells of the brain microvasculature in the study of the blood-brain barrier. J Tiss Cult Meth. 1992;14:217–224. [Google Scholar]
- 49.Mitic LL, Anderson JM. Molecular architecture of tight junctions. Annu Rev Physiol. 1998;60:121–142. doi: 10.1146/annurev.physiol.60.1.121. [DOI] [PubMed] [Google Scholar]
- 50.Miyamoto O, Auer RN. Hypoxia, hyperoxia, ischemia, and brain necrosis. Neurology. 2000;54:362–371. doi: 10.1212/wnl.54.2.362. [DOI] [PubMed] [Google Scholar]
- 51.Orr JA, Bisgard GE, Forster HV, Buss DD, Dempsey JA, Will JA. Cerebrospinal fluid alkalosis during high-altitude sojourn in unanesthetized ponies. Respir Physiol. 1975;25:23–37. doi: 10.1016/0034-5687(75)90048-1. [DOI] [PubMed] [Google Scholar]
- 52.Park TS, Gonzales ER, Gidday JM. Platelet-activating factor mediates ischemia-induced leukocyte-endothelial adherence in newborn pig brain. J Cereb Blood Flow Metab. 1999;19:417–424. doi: 10.1097/00004647-199904000-00007. [DOI] [PubMed] [Google Scholar]
- 53.Plateel M, Dehouck MP, Torpier G, Cecchelli R, Teissier E. Hypoxia increases the susceptibility to oxidant stress and the permeability of the blood-brain barrier endothelial cell monolayer. J Neurochem. 1995;65:2138–2145. doi: 10.1046/j.1471-4159.1995.65052138.x. [DOI] [PubMed] [Google Scholar]
- 54.Razandi M, Pedram A, Levin ER. Estrogen signals to the preservation of endothelial cell form and function. J Biol Chem. 2000;275:38540–38546. doi: 10.1074/jbc.M007555200. [DOI] [PubMed] [Google Scholar]
- 55.Schneeberger EE, Lynch RD. Tight junctions: their structure, composition, and function. Circ Res. 1984;55:723–733. doi: 10.1161/01.res.55.6.723. [DOI] [PubMed] [Google Scholar]
- 56.Sharkey AM, Day K, McPherson A, Malik S, Licence D, Smith SK, Charnock-Jones DS. Vascular endothelial growth factor expression in human endometrium is regulated by hypoxia. J Clin Endocrinol Metab. 2000;85:402–409. doi: 10.1210/jcem.85.1.6229. [DOI] [PubMed] [Google Scholar]
- 57.Staddon J, Ratcliffe M, Morgan L, Hirase T, Smales C, Rubin L. Protein phosphorylation and the regulation of cell-cell junctions in brain endothelial cells. Heart Vessels Suppl. 1997;12:106–109. [PubMed] [Google Scholar]
- 58.Staddon JM, Herrenknecht K, Smales C, Rubin LL. Evidence that tyrosine phosphorylation may increase tight junction permeability. J Cell Sci. 1995;108:609–619. doi: 10.1242/jcs.108.2.609. [DOI] [PubMed] [Google Scholar]
- 59.Stanimirovic D, Satoh K. Inflammatory mediators of cerebral endothelium: a role in ischemic brain inflammation. Brain Pathol. 2000;10:113–126. doi: 10.1111/j.1750-3639.2000.tb00248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stanimirovic D, Shapiro A, Wong J, Hutchison J, Durkin J. The induction of ICAM-1 in human cerebromicrovascular endothelial cells (HCEC) by ischemia-like conditions promotes enhanced neutrophil/HCEC adhesion. J Neuroimmunol. 1997;76:193–205. doi: 10.1016/s0165-5728(97)00057-x. [DOI] [PubMed] [Google Scholar]
- 61.Su Y, Block ER. Role of calpain in hypoxic inhibition of nitric oxide synthase activity in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2000;278:L1204–L1212. doi: 10.1152/ajplung.2000.278.6.L1204. [DOI] [PubMed] [Google Scholar]
- 62.Sun GY, Zhang JP, Lin TA, Lin TN, He YY, Hsu CY. Inositol trisphosphate, polyphosphoinositide turnover, and high-energy metabolites in focal cerebral ischemia and reperfusion. Stroke. 1995;26:1893–1900. doi: 10.1161/01.str.26.10.1893. [DOI] [PubMed] [Google Scholar]
- 63.Tsukamoto T, Nigam SK. Role of tyrosine phosphorylation in the reassembly of occludin and other tight junction proteins. Am J Physiol Renal Physiol. 1999;276:F737–F750. doi: 10.1152/ajprenal.1999.276.5.F737. [DOI] [PubMed] [Google Scholar]
- 64.Tsukamoto T, Nigam SK. Tight junction proteins form large complexes and associate with the cytoskeleton in an ATP depletion model for reversible junction assembly. J Biol Chem. 1997;272:16133–16139. doi: 10.1074/jbc.272.26.16133. [DOI] [PubMed] [Google Scholar]
- 65.Tsukita S, Furuse M. Occludin and claudins in tight-junction strands: leading or supporting players? Trends Cell Biol. 1999;9:268–273. doi: 10.1016/s0962-8924(99)01578-0. [DOI] [PubMed] [Google Scholar]
- 66.Utepbergenov DI, Mertsch K, Sporbert A, Tenz K, Paul M, Haseloff RF, Blasig IE. Nitric oxide protects blood-brain barrier in vitro from hypoxia/reoxygenation-mediated injury. FEBS Lett. 1998;424:197–201. doi: 10.1016/s0014-5793(98)00173-2. [DOI] [PubMed] [Google Scholar]
- 67.Yi X, Wang Y, Yu FS. Corneal epithelial tight junctions and their response to lipopolysaccharide challenge. Invest Ophthalmol Vis Sci. 2000;41:4093–4100. [PubMed] [Google Scholar]