

Abstract
The blood-brain barrier (BBB) is a critical regulator of CNS homeostasis. Additionally, the BBB is the most significant obstacle to effective CNS drug delivery. It possesses specific charcteristics (i.e., tight junction protein complexes, influx and efflux transporters) that control permeation of circulating solutes including therapeutic agents. In order to form this “barrier,” brain microvascular endothelial cells require support of adjacent astrocytes and microglia. This intricate relationship also occurs between endothelial cells and other cell types and structures of the CNS (i.e., pericytes, neurons, extracellular matrix), which implies existence of a “neurovascular unit.” Ischemic stroke can disrupt the neurovascular unit at both the structural and functional level, which leads to an increase in leak across the BBB. Recent studies have identified several pathophysiological mechanisms (i.e., oxidative stress, activation of cytokine-mediated intracellular signaling systems) that mediate changes in the neurovascular unit during ischemic stroke. This review summarizes current knowledge in this area and emphasizes pathways (i.e., oxidative stress, cytokine-mediated intracellular signaling, glial-expressed receptors/targets) that can be manipulated pharmacologically for i) preservation of BBB and glial integrity during ischemic stroke and ii) control of drug permeation and/or transport across the BBB in an effort to identify novel targets for optimization of CNS delivery of therapeutics in the setting of ischemic stroke.
Keywords: Astrocyte, Blood-Brain Barrier, Drug Delivery, Endothelial Cell, Ischemic Stroke, Tight Junction, Transporters
Introduction
The blood-brain barrier (BBB) is an essential physical and metabolic barrier that separates the central nervous system (CNS) from the systemic circulation. It is formed by a monolayer of capillary endothelial cells that have evolved to greatly limit brain uptake of circulating substances in an effort to precisely maintain cerebral homeostasis. Additionally, the BBB is the most significant obstacle to CNS drug delivery. In fact, many existing drugs have limited or no efficacy in treatment of neurological diseases primarily due to an inability to permeate the BBB and attain therapeutic concentrations within the CNS (1). A critical concept in BBB biology is that brain microvascular endothelial cells are not intrinsically capable of forming a “barrier.” In fact, formation and function of the BBB requires support of adjacent glial cells (i.e., astrocytes, microglia) as well as neurons, pericytes, and extracellular matrix (2). Cell-cell interactions and signaling occur in a co-coordinated mannerbetweenthese multiple cell types, events required forphysiological and pathological functioning of the BBB. Such an intricate relationship implies existence of a “neurovascular unit (NVU).” During ischemic stroke, various NVU cell types are triggered by pathological stimuli that disrupt the BBB. Understanding the endothelial and glial cell responses that are involved in modifying the NVU/BBB in the context of ischemic stroke provides an opportunity not only to protect BBB integrity during pathological insult but also to target these mechanisms for optimization of CNS drug delivery. Multiple studies by our group have provided rigorous evidence on how such pathways alter NVU/BBB physiology and how they can be targeted for improvement of CNS pharmacotherapy (3–13). Here, we provide a summary of the BBB and interactions with associated cell types/structures (i.e., glial cells, pericytes, neurons, extracellular matrix) of the NVU. Additionally, we review pathophysiological mechanisms in endothelial cells and glial cells that contribute to BBB/NVU dysfunction in ischemic stroke. Finally, we provide insights on how such mechanisms can be targeted in an effort to protect BBB integrity and optimize CNS drug delivery for treatment of ischemic stroke.
The Neurovascular Unit
The NVU consists of all major cell types found in the CNS including brain microvascular endothelial cells, astrocytes, microglia, pericytes, and neurons (14–16). The concept of the NVU emphasizes that normal brain function as well as dysfunction occurs via the co-coordinated interaction between these various cell types (17;18). In fact, decreased BBB functional integrity is known to occur prior to neuronal injury in stroke, an observation that suggests that NVU dysfunction is directly linked to compromise of the BBB (19–21). Below we provide an outline of these NVU constituents with an emphasis on properties that are involved in the pathophysiological response to ischemic stroke.
Components of the Neurovascular Unit
Endothelial Cells and the Blood-Brain Barrier
The CNS is the most critical and sensitive organ system in the human body. Proper neuronal and glial function necessitates precise regulation of the brain extracellular milieu. Additionally, the metabolic demands of CNS tissue are considerable with the CNS accounting for approximately 20% of oxygen consumption in humans (22). Therefore, the interface between the CNS and the systemic circulation must possess highly selective and effective mechanisms that can facilitate nutrient transport, exactly regulate ion balance, and provide a barrier to toxic substances that may be present in the systemic circulation. The requirement for a physical and metabolic barrier is further emphasized by the extreme sensitivity of CNS tissues to exogenous solutes. Therefore, brain entry of some substances must be permitted while accumulation of other substances in brain parenchyma must be excluded. This homeostatic function of the cerebral microvasculature primarily at the level of the brain microvascular endothelium, the principal cell type of the BBB.
The current understanding of its basic structure is built primarily upon work by Reese, Karnovsky, and Brightman in the late 1960s (23;24). Anatomically, BBB endothelial cells are distinguished from those in the periphery by a lack of fenestrations, minimal pinocytotic activity, and presence of tight junctions (TJs) (25). Cerebral endothelial cells are demarcated by increased mitochondrial content as compared with other endothelium in the body (26). This increased content of mitochondria is required for transport of solutes into and out of the brain thereby contributing to maintenance of CNS homeostasis. Additionally, several receptors, ion channels, and influx/efflux transport proteins are prominently expressed in brain microvascular endothelial cells. Functionally, these transport systems are similar to well-characterized systems in other tissues (i.e., D-glucose transporter, L-amino acid carrier systems, Na+/K+ ATPase), although the capacity and rate of transport can vary. Transporters involved in transcellular flux of drugs have also been identified and characterized at the BBB endothelium. Examples of such transporters known to be expressed in brain endothelial cells include efflux transporters such as P-glycoprotein (P-gp) (27–30), Multidrug Resistance Proteins 1–6 (MRP1-6 in humans; Mrp1-6 in rodents) (27;31–34), and Breast Cancer Resistance Protein (BCRP in humans; Bcrp in rodents) (29;35;36). Additionally, transporters that have been shown to facilitate drug permeation across the BBB include organic anion transporting polypeptides (OATPs in humans; Oatps in rodents) (13;30;37;38), organic anion transporters (32;39–41), monocarboxylate transporters (39;42), nucleoside transporters (43), and peptide transporters (44).
Molecular Characteristics of the BBB
a) TJ Protein Complexes
BBB endothelial cells are interconnected by TJs, large multiprotein complexes that are maintained by astrocytic trophic factors (figure 1). TJs form a continuous, almost impermeable barrier that limits paracellular flux of xenobiotics with the exception of small, lipid-soluble molecules (25). The high BBB transendothelial resistance (1,500 – 2,000 Ωcm2) further restricts the free flow of water and solutes (45). BBB TJs are formed by junction adhesion molecules (JAMs), occludin, and claudins (i.e., claudin-1, -3, and -5), transmembrane proteins linked to the cytoskeleton through interactions with accessory proteins (i.e., zonula occluden (ZO)-1, -2, and -3) (14). ZO proteins act as a scaffold for multiple intracellular signaling pathways and are involved in regulation of TJ function (46). Additionally, other protein constituents (i.e., cingulin, AF6, 7H6, EMP-1) have been localized to the TJ but their exact role has yet to be elucidated. A brief description of the primary proteins that constitute TJ protein complexes at the BBB is described below. Although endothelial cells of the BBB do also possess adherens junctions, which are ubiquitous in the vasculature and mediate inter-endothelial cell adhesion, these will not be discussed in this review.
Figure 1.
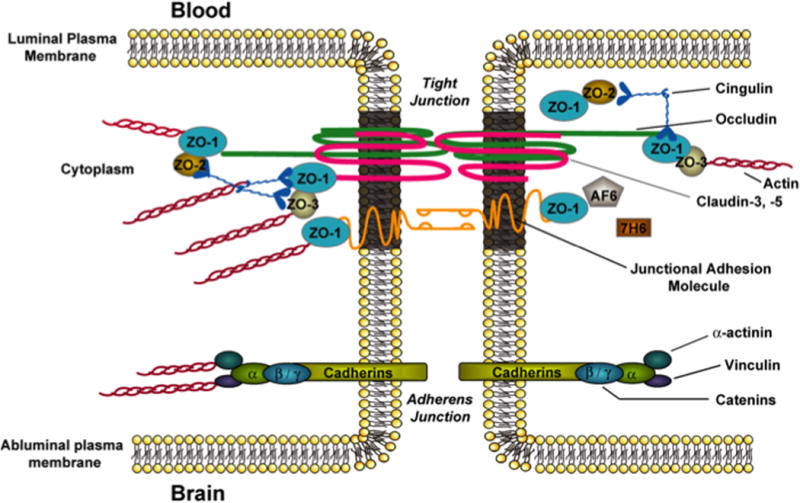
Basic molecular organization of tight junction protein complexes at the blood-brain barrier. Adapted from Ronaldson & Davis. Therapeutic Delivery. 2: 1016–1041 (2011).
Several JAM isoforms have been identified at the mammalian BBB including JAM-1, JAM-2, and JAM-3 (14;47). JAM-1 is a 40-kDa member of the IgG superfamily and is believed to mediate the early attachment of adjacent endothelial cells during development of the BBB (48). In addition to their developmental roles, JAMs regulate the transendothelial migration of leukocytes (47). Although their function in mature BBB is largely unknown, loss of JAM protein expression is directly correlated with BBB breakdown (49;50). Studies in an immortalized human brain endothelial cell line (hCMEC/d3) showed that inflammatory stimulation led to increased paracellular permeability to dextran 3000 that correlated with movement of JAM away from the tight junction, further suggesting that JAMs play a critical role in maintaining BBB functional integrity (51). Interestingly, serum levels of JAM-A were unchanged over time in 13 patients with acute ischemic stroke, suggesting that this JAM isoform is not a suitable biomarker of BBB breakdown (51).
Monomeric occludin is a 60- to 65-kDa protein that has four transmembrane domains with the carboxyl and amino terminals oriented to the cytoplasm and two extracellular loops that span the intercellular cleft (52). It is highly expressed and consistently stains in a distinct, continuous pattern along endothelial cell margins in the cerebral vasculature (10;53;54). In contrast, occludin distribution is considerably more diffuse in non-neural endothelia (55). In a previous literature review, it was suggested that while occludin is integrally localized to the TJ, it is not essential for TJ assembly and has no direct “tightening” function (56). We do not agree nor find any compelling data to support this statement. In contrast, recent studies by our group have clearly shown that occludin is a critical regulator of BBB permeability in vivo (8;54;57). This essential role for occludin has also been shown to occur via interaction between two extracellular loops on occludin monomers and homologous segments on occludin molecules localized to adjacent endothelial cells (58;59). Feldman and colleagues described this interactionthat creates a tight seal and restricts paracellular solute diffusion (59). Occludin also assembles into dimers and higher order oligomers at the TJ (8;54;57). Such occludin oligomeric assemblies are required for physiological function at the TJ, particularly as a restrictor of paracellular permeability and supports the critical role for occludin’s direct tightening function (54). Altered expression of occludin is associated with disrupted BBB function in various pathologies associated with ischemic stroke including hypoxia/aglycemia (60) and H/R stress (10).
Claudins have similar membrane topography to occludin but no sequence homology (52). Claudins are 20- to 24-kDa proteins, of which at least 24 have been identified in mammals (14). All claudins have similar sequence homology among themselves in the first and fourth transmembrane domains and extracellular loops (61). The extracellular loops of claudins interact via homophilic and heterophilic interactions between cells (62). Overexpression of claudin isoforms in fibroblasts can induce cell aggregation and formation of TJ-like strands. Conversely, occludin only localizes to TJs in cells that have already been transfected with claudins (63). Thus, it is hypothesized that claudins form the primary “seal” of the TJ. In the cerebral microvascular endothelium, various isoforms of claudins have been detected including claudin-1, -3, and -5 (3;53;64–67). In experimental models of ischemic stroke, reduced expression of claudin-5 (68;69) and disruption of interaction between claudin-5 and occludin (70) has been reported.
Proper physiological functioning of the BBB, particularly restriction of paracellular solute transport, requires association of transmembrane constituents of tight junction protein complexes with accessory proteins localized within the endothelial cell cytoplasm. These include members of the membrane-associated guanylate kinase-like (MAGUK) family. In brain microvascular endothelial cells, MAGUK proteins are involved in coordination and clustering of tight junction protein complexes to the cell membrane and in establishment of specialized domains within the membrane (71). Three MAGUK proteins have been identified at the TJ: ZO-1, -2. and -3. ZO-1 was the first protein that was shown to be directly associated with TJ complexes (72). It is a 220 kDa protein that links transmembrane proteins of the TJ (i.e., occludin, claudins) to the actin cytoskeleton (73). Previous studies have demonstrated that dissociation of ZO-1 from the junction complex is associated with increased permeability (74–76), which implies that the ZO-1-transmembrane protein interaction is critical to TJ stability and function. ZO-1 may also act as a signaling molecule that communicates the state of the TJ to the cellular interior, or vice versa. ZO-1 has been shown to localize to the nucleus under conditions of proliferation and injury (77), following Ca2+ depletion (78), and in response to nicotine (53). It has also been colocalized with transcription factors (79) and various G-proteins (80). ZO-2, a 160-kDa protein, has high sequence homology to ZO-1 (81) and is known to bind structural tight junction constituents, signaling molecules, and transcription factors (82). ZO-2 localizes to the nucleus during stress and proliferation, a property similar to ZO-1 (83;84). In cultured brain microvessel endothelial cells, ZO-2 is localized along the plasma membrane at points of cell-cell contact (76), although it may be distributed more diffusely in whole cerebral microvessels (53). Of particular note, ZO-2 may function somewhat redundantly with ZO-1 as it has been shown to facilitate formation of TJs that are morphologically intact in cultured epithelial cells lacking ZO-1 (85). More recently, ZO-3, a 130-kDa protein, has been identified at the BBB (86) but its exact role in TJ formation and/or function has not been elucidated. Using the cortical aspiration lesion experimental stroke model in rodents, Li and colleagues reported decreased ZO-1 expression in ipsilateral thalamus (87). Additionally, Jiao and colleagues reported that redistribution of ZO-1 away from the TJ in cerebral microvessels correlated with an increase in Evans blue-albumin leak across the BBB (88).
In addition to MAGUK family members, other accessory proteins have been identified at the TJ. These include cingulin, AF-6, 7H6, and EMP-1 (14;89). Cingulin is a 140- to 160-kDa protein that associates with ZOs, JAM-1, and myosin (90) and is hypothesized to mediate interactions between the cytoskeleton and the TJ. AF-6, a 180-kDa protein, interacts directly with ZO-1. This interaction is inhibited by inactivation of Ras, suggesting that disruption of ZO-1/AF-6 complex may be critical in modulation of the TJ by Ras-dependent pathways (91). The function of 7H6 at the BBB is unknown (14) but it is known to reversibly dissociate from the TJ under conditions of ATP depletion (92) as may be observed during ischemic stroke. More recently, studies have identified a novel TJ protein known as epithelial membrane protein-1 (EMP-1) (89). Bangsow and colleagues showed that EMP-1 is enriched in porcine and murine brain microvessel endothelial cells as well as rat brain microvessels and colocalizes with occludin (89). Of particular note, EMP-1 is upregulated under ischemic conditions while occludin is downregulated (89), which suggests that this protein may play a role in maintenance of barrier function during ischemic stroke and/or H/R stress.
b) Transport Proteins
For many substances, uptake into the brain and extrusion from the brain is governed by transport proteins that are endogenously and selectively expressed at the BBB endothelium. Such transport proteins that have been shown to be involved in influx and/or efflux of circulating solutes include ATP-binding cassette (ABC) transporters and solute carrier (SLC) transporters (93;94). In order to target transporters for optimization of CNS drug delivery, it is critical to understand localization (i.e., luminal versus abluminal) and functional expression of transport proteins at the BBB endothelium. Below, we summarize current knowledge on such membrane transporters that are known to be involved in determining CNS delivery of therapeutic agents. Localization of specific transport proteins known to be involved in CNS drug delivery is depicted in figure 2.
Figure 2.
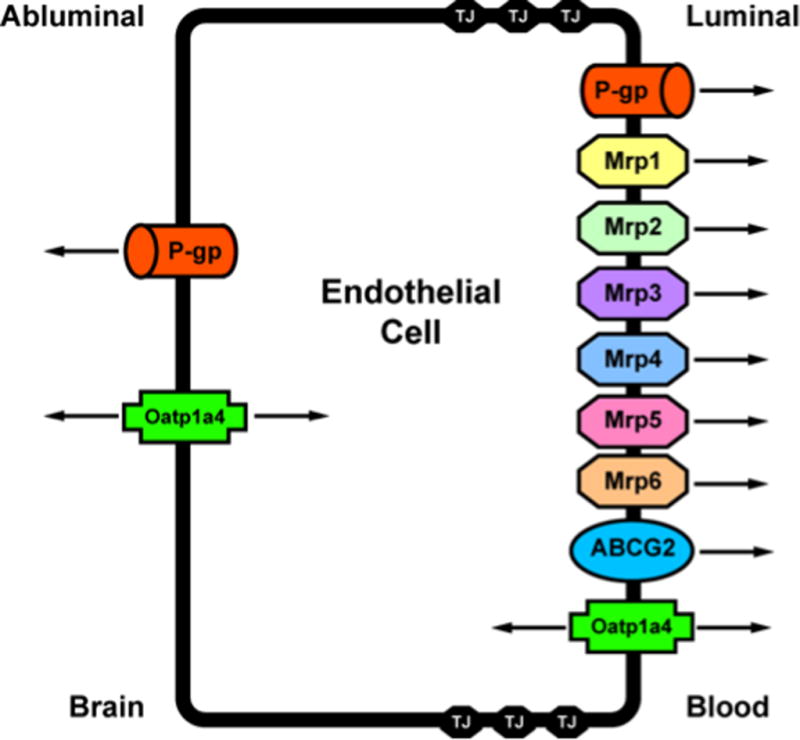
Endothelial localization of drug transporters known to be involved in transport of opioids at the blood-brain barrier. Adapted from Ronaldson & Davis. Therapeutic Delivery. 2: 1016–1041 (2011).
ABC Transporters
The ABC superfamily is among the largest and most ubiquitously expressed protein families known to date. ABC transporters are involved in translocation of opioids and their metabolites against their concentration gradient. The energy to transport xenobiotics is provided by binding and subsequent hydrolysis of ATP. In humans, 48 ABC genes have been identified and are classified according to seven subfamilies (95). ABC drug transporters, specifically P-glycoprotein (P-gp), Multidrug Resistance Proteins (MRPs in humans; Mrps in rodents) and Breast Cancer Resistance Protein (BCRP; also known as ABCG2) are known to be involved in cellular extrusion of therapeutic agents and thus constitute a considerable barrier to effective delivery of drugs to the brain. In general, P-gp transports cationic or basic and neutral compounds, whereas MRPs/Mrps are involved in cellular efflux of anionic drugs as well as their glucuronidated, sulfated, and glutathione-conjugated metabolites (96). BCRP/Bcrp has significant overlap in substrate specificity profile with P-gp and has been shown to recognize a vast array of sulfoconjugated organic anions, hydrophobic, and amphiphilic compounds (97).
P-gp is a 170-kDa ATP-dependent integral membrane protein that was originally identified in colchicine-resistant Chinese hamster ovary cells (98). It was designated as “P-glycoprotein” because of its inherent ability to affect membrane permeability of biological substances that may be potentially toxic (98). Physiologically, P-gp is believed to function as a biological defense mechanism against entry of toxic substances from the gut into the blood and for protection of vital organ systems such as the brain (99). The majority of P-gp transport substrates are weakly amphipathic and relatively hydrophobic. Additionally, many (but not all) substrates contain an aromatic ring and a positively charged tertiary nitrogen atom in their chemical structure (100). P-gp orthologues from different species have greater than 70% sequence identity (99) and are encoded by closely related genes (i.e., multidrug resistance (MDR) genes), which have two isoforms in humans (MDR1, MDR2) and three isoforms in both mice (i.e., mdr1, mdr2, mdr3) and rats (i.e., mdr1a, mdr1b, mdr2). The human MDR2 gene and the murine/rodent mdr2 gene products are exclusively involved in hepatic transport of phosphatidylcholine and will not be further discussed. In contrast, human MDR1, murine mdr1/mdr3, and rodent mdr1a/mdr1b are involved in transport of therapeutic agents in several tissues including BBB endothelium. Specifically, P-gp expression has been identified at both the luminal (101–103) and abluminal membrane (103–105) of brain microvascular endothelial cells. Abluminal localization of P-gp has been identified on perivascular astrocyte foot processes (104;105) and on the abluminal plasma membrane of the endothelial cell itself (103). Increased expression of P-gp has been reported in hippocampal microvessels isolated from stroke-prone spontaneously hypertensive rats (106), which suggests that alterations in P-gp expression and/or activity may be a critical component of the BBB response to ischemic stroke. Furthermore, this increase of an efflux transporter in an in vivo experimental stroke model may also represent a significant impediment to drug delivery to the ischemic brain.
The mammalian MRP family belongs to the ABCC group of proteins, which contains 13 members including one ion channel (i.e., CFTR), two surface receptors (i.e., SUR1 and 2) and a truncated protein that does not mediate transport (i.e., ABCC13) (31). These proteins are not involved in drug transport and will not be further discussed. Many of the functionally characterized MRP isoforms that are known to be involved in drug transport have been localized to the mammalian BBB. These include MRP1/Mrp1, MRP2/Mrp2, MRP4/Mrp4, Mrp5 and Mrp6 (33;36;107–110). The presence of multiple MRP homologues at the BBB may be a vital determinant in controlling the delivery of therapeutic agentsto the brain. Additionally, the ability of Mrp isoforms to actively efflux the endogenous antioxidant glutathione (GSH) may have significant implications in ischemic stroke. GSH is a critical factor responsible for maintenance of cellular redox balance and antioxidant defense in the brain during ischemic stroke. It has been previously demonstrated that functional expression of Mrps is upregulated in response to oxidative stress conditions, which leads to enhanced cellular efflux of GSH (111). In the ischemic brain, an upregulation of Mrp isoforms at the BBB could potentially cause reduced brain concentrations of GSH, an alteration in CNS redox status, and increased potential for neuronal and glial cell injury and death. Therefore, changes in functional expression of Mrps at the BBB in response to ischemic stroke warrants further investigation.
A third ABC superfamily member that may be involved in xenobiotic efflux is BCRP. Several recent studies have demonstrated localization of BCRP at the brain microvasculature, particularly along the luminal side of the BBB (112–114). In terms of transport activity, data from recent in vitro and in vivo studies are controversial. Although some studies have suggested that BCRP is not functional at the BBB (35;114;115) or plays a minimal role in xenobiotic efflux from the brain (116), more detailed analyses have confirmed that BCRP is a critical determinant of drug permeation across the BBB (29;117–119). The effect of ischemic stroke and/or cerebral hypoxia on BBB functional expression of BCRP is a critical point for future study.
Solute Carrier (SLC) Transporters
The second major group of drug transport proteins at the BBB endothelium is the SLC transporters. In contrast to ABC transporters, membrane transport of SLC family members is governed by either an electrochemical gradient utilizing an inorganic or organic solute as a driving force or the transmembrane concentration gradient of the substance actually being transported. To date, 319 SLC genes (i.e., SLC1 – SLC43 families) have been identified in humans (120). Of the 43 known families of SLC transporters, members of SLC21A/SLCO and SLC22 are known to be expressed at the BBB and play a critical role in determining xenobiotic permeation across the brain microvascular endothelium (121).
Of the SLC transporters known to transport drugs at the BBB, perhaps the most viable candidates for transporter targeting are members of the SLC21A/SLCO family that includes organic anion transporting polypeptides (OATPs in humans; Oatps in rodents). OATPs/Oatps have distinct substrate preferences for amphipathic solutes (122). OATPs/Oatps are well-known to transport 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors (i.e., statins), which have been recently shown to exhibit considerable neuroprotective and antioxidant activity in the CNS (123–126). For example, studies in Xenopus laevis oocytes have shown Oatp1a4-mediated uptake of pravastatin (127). More recently, studies in Oatp1a4(−/−) mice demonstrated reduced blood-to-brain transport of pitavastatin and rosuvastatin as compared to wild-type controls, which suggests that Oatp1a4 is involved in statin transport across the BBB (30). Oatp isoforms are also involved in uptake transport of opioid peptides, compounds that have recently been shown to have neuroprotective efficacy in experimental stroke models (128;129). Studies in Xenopus laevis oocytes have shown OATP1A2, the human homologue of rodent Oatp1a4, mediated uptake of opioid peptides such as [D-penicillamine2,5]-enkephalin (DPDPE) (130). Although OATP isoforms are expressed in several tissues, not all exist at the BBB. Immunofluorescence staining of human brain frontal cortex demonstrated OATP1A2 (previously known as OATP-A) localization at the level of the brain microvascular endothelium (130). In rodent brain, expression of Oatp1a4 and Oatp1c1 has been reported in capillary enriched fractions, capillary endothelial cells and/or isolated brain microvessels (13;37;38;131–133). Oatp1c1 is selectively expressed at the BBB (133) and has relatively narrow substrate specificity and primarily transports thyroxine and conjugated sterols (37;38). It has proposed that Oatp1a4, a rodent homologue of OATP1A2, is the primary drug transporting Oatp isoform expressed at the rat BBB (122). Recently, our laboratory demonstratedthat Oatp1a4 is a BBB transporter that can be effectively targeted for facilitation of effective CNS drug delivery (13).
Astrocytes
Astrocytes are the most abundant cell type in the brain. Previous studies have shown that astrocytes, localized between neuronal cell bodies and endothelial cells and ensheath over 99% of cerebral capillaries with their end-feet (14;21), are critical in the development and/or maintenance of BBB characteristics (134–139). For example, studies using human umbilical vein endothelial cells showed that these cells could develop BBB properties when co-cultured with astrocytes, which implies that astrocytes secrete trophic factors critical to maintenance of the BBB phenotype (137). Astrocytes may be involved in transient regulation of cerebral microvascular permeability (140), in particular via dynamic Ca2+ signaling between astrocytes and the endothelium via gap junctions and purinergic transmission (141;142). Recent evidence also suggests that astrocytes may play a critical role in regulating water and ion exchange across the brain microvascular endothelium (143;144). Astrocytes possess two high-affinity transporters for uptake of glutamate, termed excitatory amino-acid transporter 1 and 2 (i.e., EAAT1 (i..e, GLAST) and EAAT2 (i.e., GLT-1) (145). These transporters are criticalin removal of excess glutamate from the synapse and contribute to maintenance of excitatory neurotransmitter concentrations in the brain. Elevated brain levels of glutamate may lead to a pathological condition known as excitotoxicity, which has been implicated in neuronal damage in ischemic stroke (146). Additionally, astrocytes are known to express volume-regulated anion channels (VRACs). These channels are involved in Ca2+-independent release of anionic amino acids (i.e., glutamate, aspartate, taurine) during conditions that cause astrocyte swelling such as cerebral hypoxia (147). Astrocytes are also known to express transport proteins including P-gp (103;148;149), MRP/Mrp isoforms (111;150–153), and Bcrp (150). Studies in human glioma tissue have detected mRNA expression of various MRP and OATP isoforms (154). The expression of multiple drug transporters in astrocytes suggests that these glial cells may act as a secondary barrier to CNS drug permeation. That is, the balance of transporters in astrocytes may either sequester drugs within the astrocyte cytoplasm, thereby preventing these compounds from reaching their site of action in the brain, or concentrate drugs in brain extracellular fluid. Pharmacological agents within brain extracellular space can be effluxed by active transport mechanisms at brain barrier sites or via “sink” effects of the CSF (96). For more detailed information on transport mechanisms in astrocytes, readers are referred to a recent review (96).
Microglia
The Spanish neuroanatomist del Rio-Hortega first described microglia (155), a cell type from the monocyte lineage that represents approximately 20% of the total glial cell population within the CNS (156). Microglia are ubiquitously distributed within the CNS, with the basal ganglia and cerebellum possessing considerably greater numbers than the cerebral cortex (157). Under normal physiological conditions, microglia exist in a quiescent state lacking endocytotic and phagocytotic activity. These microglia possess a ramified morphology characterized by a small (5–10 μm) cell body and many radial cell processes extending from the cell body. Ramified microglia are thought to contribute to maintenance of homeostasis by participating in extracellular fluid cleansing and neurotransmitter deactivation (96). During disease or trauma, microglia may become activated and the degree of this activation is directly correlated to the type and severity of brain injury (158). Activated microglia are identified morphologically by their larger cell body and relatively short cytoplasmic processes. Biochemically, activated microglia are identified by upregulation of cell surface receptors such as CD14 and toll-like receptors (TLRs) (159;160). The degree of microglial activation appears to be correlated to the type and severity of brain injury (156;158). During an immune response, activated microglia may be further converted to a reactive state, which is characterized by a spheroid or rod-like morphology and presence of phagocytotic activity. Microglia activation and proliferation has been implicated in development of neuronal death in various CNS pathological states including ischemic stroke and cerebral hypoxia (161–163). Furthermore, activation of microglia is directly associated with dysfunction of the BBB characterized by changes in TJ protein expression and enhanced paracellular permeability (16). When activated, microglia produce high levels of neurotoxic mediators such as nitric oxide and peroxide as well as inflammatory cytokines (i.e., TNF-α), proteases, and complement components (158;164). Excessive production of these substances may further lead to cell injury in the CNS characterized by astrocyte activation, further microglia activation, and neuronal cell death.
Microglia express several ion channels including multiple potassium, calcium, sodium, and chloride channels (165). The expression patterns of these ion channels depend on the microglial functional state and are involved in a variety of physiological functions including proliferation, ramification, and maintenance of membrane potential, intracellular pH regulation and cell volume regulation (166–169). Glutamate receptors (170) and nutrient carrier systems such as GLUT-1 (171) are expressed in microglia. These cells also express membrane proteins involved in drug transport. Studies in a continuous rat microglia cell line (i.e., MLS-9) demonstrated functional expression of P-gp (172;173), Mrp1 (174) and Mrp4/Mrp5 (175); however, the ability of microglia to contribute to drug permeation and/or distribution in the CNS requires further study.
Pericytes
In addition to glia, pericytes also play a crucial role in maintenance of BBB homeostasis (176). Pericytes are flat, undifferentiated, contractile cells that attach at irregular intervals along capillary walls and communicate with other cell types of the neurovascular unit (176). These cells, via secretion of pericyte-derived angiopoetin, induce expression of occludin at the BBB, which suggests that pericytes are directly involved in induction and/or maintenance of barrier properties (177). Involvement of pericytes in induction of BBB properties is also exemplified by in vitro demonstration thatproper localization of endothelial proteins (i.e., P-gp, utrophin) requires co-culture with pericytes (178). Further evidence for the role of pericytes in maintenance of BBB phenotype comes from the observation that pericytes migrate away from the endothelium during hypoxia (179) or brain trauma (180), conditions that are associated with increased brain microvascular permeability. More recently, studies using adult-viable pericyte-deficient mouse mutants demonstrated that pericytes are critical in maintaining expression of BBB-specific genes in endothelial cells (i.e., transferrin receptor) and by inducing polarization of astrocyte end-feet adjacent to the cerebral microvasculature (181). Additionally, MRP isoforms (MRP1, MRP4, MRP5) have been identified in pericytes in vitro, which implies that pericytes may contribute to regulation of BBB xenobiotic permeability (182).
Neurons
There is considerable evidence for direct innervation of both brain microvascular endothelium and associated astrocyte processes. Noradrenergic (183;184), serotonergic (185), cholinergic (186;187), and GABAergic (188) neurons have been shown to make distinct connections with other cell types of the neurovascular unit. The need for direct innervation of brain microvasculature comes from the dynamic nature of neural activity and the metabolic requirements of nervous tissue, implying that the cerebral microcirculation must be highly responsive to the needs of CNS tissue. Indeed, “metabolic coupling” of regional brain activity to blood flow is the basis of functional neuroimaging (189), although the cellular mechanisms of this process are not well understood (190). Interestingly, disruption of BBB integrity induced by pathophysiological factors (i.e., inflammation, hypertension, ischemia) often accompanies changes in cerebral blood flow and perfusion pressure (191–193) and there is evidence that such BBB opening may be a selective, compensatory event rather than a simple anatomical disruption. This implies that communication between neurons and the brain microvasculature may not simply regulate blood flow, but BBB permeability as well.
Extracellular Matrix
In addition to cellular components of the neurovascular unit, the extracellular matrix of the basal lamina also interacts with the BBB endothelium. Disruption of extracellular matrix is strongly associated with increased BBB permeability in pathological states including stroke (194;195). The extracellular matrix seems to serve as an anchor for the endothelium via interaction of laminin and other matrix proteins with endothelial integrin receptors (196). Such cell-matrix interactions can stimulate a multitude of intracellular signaling pathways (197). Matrix proteins can influence expression of TJ proteins (69;198;199), indicating that although the TJ protein complexes constitute the primary impediment to paracellular diffusion, the proteins of the basal lamina are likely involved in their maintenance.
Ischemic Stroke
Overview of Ischemic Stroke
Stroke is the third most common cause of death in the United States and is the number one cause of long-term morbidity (200). On a global scale, these statistics are even more staggering as stroke is now the second leading cause of death worldwide (18). Stroke is one of the leading causes of functional disability and currently affects approximately 45 million people living in the United States (201). Of all strokes, 87% are ischemic (201). Each year, approximately 795,000 people experience either a new or recurrent stroke, which averages one incidence of stroke every 40 seconds (201). Following an ischemic stroke, the mean lifetime cost of medical and rehabilitation is estimated at $140,048 per patient, a figure that is considerably higher for people over 45 years of age (202). In the United States, the total cost of stroke therapy was $73.7 billion (201). Several factors have been identified that increase risk of stroke including history of transient ischemic attacks, hypertension, impaired glucose tolerance and diabetes mellitus, atrial fibrillation, cigarette smoking, and low serum concentrations of HDL cholesterol (201).
Stroke is characterized by a heterogeneous spectrum of conditions caused by interruption of blood flow supplying the brain (18;203;204). Such a deficit in cerebral blood flow causes an irreversibly damaged ischemic core and salvageable surrounding neural tissue known as the penumbra (205). Physiologically, energy requirements of the CNS are met by brain uptake of both glucose and oxygen, which are metabolized to enable phosphorylation of ADP to ATP. Most ATP generated within the brain is utilized to maintain intracellular homeostasis and transmembrane gradients for monovalent and divalent ions (i.e., sodium, potassium, calcium) (206). When blood flow to the brain is interrupted in a stroke, the ischemic core is rapidly deprived of oxygen and glucose. Inability to provide sufficient quantities of ATP causes collapse of ion gradients and subsequent release of neurotransmitters (i.e., dopamine, glutamate). This uncontrolled increase in extracellular dopamine/glutamate concentrations is highly toxic to neurons, an event that causes neuronal cell death and development of an infarction (206). Excess release of glutamate is particularly deleterious to the CNS due to overstimulation of glutamate receptors, activation of phospholipases/sphingomyelinases, phospholipid hydrolysis, release of arachidonic acid and ceramide, and disruption of CNS calcium homeostasis (18;206–208). Oxidative stress is also observed in the CNS at early time points following ischemic injury and is well known to contribute to cell death in the ischemic core (209). As neuronal cell damage extends to the ischemic penumbra, neuroinflammation and apoptosis become more prevalent and dramatically affect viability of salvageable brain tissue within the penumbra (209).
Cell death processes in the ischemic core occur extremely rapidly (i.e., within minutes) thereby rendering this region difficult to protect using pharmacological approaches (18). In contrast, cells within the ischemic penumbra die more slowly by active cell death mechanisms thus rendering therapeutic interventions theoretically possible (18). Therefore, the primary goal of drug therapy for acute ischemic stroke is to salvage the penumbra as much as possible and as early as possible (205). Currently, there is only one therapeutic agent approved by the FDA for acute ischemic stroke treatment, recombinant tissue plasminogen activator (r-tPA) (210). The primary goal of r-tPA therapy is to restore blood flow and oxygen supply to ischemic brain tissue; however, most cellular damage to the brain occurs when cerebral perfusion is re-established (i.e., reoxygenation) (211). Such hypoxia/reoxygenation (H/R) stress/injury is directly associated with neuronal apoptosis characterized by cytochrome c release, caspase-3 activation and internucleosomal DNA fragmentation (212). Pathophysiological mechanisms that can cause neuronal apoptosis during H/R include oxidative stress secondary to increased production of reactive oxygen species (ROS) (213). ROS contribute to brain injury by interacting with proteins, lipids, and nucleic acids as well as via activation of redox-sensitive signaling pathways. Such responses are characterized by increased CNS production of hydrogen peroxide (214), upregulation of the cellular stress marker heat shock protein-70 (10), and increased nuclear expression of hypoxia-sensitive transcription factors such as hypoxia-inducible factor-1 and nuclear factor-κB (10;215). The H/R component of stroke is also associated with decreased brain concentrations of the endogenous antioxidant GSH (214), an effect that is further indicative of oxidative stress. These molecular events associated with H/R injury emphasize a critical need in stroke therapy for discovery of new therapeutic agents that can be administered alone or in conjunction with r-tPA for “rescue” of salvageable neural tissue.
The Neurovascular Unit in Ischemic Stroke
Disruption of the Blood-Brain Barrier
A critical facet of early neurovascular damage is manifested as significant perturbations in BBB function. BBB homeostasis is remarkably dependent on interactions between endothelial cells, astrocytes, and extracellular matrix (216;217). Perturbation of the extracellular matrix (i.e., type IV collagen, heparan sulfate proteoglycan, laminin, fibronectin, perlecan) disrupts cell-matrix and cell-cell signaling mechanisms critical to proper functioning of the NVU (195). Many proteinases, in particular the matrix metalloproteinases (MMPs), contribute to breakdown of the extracellular matrix and disruption of the BBB in stroke (218). This includes MMPs that are activated by a HIF-1α-dependent mechanism (i.e., MMP2) and MMPs whose activation is associated with upregulation of pro-inflammatory cytokines (i.e., TNF-α, IL-1β) such as MMP3 and MMP9 (217). Involvement of MMPs in BBB disruption following ischemic stroke has been demonstrated in many experimental stroke models (219–223). Furthermore, a recent clinical study demonstrated that MMP9 levels were significantly elevated in patients with acute ischemic stroke (224). MMPs degrade the extracellular matrix that comprises the basal lamina and thereby directly compromise the BBB. Additionally, activity of MMPs opens the BBB by directly degrading TJ constituent proteins such as claudin-5 and occludin (69). MMP-mediated opening of the BBB in ischemic stroke may be regulated by nitric oxide (NO) signaling. Specifically, pharmacological inhibition of nitric oxide synthase (NOS) reduced activity of MMP2/MMP9 and prevented disruption of the BBB in a rodent model of focal cerebral ischemia (225). Damage and subsequent opening of the BBB is a key event in development of intracerebral hemorrhage and brain edema following an ischemic stroke.
Experimental models of focal cerebral ischemia have provided considerable information on solute leak across the BBB following an ischemic stroke. Using the transient middle cerebral artery occlusion (MCAO) rodent model, Pfefferkorn and Rosenberg demonstrated increased leak of sucrose, a vascular marker that does not typically cross the BBB (226), in the ischemic hemisphere (227). However, BBB disruption following an ischemic insult is much more profound than to allow leak of small molecules only. Recently, it was shown that BBB disruption following focal cerebral ischemia was sufficient to allow blood-to-brain leak of Evan’s blue dye (88). Evan’s blue dye, when unconjugated to plasma proteins, is a relatively small molecule with a molecular weight of 960.8 Da. It is well established that Evan’s blue dye irreversibly binds to serum albumin in vivo. This leads to the formation of a very large, solute-protein complex (i.e., in excess of 60,000 Da) that can only traverse the BBB under considerable pathological stress such as that observed during an ischemic stroke (228;229). Of particular note, Jiao and colleagues observed redistribution of critical TJ proteins occludin, claudin-5, and ZO-1 following 2 h of focal cerebral ischemia, an event that directly correlated with increased blood-to-brain flux of Evan’s blue-albumin (88). Of particular note, these researchers observed that changes in BBB TJ constituent proteins follow a distinct biphasic pattern, which they observed at 3 h and 72 h following ischemic insult (88). Reorganization of TJ proteins following focal cerebral ischemia is mediated by vascular endothelial growth factor (VEGF) (230–232) and NO (5;233).
Reorganization of TJ protein complexes and associated leak across the BBB following focal ischemiaenables considerable movement of vascular fluid across the microvascular endothelium and development of vasogenic edema (234–236). Recent studies using the MCAO model have shown that water movement across the BBB is exacerbated by enhanced blood-to-brain movement of sodium. This phenomenon dramatically alters oncotic pressure across the brain microvascular leading to enhanced movement of water into brain parenchyma. Alterations in sodium gradients across the microvascular endothelium is facilitated by increased functional expression of Na-K-Cl cotransporter, which is typically expressed at the luminal aspect of the BBB (237), as well as Na-H exchangers NHE1 and/or NHE2 (238). MCAO studies in spontaneously hypertensive rats demonstrated that the NHE1 transporter is a critical regulator of ischemic-induced infarct volume (6). Disruption of sodium gradients across the BBB during ischemic stroke may also involve upregulation of sodium-dependent glucose transporters such as sodium-glucose cotransporter (SGLT). Specifically, pharmacological inhibition of SGLT in MCAO rats significantly reduced infarct and edema ratios, which implies that this transporter may be a critical determinant of stroke outcome (239).
Functional BBB integrity can also be disrupted by production of reactive oxygen species (ROS) and subsequent oxidative stress. Production of highly potent ROS such as superoxide anion is a well-established component of global and focal ischemia (234;240). Biological activity of superoxide anion, a by-product of normal physiological processes, is tightly controlled by superoxide dismutase (SOD) enzymes. During cerebral ischemia, superoxide is produced at such high levels that the ability of SODs to metabolize it is overwhelmed. This phenomenon is supported by the observation that pharmacological inhibition of SOD markedly reduced cerebral edema and infarct size in transgenic mice engineered to overexpress SOD (240). Increased levels of superoxide also contribute to ischemic injury to the BBB endothelium (56;241;242). BBB damage can be further intensified by conjugation of superoxide and NO to form peroxynitrite, a potent cytotoxic and proinflammatory molecule. Peroxynitrite is well-known to induce cellular damage by its ability to nitrosylate tyrosine, leading to functional modifications of critical proteins (243). Breakdown of peroxynitrite into nitrogen dioxide and hydroxyl radicals is also known to contribute to endothelial cell dysfunction and BBB disruption in cerebral ischemia (234). Overall, oxidative stress contributes to endothelial dysfunction and BBB disruption by promoting redistribution and/or disappearance of critical TJ proteins such as claudin-5 and occludin (244).
Inflammatory stimuli are also critical mediators of BBB dysfunction in the setting of ischemic stroke. Previous research has demonstrated that inflammatory responses in focal cerebral ischemia are primarily mediated through pro-inflammatory cytokines TNF-α and IL-1β, which appear within 2 -6 h following ischemic insult (245). Signaling mediated by these cytokines induces an upregulation of adhesion molecules and subsequent transmigration of activated neutrophils, lymphocytes or monocytes into brain parenchyma (246). Under normal conditions, expression of vascular adhesion molecules such as ICAM-1 and VCAM-1 are barely detectable in brain microvessel endothelial cells; however, their expression is dramatically increased in response to pathological stressors such as ischemic stroke (246–249). Pro-inflammatory mediators are well known to alter functional expression of endogenous BBB transporters. For example, TNF-α increased expression of P-gp but decreased BCRP expression in hCMEC/d3 cells (250). Similarly, TNF-α was shown to increase mdr1b promoter activity in an immortalized rat brain endothelial cell line (RBE4) (251). Pro-inflammatory stimuli (i.e., lipopolysachharide-induced production and secretion of TNF-α and IL-1β) was also shown to alter expression of critical TJ proteins occludin and ZO-1 (252), suggesting that inflammation may play a role in exacerbating paracellular leak in the context of ischemic stroke.
Ischemic Stroke and Glial Support of the BBB
It is well established in the literature that the glial response to an ischemic insult is highly complex and multifaceted; however, it is known that injury and/or activation of astrocytes at the NVU leads to compromise of the BBB. Studies using male Fisher F344 rats injected with 3-chloropropanediol, an astrocyte-selective toxin, has provided evidence that astrocyte injury and deathis a critical component ofBBB disruption. Specifically, focal astrocyte loss leads to disassembly of BBB TJ protein complexes characterized by decreased expression of TJ constituent proteins and a corresponding increase in paracellular solute leak (138;139). In the inferior colliculus, focal astrocyte loss demarcated by reduced GFAP immunoreactivity directly corresponded with decreased paracellular localization of critical TJ proteins claudin-5, occludin, and ZO-1 (139). At the same time points that TJ proteins were downregulated, an increase in leak of dextran (10 kDa) and fibrinogen was observed (139), which suggests significant disruption of the BBB due to astrocyte cell death. The results of this study and others (134;253;254) illustrate the requirement of intercellular interactions/communication between astrocytes and endothelial cells for maintenance of BBB integrity. Additionally, astrocytes have many other features that contribute to BBB physiology. For example, astrocytes are well known to express the water channel aquaporin 4 (AQP4) at end-feet localized adjacent to brain microvascular endothelium, which contributes to endothelial cell polarity and brain water volume (255). Astrocytes secrete VEGF and fibroblast growth factor-2 (FGF-2), which promote angiogenesis and regulate biological transport at the BBB (21;256).
Astrocytes are critical contributors in the brain immunological response during ischemic stroke. In the normal non-pathological brain, astrocytes maintain focal contacts with neighboring microglia and maintain these cells in a dormant, ramified state (158). This property of astrocytes has been shown in vitro when microglia are cultured on a monolayer of astrocytes (257;258) or exposed to astrocyte-conditioned medium (259). Regulation of microglia by astrocytes is prevented by inflammatory signaling thus enabling microglia to elicit an immune response (158). Astrocytes can also directly contribute to brain immunological responses via upregulation of adhesion molecules (i.e., ICAM-1, VCAM) at their cell surface, an event that contributes to CNS targeting of leukocytes (260;261). This enhancement is also facilitated by astrocytic secretion of chemokines such as macrophage inflammatory protein-1α/β (MIP-1α/β), monocyte chemoattractant protein-1 (MCP-1) and regulated upon activation normal T-cell expressed and secreted (RANTES). Production and secretion of these chemokines by astrocytes is well known to occur in response to ischemic stroke (262–264) and to increased brain parenchymal concentrations of tPA (265). Of particular note, MCP-1 secretion by astrocytes was shown to coincide with a significant increase in FITC-albumin leak in an in vitro co-culture of endothelial cells and astrocytes subjected to 5 h oxygen-glucose deprivation, suggesting that MCP-1 may be a critical factor involved in BBB opening following an ischemic stroke (263). Astrocytes also synthesize several pro- and anti-inflammatory cytokines including interleukins (i.e., IL-1α, IL-1β, IL-4–8, IL-10), TNF-α, and interferon-γ (158;202;266–268) as well as transforming growth factor-β (TGF-β) (269;270). Cytokines such as TNF-α, IL-1α, IL-1β, and interferon-γ can directly trigger the endothelium and activate processes involved in BBB disruption (202;268;271;272). Although secretion of TGF-β1 may play a neuroprotective role in brain parenchyma (270), its effects on the endothelium are much more deleterious. Specifically, excessive endothelial stimulation by TGF-β1 affects the angiogenic response to ischemic stroke by causing formation of defective capillaries (140). Histologically, these capillaries differ considerably from normal brain microvasculature because they lack pericytes, consist of fewer endothelial cells, and are shorter in length (140). This observation suggests that pharmacological inhibition/targeting of TGF-β signaling at the level of the brain microvascular endothelium may be an efficacious approach for protection of BBB integrity and/or preservation ofthe angiogenic response to ischemic stroke.
Inflammatory signaling by astrocytes is a critical event in exacerbation of CNS oxidative stress during ischemic stroke. Previous studies have shown that astrocytic production of proinflammatory cytokines can induce deleterious processes in astrocytes themselves via upregulation of inducible nitric oxide synthase (iNOS) (273). Upregulation of iNOS leads to a significant enhancement in NO production, which can react with superoxide to produce peroxynitrite. Increased exposure of astrocytes to peroxynitrite can lead to rapid astrocyte proliferation and hypertrophy (i.e., reactive astrocytosis) and astrocyte apoptosis (274–277). This has been demonstrated in the rodent MCAO model where increased expression of iNOS in brain parenchyma correlated with enhanced astrocytic expression of GFAP, a hallmark of reactive astrocytosis (276). Reactive astrocytosis is associated with disruption of TJs between adjacent brain microvessel endothelial cells and increased BBB permeability (278).
Although astrocyte injury is a critical determinant of BBB dysfunction in the setting of ischemic stroke, endothelial damage may also be induced via immune stimulated microglia (279). This is supported by the observation that minocycline, a pharmacological inhibitor of activated microglia, dramatically reduced cell death in cultures of murine endothelial cells exposed to activated microglia in vitro (279). Activated microglia produce proinflammatory cytokines (i.e., TNF-α, IL-1β, IL-6) in response to cerebral ischemia (280–282), all of which can trigger BBB disruption. In the setting of ischemic stroke, cytokine production in microglia is mediated by NF-kB signaling (283). Inflammatory signaling in microglia may also involve cyclooxygenase-2 (COX2), which is inducible in response to ischemic injury and contributes to opening of the BBB (217). In neuroinflammation, COX2 activates sphingomyelinases leading to release of ceramides, an event that leads to activation of p38 mitogen activated protein kinase (MAPK) (284). Activation of the p38 MAPK pathway is also associated with production and secretion of proinflammatory cytokines in microglia (285;286). This is supported by the observation that pharmacological treatment with SB203580, a specific p38 MAPK inhibitor, attenuates production and secretion of TNF-α in primary cultures of rat microglia (285). More recently, administration of SB203580 following MCAO was shown to suppress microglia secretion of TNF-α and IL-1β as well as decreased microglial expression of COX2, results that point to a critical role for microglia in the inflammatory response to ischemic stroke (286).
Targeting the Neurovascular Unit in Ischemic Stroke
Targeting the Tight Junction
The studies reviewed above clearly demonstrate that the BBB and associated glial support network of the NVU may be compromised in response to ischemic stroke. A critical “component” of ischemic stroke is cerebral hypoxia and subsequent brain injury resulting from reoxygenation/reperfusion (i.e., H/R stress). Over the past several years, our laboratory has studied BBB changes associated with H/R stress in an in vivo rodent model (3;7;8;10;11;215;287). This in vivo system employs an acute, moderate hypoxic insult of inhaled 6% O2 for 1 h followed by reoxygenation under normal atmospheric conditions (i.e., 21% O2) for various time points. This approach offers several advantages for the study of NVU biology in the context of cerebral ischemia/hypoxia. Firstly, the in vivo nature of the model allows interaction of all cellular components of the NVU (i.e., endothelial cells, astrocytes, microglia, pericytes, neurons) and systemic circulation mediators, which in vitro systems are unable to accurately represent (7). Secondly, the degree of hypoxic stress does not induce necrotic damage of the endothelium, often associated with other in vivo focal ischemia models, allowing us to study a dynamically regulated and recoverable BBB (7). Changes in BBB integrity under H/R conditions were demarcated by enhanced brain accumulation of 14C-sucrose (3;7;10;215), a vascular marker that does not typically cross the brain microvascular endothelium. Additionally, H/R stress also increased vascular leak to dextrans (molecular weight range 4-kDa to 10 kDa) in hippocampal and cortical microvessels (11). These alterations in BBB permeability in animals subjected to H/R stress were directly associated with an increase in expression of HIF-1α and NF-kB in nuclear fractions isolated from intact microvessels (215). In our studies, changes in brain solute uptake is not likely attributed to altered cerebral blood flow because we have previously shown that blood flow changes are negligible in our in vivo H/R model (3). Changes in BBB permeability to 14C-sucrose and dextrans were directly correlated with altered organization and/or expression of constituent TJ proteins including occludin (3;10;11), claudin-5 (11), and ZO-1 (11). Of paramount significance was the observation that H/R stress disrupted disulfide-bonded occludin oligomeric assemblies, thereby preventing monomeric occludin from forming an impermeable physical barrier to paracellular transport (8). TJ protein organization and expression were found to be regulated by protein kinase C (PKC) signaling involving nPKC-θ and aPKC-ζ isoforms (11). These changes in TJ organization and expression and BBB solute leak also correlated with a significant increase in brain water content following H/R, providing further evidence that disruption of the BBB under conditions of cerebral ischemia contributes to vasogenic edema (7).
Production of ROS and subsequent oxidative stress has been previously shown to alter BBB expression of claudin-5 and occludin leading to increased paracellular solute leak (244). Therefore, we hypothesized that oxidative stress associated changes in BBB permeability and occludin expression could be attenuated with the use of an antioxidant drug. In order to conduct these studies, we utilized 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPOL), a stable, membrane-permeable, water-soluble nitroxide antioxidant. TEMPOL shows SOD-like activity towards the superoxide anion as well as reactivity with hydroxyl radicals (288), nitrogen dioxide, and the carbonate radical (289). TEMPOL readily crosses the BBB (290) and has been previously shown to provide neuroprotection as a free radical scavenger in several models of brain injury and ischemia (291–294). Using the dual artery in situ brain perfusion technique, we demonstrated that administration of TEMPOL 10 min before H/R treatment significantly attenuated CNS uptake of 14C-sucrose as compared to animals subjected to H/R only (10). This reduction in 14C-sucrose leak was associated with a preservation of occludin localization and occludin oligomerization at the TJ (10). Specifically, TEMPOL inhibits breakage of disulfide bonds on occludin monomers and thus prevents breakdown of occludin oligomeric assemblies and subsequent blood-to-brain leak of circulating solutes (figure 3) (10). Restoration of BBB functional integrity coincided with a decrease in nuclear translocation of HIF-1α and a decrease in microvascular expression of the cellular stress marker heat shock protein 70 (hsp70) in rats subjected to H/R stress and administered TEMPOL (10). Taken together, these observations provide evidence that the TJ can be targeted pharmacologically during ischemic stroke for the purpose of reducing both oxidative stress associated injury to the brain microvascular endothelium and blood-to-brain solute leak. Future studies will focus on how TEMPOL can be utilized to modulate CNS drug delivery in the setting of ischemic stroke and H/R injury.
Figure 3.
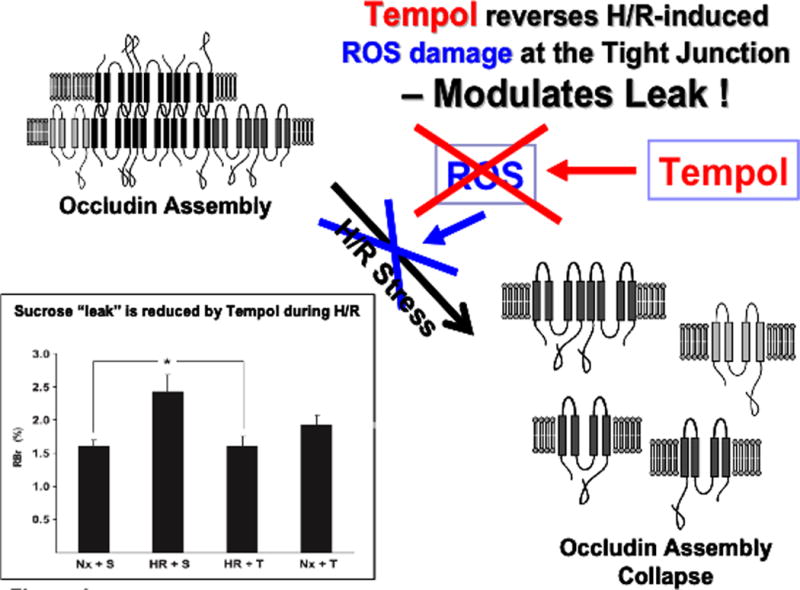
Effect of TEMPOL on H/R-mediated disruption of the tight junction. ROS and subsequent oxidative stress are known to disrupt assembly of critical TJ proteins such as occludin. Our results show that administration of TEMPOL, by scavenging ROS, prevents disruption of occludin oligomeric assemblies. Furthermore, TEMPOL attenuates the increase in sucrose leak across the BBB observed in animals subjected to H/R stress. Taken together, our studies with TEMPOL demonstrate that the TJ can be targeted pharmacologically in an effort to preserve BBB functional integrity during ischemic stroke.
Targeting Endogenous BBB Transporters
The ability of a pharmacological agent to cross the BBB endothelium and achieve efficacious concentrations within the CNS is dependent upon multiple mechanisms of transport. Such mechanisms include uptake into the brain via an influx transporter and/or extrusion from the CNS mediated by an efflux transporter. For many drugs, it is this discrete balance between influx and efflux that determines if a pharmacological agent will accumulate within the brain extracellular milieu and be able to elicit a therapeutic effect. The complexity of drug transport biology at the BBB is further underscored by the observation that functional expression of such transport proteins may be dramatically altered by pathophysiological stressors (13;295–298). A thorough understanding of the regulation and functional expression of endogenous BBB transporters in both health and disease is critical for optimization of pharmacotherapy. Furthermore, such information will enable efficient targeting of transporters and/or transporter regulatory mechanisms, thus allowing endogenous BBB transport systems to be exploited for purposes of improving CNS drug delivery.
Perhaps the most critical transporter determinant of solute permeation to the CNS is P-gp. Recently, it has been reported that P-gp plays a critical functional role in regulating brain injury associated with focal cerebral ischemia. Using MCAO, Murozono and colleagues showed a significant reduction in infarction size in mdr1a-knockout mice as compared to wild type controls (299). Similarly, pharmacological inhibition of P-gp with cyclosporine A (1 mg/kg) dramatically reduced infarctionvolume in ischemic brain tissue as compared to vehicle controls (300). Taken together, these studies indicate that P-gp actively controls CNS concentrations of neuronal modulatory substances such as cytokines and neuronal peptides (299). In terms of drug delivery, much effort has focused on blocking P-gp activity as a means of enhancing brain delivery of therapeutic compounds. In fact, several clinical trials have attempted to incorporate pharmacological inhibitors of P-gp into therapeutic regimens; however, these trials have not been encouraging because of systemic toxicity associated with high inhibitor doses necessary for effective transporter inhibitor (301–303). Therefore, pharmacological approaches that involve inhibition of P-gp mediated transport should be employed with extreme caution to avoid an unwanted elevation in CNS drug concentrations and subsequent toxicity as well as unexpected adverse drug reactions.
An alternative approach for optimizing delivery of drugs to the CNS is to focus on influx processes and target endogenous BBB transporters known to be involved in blood-to-brain transport of drugs. An intriguing candidate is Oatp1a4, which is known to transportboth HMG-CoA reductase inhibitors (i.e., statins) and opioids peptides. In the context of ischemic stroke pharmacotherapy, there is considerable interest in neuroprotective/antioxidant properties of statins. Recent evidence suggests that statins can act as free-radical scavengers independent of their well-documented effects on cholesterol biosynthesis (125;304). For example, studies in dogs demonstrated that high-dose atorvastatin (80 mg/day) reduced markers of oxidative and nitrosative stress (i.e., protein carbonyls, 4-hydroxy-2-nonenal, 3-nitrotyrosine) and increased the GSH:GSSG ratio in the brain but not in the periphery, suggesting efficacy as a neuroprotectant and CNS antioxidant (125;126). Although statins are associated with neurotoxic effects, these drugs generally do not compromise neuronal cell viability at concentrations below 1 μM (124). Additionally, there is evidence that opioid receptor agonists such as opioid peptide analgesics may have efficacy in treatment of ischemic stroke. For example, opioid peptides that selectively bind to the mu-opioid receptor (i.e., Tyr-D-Ala’, N-CH, -Phe4, Glyol-Enkephalin (DAMGO)), delta-opioid receptor (i.e., [D-penicillamine2,5]-enkephalin (DPDPE)), and kappa-opioid receptor (i.e., U50 488) all reduced water uptake in rat hippocampal slices in situ (129), suggesting that these drugs may be effective as stroke therapeutics.
Recently, we reported for the first time increased functional expression of Oatp1a4 at the BBB in rats subjected to a pathological stressor (i.e., peripheral inflammatory pain) (13). Evidence for increased Oatp1a4 transport at the BBB included i) increased brain accumulation of taurocholate, a selective Oatp substrate (305); ii) attenuation of taurocholate uptake by Oatp transport inhibitors (i.e., digoxin, estrone-3-sulfate, fexofenadine); iii) increased in KIN for taurocholate in response to pathological stress, which implies increased blood-to-brain transport; and iv) an increase in taurocholate accumulation within brain interstitial fluid but no change in taurocholate sequestration within the BBB endothelium itself (13). In order to determine if Oatp1a4 could effectively facilitate CNS drug delivery, we studied BBB transport of the opioid peptide DPDPE. Brain uptake of DPDPE is governed by multiple mechanisms in addition to Oatp1a4-mediated transport including transcytosis (306) and P-gp-mediated efflux (307). Although we showed increased Oatp1a4 functional expression at the BBB in animals subjected to peripheral inflammatory pain, we did not see any change in blood-to-brain DPDPE transport (13). In light of our previous work with P-gp (298), we proposed that Oatp1a4 influx transport was negated by P-gp efflux. This implies that the relative contribution of Oatp1a4 to overall brain uptake of DPDPE could only be determined in the absence of P-gp mediated transport activity. When we inhibited P-gp efflux transport using reversin 205, a selective P-gp inhibitory peptide (308), we observed that the relative contribution of Oatp1a4 to brain uptake of DPDPE increased from 56% in saline controls to 71% in animals subjected to peripheral inflammatory pain (13). These data are particularly critical because they showed, for the first time, that Oatp1a4 can be targeted for delivering drugs such as opioid peptides to the brain.
In order to successfully target a transporter system for optimization of CNS drug delivery, it is crucial to determine how a transport of interest is regulated at the molecular level. In the context of ischemic stroke, this includes identification and characterization of biological mechanisms that enable peripheral pain to “transmit” signals upstream and alter BBB drug transporters. Of particular interest is the TGF-β signaling pathway. TGF-β signaling regulates multiple cellular processes including vascular remodeling (309). The TGF-βs are a family of pleiotropic cytokines that modulate cellular function by binding to a heterotetrameric complex of type I and type II serine/threonine kinase receptors (310). The type I receptors, also known as activin receptor-like kinases (ALKs), propagate intracellular signals through phosphorylation of specific Smad proteins (i.e., receptor-regulated (R)-Smads) (figure 4). Phosphorylated (R)-Smads form complexes with the common Smad (i.e., Smad4) enabling them to be translocated to the nucleus and regulate transcription of target genes (310).
Figure 4.
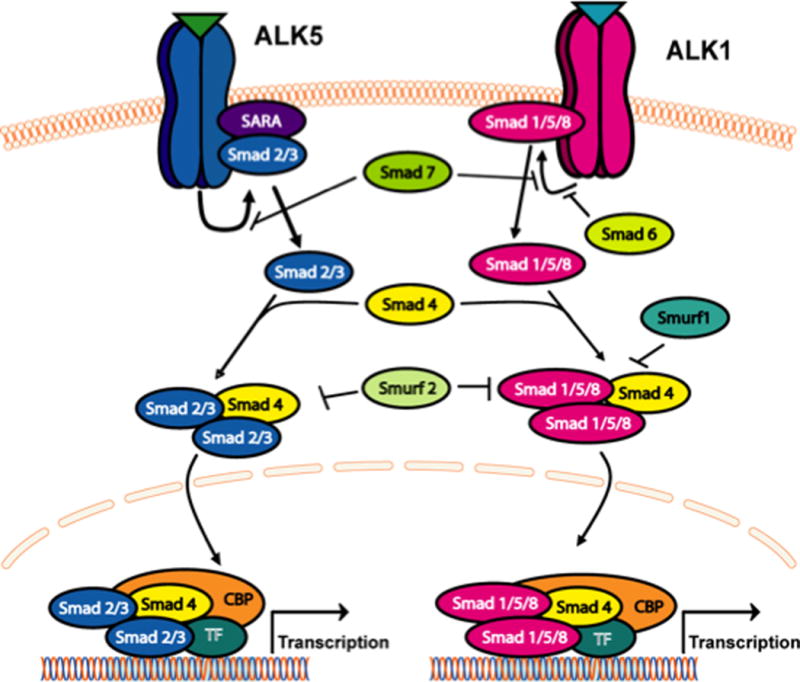
The transforming growth factor-β (TGF-β) signalling pathway. Intracellular signalling molecules associated with TGF-β signalling at the blood-brain barrier. Signals elicited by the TGF-β pathway involve two cell surface receptors at the brain microvascular endothelium, which are designated activin receptor-like kinase (ALK)-1 and ALK-5. ALK1 transduces signals via phosphorylation of Smad proteins -1, -5, and -8 while ALK5 signals by phosphorylation of Smad2 and Smad3. Once phosphorylated, these Smad proteins bind to the common Smad (i.e., Smad4), thereby forming a protein complex that can translocate to the nucleus and affect transcription.
In the majority of tissues, TGF-β signaling is mediated by ALK5 (311); however, studies in cultured human endothelial cells (312) and in isolated arterial endothelium from ALK1-deficient mice (313) have indicated that ALK1 is also involved in vascular TGF-β signaling. TGF-β regulates the endothelial cell activation state through a precise balance between ALK1 and ALK5 signaling processes (312;314). Whereas the ALK1 pathway leads to endothelial activation characterized by increased permeability, ALK5-mediated signaling promotes vascular resolution that is demarcated by decreased permeability (311;312). Such effects on vascular permeability may be due to the ability of TGF-β signaling to alter expression of tight junction constituent proteins. For example, claudin-5 expression was increased by pharmacological ALK5 inhibition in embryonic stem cells, suggesting the involvement of TGF-β/ALK5 signaling in the regulation of tight junction constituent proteins (315). Similarly, we demonstrated that ALK5 inhibition using SB431542 increased expression of claudin-3, claudin-5, occludin monomers, and ZO-1 in vivo (9). Additionally, studies using human glioblastoma cells cocultured with human brain endothelial cells showed that activation of TGF-β-mediated signaling decreased endothelial expression of occludin, claudin-1, and claudin-5 (316).
The TGF-β1 isoform is dramatically induced in the CNS in response to pathological conditions that cause acute and chronic brain injury such as ischemic stroke (269;270). Recently, our laboratory demonstrated that pharmacological inhibition of TGF-β signaling led to increased microvascular expression and activity of Oatp1a4 at the BBB (13). Of particular interest was the observation that this blockade of TGF-β/ALK5 signaling using SB431542 increased Oatp1a4 functional expression in saline control animals (13). A crucial consideration in interpretation of our data is contribution of paracellular diffusion to brain uptake of taurocholate. Our laboratory has previously reported that pharmacological inhibition of TGF-β/ALK5 signaling also increased BBB permeability via the paracellular route for solutes such as sucrose (9). The molecular weight of taurocholate (537.7 Da) is greater than sucrose (342 Da), suggesting a lesser degree of paracellular diffusion. Since our data showed no statistical difference in taurocholate uptake in the presence and absence of Oatp1a4 inhibitors in animals subjected to a pathological stressor known to open the BBB (i.e., pain/inflammation), we conclude that paracellular diffusion was not a significant factor in our study. This is not to say that paracellular drug transport is not a significant factor in determining blood-to-brain delivery of other drugs in the setting of a pathophysiological response. Therefore, it is critical to correct for paracellular transport in any study on the effect of targeting TGF-β signaling for optimization of CNS drug delivery. Although studies in immortalized mouse brain endothelial cells (MBE4) have shown involvement of ALK5-mediated signaling in P-gp regulation (317), we are the first to report TGF-β/ALK5 signaling regulation of an endogenous BBB drug uptake transporter. Our work on TGF-β/ALK5 signaling demonstrated that this pathway can regulate permeability at the BBB by increasing functional expression of an influx transporter. Furthermore, these studies highlight the potential of the TGF-β/ALK5 pathway as a pharmacological target that can be utilized for optimization of drug delivery to the CNS, particularly for treatment of ischemic stroke.
Targeting Glial Support of the BBB
In addition to the BBB endothelium, glial cells (i.e., astrocytes, microglia) also have considerable potential as a therapeutic target for treatment of ischemic stroke. As described above, glia play a crucial role in regulating BBB functional integrity in health and diseasethrough release of trophic factors that maintain TJ protein complexes, release of factors that promote angiogenesis, pro-inflammatory signaling, and production of ROS. Pharmacological manipulation of glial cell biology represents a therapeutic approach that may enable control of BBB/NVU pathophysiological mechanisms during ischemic stroke and/or H/R injury. A brief description of such promising glial-based strategies for treatment of ischemic stroke is discussed below.
An opportunity for cellular protection of glia in ischemic stroke involves targeting the proteinase-activated receptor (PAR) pathway. To date, four members of the PAR family (i.e., PAR-1, PAR-2, PAR-3, PAR-4) have been cloned and characterized (318). Both PAR-1 and PAR-2 are expressed on the cell surface of astrocytes (319–322) and microglia (321;323) as well as on the endothelial cell surface (324–326). PAR-1 has been implicated in cytoprotective mechanisms (327;328) while PAR-2 is involved in regulation of inflammatory responses (321;329). Recent research has focused on pharmaceutical development of agonists targeted to the PAR-1 receptor such as activated protein C (APC) (328;330–332). In a mouse model of transient cerebral ischemia, APC was shown to reduce ischemic brain damage and promote neovasacularization and neurogenesis, suggesting that pharmacological targeting of the PAR-1 receptor may be an efficacious approach for treatment of ischemic stroke (330). Using a murine model of cerebral venous sinus thrombosis, Nagai and colleagues demonstrated that neutralization of APC resulted in BBB dysfunction and brain edema, which suggests that the protein C pathway via the PAR-1 receptor is involved in cytoprotection against deleterious pathological responses (331). Brain vascular perfusion studies demonstrated that brain accumulation of APC was reduced by 64% in mice lacking the endothelial protein-C receptor (EPCR), suggesting that CNS delivery of APC is dependent upon saturable EPCR-mediated transport at the BBB (333). Although native APC exhibits cytoprotection in stroke models, its use is limited by bleeding complications (334); however, a mutant form of APC termed 3K3A-APC has been discovered that exhibits considerable cytoprotective efficacy without complications of bleeding (328). Specifically, studies in human brain endothelial cells in vitro showed that 3K3A-APC protected these cells from oxygen-glucose deprivation to a significantly greater degree than APC (328). Furthermore, 3K3A-APC improved functional outcome and reduced infarction size at a level that was significantly better than APC in the in vivo murine distal MCAO model (328), which implies that 3K3A-APC offers a safer and more efficacious alternative to APC in pharmacological targeting of the PAR-1 receptor. In the case of the PAR-2 receptor, a small molecule PAR-2 antagonist (i.e., N1-3-methylbutyryl-N4-6-aminohexanoyl-piperazine; ENMD-1068) has been shown to attenuate inflammatory responses in a dose-dependent manner (335). More recently, this same PAR-2 antagonist reduced synovitis in rheumatoid and osteoarthritis patients (336). Currently, there is no report in the literature on the effect of PAR-1 activation or PAR-2 inhibition on regulating glial cell biology in the setting of stroke; however, this receptor may represent a novel target that can be exploited for protection of BBB/NVU glial support networks in the ischemic brain.
Minocycline is a tetracycline with anti-inflammatory properties that directly inhibit microglial activation. Minocycline easily crosses the BBB, has a good safety profile, and a delayed therapeutic window thus rendering it an ideal candidate drug for treatment of ischemic stroke (337). Blocking microglial activation may limit BBB disruption and reduce vasogenic edema in the context of ischemic stroke. For example, Yenari and colleagues reported that, in vivo, minocycline reduced infarction volume and neurological deficits as well as prevented BBB disruption and hemorrhage in a murine experimental stroke model (279). In vitro, inhibition of microglial activation with minocycline limited ischemic damage in cultured endothelial cells and reduced superoxide release following oxygen-glucose deprivation (279). Currently, minocycline has been incorporated into two clinical trials involving stroke patients. Results of these studies demonstrated that minocycline administration, both alone and in combination with tPA, improved functional neurological outcome following as ischemic stroke (337).
Toll-like receptors (TLRs) are widely expressed in the human CNS, particularly by astrocytes and microglia (338). Targeting these receptors has emerged as a promising goal for therapeutic control of ischemic stroke, primarily because TLRs are involved in all aspects of BBB dysfunction and NVU ischemic injury (339). While mRNA for TLRs 1 through 10 have been detected in murine microglia (340), all except TLR10 have been reported in human microglia (341). Astrocytes possess a much more limited complement of TLRs since mRNA for TLRs 2, 4, 5, and 9 have been detected in murine astrocytes (342) and only TLR3 mRNA in human astrocytes (343). While the large number of TLR receptors expressed on glial cells suggests a plethora of potential therapeutic targets for modification of glial pathology in the ischemic brain, much work needs to be done on understanding pharmacokinetics of TLR ligand binding and interactions between the TLR and the Toll/IL-1 receptor (TIR) before TLR based stroke therapeutics can reach development (339).
Conclusions
The field of BBB biology, particularly the study of TJ protein complexes and endogenous transport systems, has rapidly advanced over the past two decades. For example, it is now well-established that TJ protein complexes are dynamic in nature and can organize and reorganize in response to ischemic stroke. These changes in TJ protein complexes can lead to increased BBB permeability to small molecule drugs via the paracellular route. Additionally, many previous studies reported on the controversial ability of transporters (i.e., Oatp1a4) to act as facilitators of brain drug uptake (figure 5). Now, it is beginning to be appreciated that endogenous BBB transporters can facilitate uptake of therapeutics from blood to the brain, thereby rendering these transport proteins potential targets for optimizing CNS drug delivery. Furthermore, molecular machinery involved in regulating endogenous BBB transport systems (i.e., TGF-β/ALK5 signaling, nuclear receptor systems) are just now being identified and characterized. These critical discoveries have identified multiple molecular targets that can be exploited for optimization of CNS delivery of therapeutic agents. Perhaps targeting of novel drugs to influx transporters such as Oatp1a4will lead to significant advancements in the treatment of ischemic stroke. Identification and characterization of intracellular signaling pathways that can regulate functional expression of uptake transporters provides yet another approach for pharmacological control of transporter systems in an effort to deliver therapeutics to the CNS. Additionally, identification and characterization of novel targets on glial cells (i.e., astrocytes, microglia) provide yet another opportunity for the design and development of therapeutics aimed at protecting the BBB/NVU during ischemic injury and, by extension, controlling CNS drug delivery. Future work will continue to provide more insight on the interplay of TJ protein complexes, transporters, and intracellular signaling pathways at the BBB/NVU and how these systems can be effectively targeted. Ultimately, data derived from these studies will enable achievement of more precise and more effective drug concentrations within the CNS and improved treatment for ischemic stroke.
Figure 5. Opportunities for targeting the BBB to optimize CNS drug delivery.
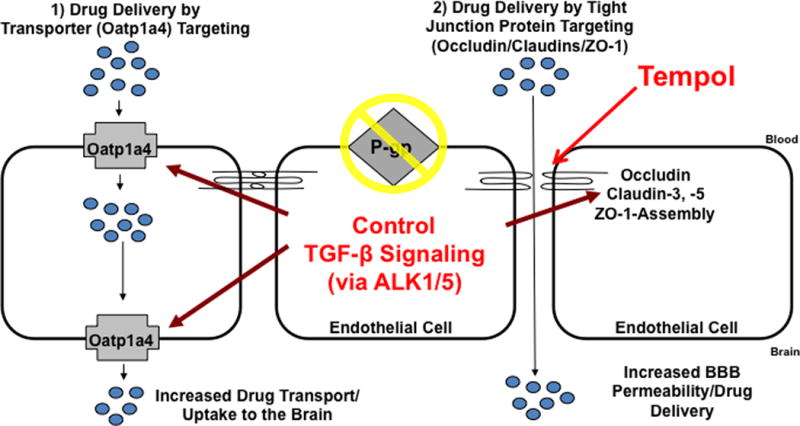
Results from our recent studies demonstrate that CNS drug delivery can be modified by targeting specific molecular structures of the BBB during pathophysiological stress such as H/R. TJ functional integrity can be maintained by scavenging ROS with an antioxidant drug such as TEMPOL. Targeting the TJ provides an opportunity to prevent blood-to-brain solute leak during ischemic stroke, thereby enabling control of CNS drug concentrations. In situations where increased uptake of therapeutics into the brain may be desirable, transporters such as Oatp1a4 can be targeted. Oatp1a4 facilitates brain delivery of drugs that may exhibit efficacy in treatment of ischemic stroke such as statins and opioid peptide analgesics. The TGF-β signalling pathway offers an opportunity to control both TJs and transporters by targeting TGF-β receptors (i.e., ALK1, ALK5) with small molecule therapeutics such as SB431542. Although P-gp is also a critical determinant of CNS drug delivery, caution must be exercised when targeting this transporter to enable greater uptake of therapeutic agents into brain parenchyma. This warning arises from evidence obtained from several laboratories including our own that have shown that enhanced brain delivery of drugs can lead to CNS toxicity and unexpected adverse drug reactions.
Acknowledgments
This work was supported by National Institutes of Health Grants R01-NS42652 and R01-DA11271 to TPD and American Heart Association Beginning Grant-in-Aid and American Association of Pharmaceutical Scientists’ (AAPS) New Investigator Grant in Pharmacokinetics, Pharmacodynamics, and Drug Metabolism to PTR.
Footnotes
Disclosure
Part of the information that appears in this article has been adapted from Ronaldson & Davis. Therapeutic Delivery. 2: 1015–1041 (2011).
Reference List
- 1.Hartz AM, Bauer B. Regulation of ABC transporters at the blood-brain barrier: new targets for CNS therapy. Mol Interv. 2010;10(5):293–304. doi: 10.1124/mi.10.5.6. [DOI] [PubMed] [Google Scholar]
- 2.Del Zoppo GJ. The neurovascular unit in the setting of stroke. J Intern Med. 2010;267(2):156–171. doi: 10.1111/j.1365-2796.2009.02199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Witt KA, Mark KS, Hom S, Davis TP. Effects of hypoxia-reoxygenation on rat blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol. 2003;285(6):H2820–H2831. doi: 10.1152/ajpheart.00589.2003. [DOI] [PubMed] [Google Scholar]
- 4.Brown RC, Mark KS, Egleton RD, Davis TP. Protection against hypoxia-induced blood-brain barrier disruption: changes in intracellular calcium. Am J Physiol Cell Physiol. 2004;286(5):C1045–C1052. doi: 10.1152/ajpcell.00360.2003. [DOI] [PubMed] [Google Scholar]
- 5.Mark KS, Burroughs AR, Brown RC, Huber JD, Davis TP. Nitric oxide mediates hypoxia-induced changes in paracellular permeability of cerebral microvasculature. Am J Physiol Heart Circ Physiol. 2004;286(1):H174–H180. doi: 10.1152/ajpheart.00669.2002. [DOI] [PubMed] [Google Scholar]
- 6.Hom S, Fleegal MA, Egleton RD, Campos CR, Hawkins BT, Davis TP. Comparative changes in the blood-brain barrier and cerebral infarction of SHR and WKY rats. Am J Physiol Regul Integr Comp Physiol. 2007;292(5):R1881–R1892. doi: 10.1152/ajpregu.00761.2005. [DOI] [PubMed] [Google Scholar]
- 7.Witt KA, Mark KS, Sandoval KE, Davis TP. Reoxygenation stress on blood-brain barrier paracellular permeability and edema in the rat. Microvasc Res. 2008;75(1):91–96. doi: 10.1016/j.mvr.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCaffrey G, Willis CL, Staatz WD, Nametz N, Quigley CA, Hom S, et al. Occludin oligomeric assemblies at tight junctions of the blood-brain barrier are altered by hypoxia and reoxygenation stress. J Neurochem. 2009;110(1):58–71. doi: 10.1111/j.1471-4159.2009.06113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ronaldson PT, Demarco KM, Sanchez-Covarrubias L, Solinsky CM, Davis TP. Transforming growth factor-beta signaling alters substrate permeability and tight junction protein expression at the blood-brain barrier during inflammatory pain. J Cereb Blood Flow Metab. 2009;29(6):1084–1098. doi: 10.1038/jcbfm.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lochhead JJ, McCaffrey G, Quigley CE, Finch J, Demarco KM, Nametz N, et al. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab. 2010;30(9):1625–1636. doi: 10.1038/jcbfm.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willis CL, Meske DS, Davis TP. Protein kinase C activation modulates reversible increase in cortical blood-brain barrier permeability and tight junction protein expression during hypoxia and posthypoxic reoxygenation. J Cereb Blood Flow Metab. 2010;30(11):1847–1859. doi: 10.1038/jcbfm.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lochhead JJ, McCaffrey G, Sanchez-Covarrubia L, Finch JD, Demarco KM, Quigley CE, et al. Tempol Modulates Changes in Xenobiotic Permeability and Occludin Oligomeric Assemblies at the Blood-Brain Barrier during Inflammatory Pain. Am J Physiol Heart Circ Physiol. 2011 doi: 10.1152/ajpheart.00889.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ronaldson PT, Finch JD, Demarco KM, Quigley CE, Davis TP. Inflammatory pain signals an increase in functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier. J Pharmacol Exp Ther. 2011;336(3):827–839. doi: 10.1124/jpet.110.174151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57(2):173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 15.Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006;1(3):223–236. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- 16.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57(2):178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 17.Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009;118(1):103–113. doi: 10.1007/s00401-009-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arai K, Lok J, Guo S, Hayakawa K, Xing C, Lo EH. Cellular mechanisms of neurovascular damage and repair after stroke. J Child Neurol. 2011;26(9):1193–1198. doi: 10.1177/0883073811408610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee MY, Kuan YH, Chen HY, Chen TY, Chen ST, Huang CC, et al. Intravenous administration of melatonin reduces the intracerebral cellular inflammatory response following transient focal cerebral ischemia in rats. J Pineal Res. 2007;42(3):297–309. doi: 10.1111/j.1600-079X.2007.00420.x. [DOI] [PubMed] [Google Scholar]
- 20.DiNapoli VA, Huber JD, Houser K, Li X, Rosen CL. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol Aging. 2008;29(5):753–764. doi: 10.1016/j.neurobiolaging.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vangilder RL, Rosen CL, Barr TL, Huber JD. Targeting the neurovascular unit for treatment of neurological disorders. Pharmacol Ther. 2011;130(3):239–247. doi: 10.1016/j.pharmthera.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77(3):731–758. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- 23.Reese TS, Karnovsky MJ. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol. 1967;34(1):207–217. doi: 10.1083/jcb.34.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brightman MW, Reese TS. Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol. 1969;40(3):648–677. doi: 10.1083/jcb.40.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abbott NJ. Dynamics of CNS barriers: evolution, differentiation, and modulation. Cell Mol Neurobiol. 2005;25(1):5–23. doi: 10.1007/s10571-004-1374-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oldendorf WH, Cornford ME, Brown WJ. The large apparent work capability of the blood-brain barrier: a study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol. 1977;1(5):409–417. doi: 10.1002/ana.410010502. [DOI] [PubMed] [Google Scholar]
- 27.Roberts LM, Black DS, Raman C, Woodford K, Zhou M, Haggerty JE, et al. Subcellular localization of transporters along the rat blood-brain barrier and blood-cerebral-spinal fluid barrier by in vivo biotinylation. Neuroscience. 2008;155(2):423–438. doi: 10.1016/j.neuroscience.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 28.Yousif S, Saubamea B, Cisternino S, Marie-Claire C, Dauchy S, Scherrmann JM, et al. Effect of chronic exposure to morphine on the rat blood-brain barrier: focus on the P-glycoprotein. J Neurochem. 2008;107(3):647–657. doi: 10.1111/j.1471-4159.2008.05647.x. [DOI] [PubMed] [Google Scholar]
- 29.Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y. Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther. 2010;333(3):788–796. doi: 10.1124/jpet.109.162321. [DOI] [PubMed] [Google Scholar]
- 30.Ose A, Kusuhara H, Endo C, Tohyama K, Miyajima M, Kitamura S, et al. Functional characterization of mouse organic anion transporting peptide 1a4 in the uptake and efflux of drugs across the blood-brain barrier. Drug Metab Dispos. 2010;38(1):168–176. doi: 10.1124/dmd.109.029454. [DOI] [PubMed] [Google Scholar]
- 31.Dallas S, Miller DS, Bendayan R. Multidrug resistance-associated proteins: expression and function in the central nervous system. Pharmacol Rev. 2006;58(2):140–161. doi: 10.1124/pr.58.2.3. [DOI] [PubMed] [Google Scholar]
- 32.Hawkins BT, Ocheltree SM, Norwood KM, Egleton RD. Decreased blood-brain barrier permeability to fluorescein in streptozotocin-treated rats. Neurosci Lett. 2007;411(1):1–5. doi: 10.1016/j.neulet.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bauer B, Hartz AM, Lucking JR, Yang X, Pollack GM, Miller DS. Coordinated nuclear receptor regulation of the efflux transporter, Mrp2, and the phase-II metabolizing enzyme, GSTpi, at the blood-brain barrier. J Cereb Blood Flow Metab. 2008;28(6):1222–1234. doi: 10.1038/jcbfm.2008.16. [DOI] [PubMed] [Google Scholar]
- 34.Dauchy S, Miller F, Couraud PO, Weaver RJ, Weksler B, Romero IA, et al. Expression and transcriptional regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral microvascular endothelial cells. Biochem Pharmacol. 2009;77(5):897–909. doi: 10.1016/j.bcp.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 35.Lee G, Babakhanian K, Ramaswamy M, Prat A, Wosik K, Bendayan R. Expression of the ATP-binding cassette membrane transporter, ABCG2, in human and rodent brain microvessel endothelial and glial cell culture systems. Pharm Res. 2007;24(7):1262–1274. doi: 10.1007/s11095-007-9244-1. [DOI] [PubMed] [Google Scholar]
- 36.Uchida Y, Ohtsuki S, Katsukura Y, Ikeda C, Suzuki T, Kamiie J, et al. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J Neurochem. 2011;117(2):333–345. doi: 10.1111/j.1471-4159.2011.07208.x. [DOI] [PubMed] [Google Scholar]
- 37.Westholm DE, Salo DR, Viken KJ, Rumbley JN, Anderson GW. The blood-brain barrier thyroxine transporter organic anion-transporting polypeptide 1c1 displays atypical transport kinetics. Endocrinology. 2009;150(11):5153–5162. doi: 10.1210/en.2009-0769. [DOI] [PubMed] [Google Scholar]
- 38.Westholm DE, Stenehjem DD, Rumbley JN, Drewes LR, Anderson GW. Competitive inhibition of organic anion transporting polypeptide 1c1-mediated thyroxine transport by the fenamate class of nonsteroidal antiinflammatory drugs. Endocrinology. 2009;150(2):1025–1032. doi: 10.1210/en.2008-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kusch-Poddar M, Drewe J, Fux I, Gutmann H. Evaluation of the immortalized human brain capillary endothelial cell line BB19 as a human cell culture model for the blood-brain barrier. Brain Res. 2005;1064(1–2):21–31. doi: 10.1016/j.brainres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 40.Ose A, Ito M, Kusuhara H, Yamatsugu K, Kanai M, Shibasaki M, et al. Limited brain distribution of [3R,4R,5S]-4-acetamido-5-amino-3-(1-ethylpropoxy)-1-cyclohexene-1-carboxyl ate phosphate (Ro 64-0802), a pharmacologically active form of oseltamivir, by active efflux across the blood-brain barrier mediated by organic anion transporter 3 (Oat3/Slc22a8) and multidrug resistance-associated protein 4 (Mrp4/Abcc4) Drug Metab Dispos. 2009;37(2):315–321. doi: 10.1124/dmd.108.024018. [DOI] [PubMed] [Google Scholar]
- 41.Miyajima M, Kusuhara H, Fujishima M, Adachi Y, Sugiyama Y. Organic anion transporter 3 mediates the efflux transport of an amphipathic organic anion, dehydroepiandrosterone sulfate, across the blood-brain barrier in mice. Drug Metab Dispos. 2011;39(5):814–819. doi: 10.1124/dmd.110.036863. [DOI] [PubMed] [Google Scholar]
- 42.Batrakova EV, Zhang Y, Li Y, Li S, Vinogradov SV, Persidsky Y, et al. Effects of pluronic P85 on GLUT1 and MCT1 transporters in the blood-brain barrier. Pharm Res. 2004;21(11):1993–2000. doi: 10.1023/b:pham.0000048189.79606.6e. [DOI] [PubMed] [Google Scholar]
- 43.Chishty M, Begley DJ, Abbott NJ, Reichel A. Interaction of nucleoside analogues with nucleoside transporters in rat brain endothelial cells. J Drug Target. 2004;12(5):265–272. doi: 10.1080/10611860410001731398. [DOI] [PubMed] [Google Scholar]
- 44.Dogrukol-Ak D, Kumar VB, Ryerse JS, Farr SA, Verma S, Nonaka N, et al. Isolation of peptide transport system-6 from brain endothelial cells: therapeutic effects with antisense inhibition in Alzheimer and stroke models. J Cereb Blood Flow Metab. 2009;29(2):411–422. doi: 10.1038/jcbfm.2008.131. [DOI] [PubMed] [Google Scholar]
- 45.Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study. J Physiol. 1990;429:47–62. doi: 10.1113/jphysiol.1990.sp018243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haskins J, Gu L, Wittchen ES, Hibbard J, Stevenson BR. ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J Cell Biol. 1998;141(1):199–208. doi: 10.1083/jcb.141.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Del Maschio A, De Luigi A, Martin-Padura I, Brockhaus M, Bartfai T, Fruscella P, et al. Leukocyte recruitment in the cerebrospinal fluid of mice with experimental meningitis is inhibited by an antibody to junctional adhesion molecule (JAM) J Exp Med. 1999;190(9):1351–1356. doi: 10.1084/jem.190.9.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dejana E, Lampugnani MG, Martinez-Estrada O, Bazzoni G. The molecular organization of endothelial junctions and their functional role in vascular morphogenesis and permeability. Int J Dev Biol. 2000;44(6):743–748. [PubMed] [Google Scholar]
- 49.Yeung D, Manias JL, Stewart DJ, Nag S. Decreased junctional adhesion molecule-A expression during blood-brain barrier breakdown. Acta Neuropathol. 2008;115(6):635–642. doi: 10.1007/s00401-008-0364-4. [DOI] [PubMed] [Google Scholar]
- 50.Hoffman WH, Stamatovic SM, Andjelkovic AV. Inflammatory mediators and blood brain barrier disruption in fatal brain edema of diabetic ketoacidosis. Brain Res. 2009;1254:138–148. doi: 10.1016/j.brainres.2008.11.100. [DOI] [PubMed] [Google Scholar]
- 51.Haarmann A, Deiss A, Prochaska J, Foerch C, Weksler B, Romero I, et al. Evaluation of soluble junctional adhesion molecule-A as a biomarker of human brain endothelial barrier breakdown. PLoS One. 2010;5(10):e13568. doi: 10.1371/journal.pone.0013568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123(6 Pt 2):1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hawkins BT, Abbruscato TJ, Egleton RD, Brown RC, Huber JD, Campos CR, et al. Nicotine increases in vivo blood-brain barrier permeability and alters cerebral microvascular tight junction protein distribution. Brain Res. 2004;1027(1–2):48–58. doi: 10.1016/j.brainres.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 54.McCaffrey G, Seelbach MJ, Staatz WD, Nametz N, Quigley C, Campos CR, et al. Occludin oligomeric assembly at tight junctions of the blood-brain barrier is disrupted by peripheral inflammatory hyperalgesia. J Neurochem. 2008;106(6):2395–2409. doi: 10.1111/j.1471-4159.2008.05582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hirase T, Staddon JM, Saitou M, Ando-Akatsuka Y, Itoh M, Furuse M, et al. Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci. 1997;110(Pt 14):1603–1613. doi: 10.1242/jcs.110.14.1603. [DOI] [PubMed] [Google Scholar]
- 56.Blasig IE, Bellmann C, Cording J, Del Vecchio G, Zwanziger D, Huber O, et al. Occludin protein family: oxidative stress and reducing conditions. Antioxid Redox Signal. 2011;15(5):1195–1219. doi: 10.1089/ars.2010.3542. [DOI] [PubMed] [Google Scholar]
- 57.McCaffrey G, Staatz WD, Quigley CA, Nametz N, Seelbach MJ, Campos CR, et al. Tight junctions contain oligomeric protein assembly critical for maintaining blood-brain barrier integrity in vivo. J Neurochem. 2007;103(6):2540–2555. doi: 10.1111/j.1471-4159.2007.04943.x. [DOI] [PubMed] [Google Scholar]
- 58.Lacaz-Vieira F, Jaeger MM, Farshori P, Kachar B. Small synthetic peptides homologous to segments of the first external loop of occludin impair tight junction resealing. J Membr Biol. 1999;168(3):289–297. doi: 10.1007/s002329900518. [DOI] [PubMed] [Google Scholar]
- 59.Feldman GJ, Mullin JM, Ryan MP. Occludin: structure, function and regulation. Adv Drug Deliv Rev. 2005;57(6):883–917. doi: 10.1016/j.addr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 60.Brown RC, Davis TP. Hypoxia/aglycemia alters expression of occludin and actin in brain endothelial cells. Biochem Biophys Res Commun. 2005;327(4):1114–1123. doi: 10.1016/j.bbrc.2004.12.123. [DOI] [PubMed] [Google Scholar]
- 61.Heiskala M, Peterson PA, Yang Y. The roles of claudin superfamily proteins in paracellular transport. Traffic. 2001;2(2):93–98. doi: 10.1034/j.1600-0854.2001.020203.x. [DOI] [PubMed] [Google Scholar]
- 62.Furuse M, Sasaki H, Tsukita S. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J Cell Biol. 1999;147(4):891–903. doi: 10.1083/jcb.147.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kubota K, Furuse M, Sasaki H, Sonoda N, Fujita K, Nagafuchi A, et al. Ca(2+)-independent cell-adhesion activity of claudins, a family of integral membrane proteins localized at tight junctions. Curr Biol. 1999;9(18):1035–1038. doi: 10.1016/s0960-9822(99)80452-7. [DOI] [PubMed] [Google Scholar]
- 64.Wolburg H, Wolburg-Buchholz K, Kraus J, Rascher-Eggstein G, Liebner S, Hamm S, et al. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol. 2003;105(6):586–592. doi: 10.1007/s00401-003-0688-z. [DOI] [PubMed] [Google Scholar]
- 65.Nag S, Venugopalan R, Stewart DJ. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol. 2007;114(5):459–469. doi: 10.1007/s00401-007-0274-x. [DOI] [PubMed] [Google Scholar]
- 66.Forster C, Burek M, Romero IA, Weksler B, Couraud PO, Drenckhahn D. Differential effects of hydrocortisone and TNFalpha on tight junction proteins in an in vitro model of the human blood-brain barrier. J Physiol. 2008;586(7):1937–1949. doi: 10.1113/jphysiol.2007.146852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang P, Liu Y, Shang X, Xue Y. CRM197-induced blood-brain barrier permeability increase is mediated by upregulation of caveolin-1 protein. J Mol Neurosci. 2011;43(3):485–492. doi: 10.1007/s12031-010-9471-5. [DOI] [PubMed] [Google Scholar]
- 68.McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. 2008;28(38):9451–9462. doi: 10.1523/JNEUROSCI.2674-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang Y, Rosenberg GA. MMP-mediated disruption of claudin-5 in the blood-brain barrier of rat brain after cerebral ischemia. Methods Mol Biol. 2011;762:333–345. doi: 10.1007/978-1-61779-185-7_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaur J, Tuor UI, Zhao Z, Barber PA. Quantitative MRI reveals the elderly ischemic brain is susceptible to increased early blood-brain barrier permeability following tissue plasminogen activator related to claudin 5 and occludin disassembly. J Cereb Blood Flow Metab. 2011;31(9):1874–1885. doi: 10.1038/jcbfm.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gonzalez-Mariscal L, Betanzos A, Avila-Flores A. MAGUK proteins: structure and role in the tight junction. Semin Cell Dev Biol. 2000;11(4):315–324. doi: 10.1006/scdb.2000.0178. [DOI] [PubMed] [Google Scholar]
- 72.Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol. 1986;103(3):755–766. doi: 10.1083/jcb.103.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem. 1998;273(45):29745–29753. doi: 10.1074/jbc.273.45.29745. [DOI] [PubMed] [Google Scholar]
- 74.Abbruscato TJ, Lopez SP, Mark KS, Hawkins BT, Davis TP. Nicotine and cotinine modulate cerebral microvascular permeability and protein expression of ZO-1 through nicotinic acetylcholine receptors expressed on brain endothelial cells. J Pharm Sci. 2002;91(12):2525–2538. doi: 10.1002/jps.10256. [DOI] [PubMed] [Google Scholar]
- 75.Fischer S, Wobben M, Marti HH, Renz D, Schaper W. Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc Res. 2002;63(1):70–80. doi: 10.1006/mvre.2001.2367. [DOI] [PubMed] [Google Scholar]
- 76.Mark KS, Davis TP. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2002;282(4):H1485–H1494. doi: 10.1152/ajpheart.00645.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gottardi CJ, Arpin M, Fanning AS, Louvard D. The junction-associated protein, zonula occludens-1, localizes to the nucleus before the maturation and during the remodeling of cell-cell contacts. Proc Natl Acad Sci U S A. 1996;93(20):10779–10784. doi: 10.1073/pnas.93.20.10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Riesen FK, Rothen-Rutishauser B, Wunderli-Allenspach H. A ZO1-GFP fusion protein to study the dynamics of tight junctions in living cells. Histochem Cell Biol. 2002;117(4):307–315. doi: 10.1007/s00418-002-0398-y. [DOI] [PubMed] [Google Scholar]
- 79.Balda MS, Matter K. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J. 2000;19(9):2024–2033. doi: 10.1093/emboj/19.9.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meyer TN, Schwesinger C, Denker BM. Zonula occludens-1 is a scaffolding protein for signaling molecules. Galpha (12) directly binds to the Src homology 3 domain and regulates paracellular permeability in epithelial cells. J Biol Chem. 2002;277(28):24855–24858. doi: 10.1074/jbc.C200240200. [DOI] [PubMed] [Google Scholar]
- 81.Gumbiner B, Lowenkopf T, Apatira D. Identification of a 160-kDa polypeptide that binds to the tight junction protein ZO-1. Proc Natl Acad Sci U S A. 1991;88(8):3460–3464. doi: 10.1073/pnas.88.8.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Betanzos A, Huerta M, Lopez-Bayghen E, Azuara E, Amerena J, Gonzalez-Mariscal L. The tight junction protein ZO-2 associates with Jun, Fos and C/EBP transcription factors in epithelial cells. Exp Cell Res. 2004;292(1):51–66. doi: 10.1016/j.yexcr.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 83.Islas S, Vega J, Ponce L, Gonzalez-Mariscal L. Nuclear localization of the tight junction protein ZO-2 in epithelial cells. Exp Cell Res. 2002;274(1):138–148. doi: 10.1006/excr.2001.5457. [DOI] [PubMed] [Google Scholar]
- 84.Traweger A, Fuchs R, Krizbai IA, Weiger TM, Bauer HC, Bauer H. The tight junction protein ZO-2 localizes to the nucleus and interacts with the heterogeneous nuclear ribonucleoprotein scaffold attachment factor-B. J Biol Chem. 2003;278(4):2692–2700. doi: 10.1074/jbc.M206821200. [DOI] [PubMed] [Google Scholar]
- 85.Umeda K, Matsui T, Nakayama M, Furuse K, Sasaki H, Furuse M, et al. Establishment and characterization of cultured epithelial cells lacking expression of ZO-1. J Biol Chem. 2004;279(43):44785–44794. doi: 10.1074/jbc.M406563200. [DOI] [PubMed] [Google Scholar]
- 86.Takenaga Y, Takagi N, Murotomi K, Tanonaka K, Takeo S. Inhibition of Src activity decreases tyrosine phosphorylation of occludin in brain capillaries and attenuates increase in permeability of the blood-brain barrier after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2009;29(6):1099–1108. doi: 10.1038/jcbfm.2009.30. [DOI] [PubMed] [Google Scholar]
- 87.Li JJ, Xing SH, Zhang J, Hong H, Li YL, Dang C, et al. Decrease of tight junction integrity in the ipsilateral thalamus during the acute stage after focal infarction and ablation of the cerebral cortex in rats. Clin Exp Pharmacol Physiol. 2011;38(11):776–782. doi: 10.1111/j.1440-1681.2011.05591.x. [DOI] [PubMed] [Google Scholar]
- 88.Jiao H, Wang Z, Liu Y, Wang P, Xue Y. Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. J Mol Neurosci. 2011;44(2):130–139. doi: 10.1007/s12031-011-9496-4. [DOI] [PubMed] [Google Scholar]
- 89.Bangsow T, Baumann E, Bangsow C, Jaeger MH, Pelzer B, Gruhn P, et al. The epithelial membrane protein 1 is a novel tight junction protein of the blood-brain barrier. J Cereb Blood Flow Metab. 2008;28(6):1249–1260. doi: 10.1038/jcbfm.2008.19. [DOI] [PubMed] [Google Scholar]
- 90.Citi S, Sabanay H, Kendrick-Jones J, Geiger B. Cingulin: characterization and localization. J Cell Sci. 1989;93(Pt 1):107–122. doi: 10.1242/jcs.93.1.107. [DOI] [PubMed] [Google Scholar]
- 91.Yamamoto T, Harada N, Kawano Y, Taya S, Kaibuchi K. In vivo interaction of AF-6 with activated Ras and ZO-1. Biochem Biophys Res Commun. 1999;259(1):103–107. doi: 10.1006/bbrc.1999.0731. [DOI] [PubMed] [Google Scholar]
- 92.Zhong Y, Enomoto K, Isomura H, Sawada N, Minase T, Oyamada M, et al. Localization of the 7H6 antigen at tight junctions correlates with the paracellular barrier function of MDCK cells. Exp Cell Res. 1994;214(2):614–620. doi: 10.1006/excr.1994.1299. [DOI] [PubMed] [Google Scholar]
- 93.Li JY, Boado RJ, Pardridge WM. Blood-brain barrier genomics. J Cereb Blood Flow Metab. 2001;21(1):61–68. doi: 10.1097/00004647-200101000-00008. [DOI] [PubMed] [Google Scholar]
- 94.Li JY, Boado RJ, Pardridge WM. Rat blood-brain barrier genomics. II. J Cereb Blood Flow Metab. 2002;22(11):1319–1326. doi: 10.1097/01.WCB.0000040944.89393.0f. [DOI] [PubMed] [Google Scholar]
- 95.Leslie EM, Deeley RG, Cole SP. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol. 2005;204(3):216–237. doi: 10.1016/j.taap.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 96.Ronaldson PT, Persidsky Y, Bendayan R. Regulation of ABC membrane transporters in glial cells: relevance to the pharmacotherapy of brain HIV-1 infection. Glia. 2008;56(16):1711–1735. doi: 10.1002/glia.20725. [DOI] [PubMed] [Google Scholar]
- 97.Robey RW, To KK, Polgar O, Dohse M, Fetsch P, Dean M, et al. ABCG2: a perspective. Adv Drug Deliv Rev. 2009;61(1):3–13. doi: 10.1016/j.addr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455(1):152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 99.Gottesman MM, Hrycyna CA, Schoenlein PV, Germann UA, Pastan I. Genetic analysis of the multidrug transporter. Annu Rev Genet. 1995;29:607–649. doi: 10.1146/annurev.ge.29.120195.003135. [DOI] [PubMed] [Google Scholar]
- 100.Sharom FJ. Shedding light on drug transport: structure and function of the P-glycoprotein multidrug transporter (ABCB1) Biochem Cell Biol. 2006;84(6):979–992. doi: 10.1139/o06-199. [DOI] [PubMed] [Google Scholar]
- 101.Beaulieu E, Demeule M, Ghitescu L, Beliveau R. P-glycoprotein is strongly expressed in the luminal membranes of the endothelium of blood vessels in the brain. Biochem J. 1997;326(Pt 2):539–544. doi: 10.1042/bj3260539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Virgintino D, Robertson D, Errede M, Benagiano V, Girolamo F, Maiorano E, et al. Expression of P-glycoprotein in human cerebral cortex microvessels. J Histochem Cytochem. 2002;50(12):1671–1676. doi: 10.1177/002215540205001212. [DOI] [PubMed] [Google Scholar]
- 103.Bendayan R, Ronaldson PT, Gingras D, Bendayan M. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J Histochem Cytochem. 2006;54(10):1159–1167. doi: 10.1369/jhc.5A6870.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Golden PL, Pardridge WM. P-Glycoprotein on astrocyte foot processes of unfixed isolated human brain capillaries. Brain Res. 1999;819(1–2):143–146. doi: 10.1016/s0006-8993(98)01305-5. [DOI] [PubMed] [Google Scholar]
- 105.Schlachetzki F, Pardridge WM. P-glycoprotein and caveolin-1alpha in endothelium and astrocytes of primate brain. Neuroreport. 2003;14(16):2041–2046. doi: 10.1097/00001756-200311140-00007. [DOI] [PubMed] [Google Scholar]
- 106.Ueno M, Nakagawa T, Huang CL, Ueki M, Kusaka T, Hosomi N, et al. The expression of P-glycoprotein is increased in vessels with blood-brain barrier impairment in a stroke-prone hypertensive model. Neuropathol Appl Neurobiol. 2009;35(2):147–155. doi: 10.1111/j.1365-2990.2008.00966.x. [DOI] [PubMed] [Google Scholar]
- 107.Miller DS, Nobmann SN, Gutmann H, Toeroek M, Drewe J, Fricker G. Xenobiotic transport across isolated brain microvessels studied by confocal microscopy. Mol Pharmacol. 2000;58(6):1357–1367. doi: 10.1124/mol.58.6.1357. [DOI] [PubMed] [Google Scholar]
- 108.Leggas M, Adachi M, Scheffer GL, Sun D, Wielinga P, Du G, et al. Mrp4 confers resistance to topotecan and protects the brain from chemotherapy. Mol Cell Biol. 2004;24(17):7612–7621. doi: 10.1128/MCB.24.17.7612-7621.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang Y, Schuetz JD, Elmquist WF, Miller DW. Plasma membrane localization of multidrug resistance-associated protein homologs in brain capillary endothelial cells. J Pharmacol Exp Ther. 2004;311(2):449–455. doi: 10.1124/jpet.104.068528. [DOI] [PubMed] [Google Scholar]
- 110.Bandler PE, Westlake CJ, Grant CE, Cole SP, Deeley RG. Identification of regions required for apical membrane localization of human multidrug resistance protein 2. Mol Pharmacol. 2008;74(1):9–19. doi: 10.1124/mol.108.045674. [DOI] [PubMed] [Google Scholar]
- 111.Ronaldson PT, Bendayan R. HIV-1 viral envelope glycoprotein gp120 produces oxidative stress and regulates the functional expression of multidrug resistance protein-1 (Mrp1) in glial cells. J Neurochem. 2008;106(3):1298–1313. doi: 10.1111/j.1471-4159.2008.05479.x. [DOI] [PubMed] [Google Scholar]
- 112.Cooray HC, Blackmore CG, Maskell L, Barrand MA. Localisation of breast cancer resistance protein in microvessel endothelium of human brain. Neuroreport. 2002;13(16):2059–2063. doi: 10.1097/00001756-200211150-00014. [DOI] [PubMed] [Google Scholar]
- 113.Hori S, Ohtsuki S, Tachikawa M, Kimura N, Kondo T, Watanabe M, et al. Functional expression of rat ABCG2 on the luminal side of brain capillaries and its enhancement by astrocyte-derived soluble factor(s) J Neurochem. 2004;90(3):526–536. doi: 10.1111/j.1471-4159.2004.02537.x. [DOI] [PubMed] [Google Scholar]
- 114.Lee YJ, Kusuhara H, Jonker JW, Schinkel AH, Sugiyama Y. Investigation of efflux transport of dehydroepiandrosterone sulfate and mitoxantrone at the mouse blood-brain barrier: a minor role of breast cancer resistance protein. J Pharmacol Exp Ther. 2005;312(1):44–52. doi: 10.1124/jpet.104.073320. [DOI] [PubMed] [Google Scholar]
- 115.van Herwaarden AE, Jonker JW, Wagenaar E, Brinkhuis RF, Schellens JH, Beijnen JH, et al. The breast cancer resistance protein (Bcrp1/Abcg2) restricts exposure to the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Cancer Res. 2003;63(19):6447–6452. [PubMed] [Google Scholar]
- 116.Zhao R, Raub TJ, Sawada GA, Kasper SC, Bacon JA, Bridges AS, et al. Breast cancer resistance protein interacts with various compounds in vitro, but plays a minor role in substrate efflux at the blood-brain barrier. Drug Metab Dispos. 2009;37(6):1251–1258. doi: 10.1124/dmd.108.025064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.de Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O. P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res. 2007;13(21):6440–6449. doi: 10.1158/1078-0432.CCR-07-1335. [DOI] [PubMed] [Google Scholar]
- 118.Zhou L, Schmidt K, Nelson FR, Zelesky V, Troutman MD, Feng B. The effect of breast cancer resistance protein and P-glycoprotein on the brain penetration of flavopiridol, imatinib mesylate (Gleevec), prazosin, and 2-methoxy-3-(4-(2-(5-methyl-2-phenyloxazol-4-yl)ethoxy)phenyl)propanoic acid (PF-407288) in mice. Drug Metab Dispos. 2009;37(5):946–955. doi: 10.1124/dmd.108.024489. [DOI] [PubMed] [Google Scholar]
- 119.Agarwal S, Sane R, Ohlfest JR, Elmquist WF. The role of the breast cancer resistance protein (ABCG2) in the distribution of sorafenib to the brain. J Pharmacol Exp Ther. 2011;336(1):223–233. doi: 10.1124/jpet.110.175034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sugiura T, Kato Y, Tsuji A. Role of SLC xenobiotic transporters and their regulatory mechanisms PDZ proteins in drug delivery and disposition. Cancer Res. 2006;116(2):238–246. doi: 10.1016/j.jconrel.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 121.Kusuhara H, Sugiyama Y. Active efflux across the blood-brain barrier: role of the solute carrier family. NeuroRx. 2005;2(1):73–85. doi: 10.1602/neurorx.2.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/ SLC21 family: phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004;447(5):653–665. doi: 10.1007/s00424-003-1168-y. [DOI] [PubMed] [Google Scholar]
- 123.Ponce J, de la Ossa NP, Hurtado O, Millan M, Arenillas JF, Davalos A, et al. Simvastatin reduces the association of NMDA receptors to lipid rafts: a cholesterol-mediated effect in neuroprotection. Stroke. 2008;39(4):1269–1275. doi: 10.1161/STROKEAHA.107.498923. [DOI] [PubMed] [Google Scholar]
- 124.Wood WG, Eckert GP, Igbavboa U, Muller WE. Statins and neuroprotection: a prescription to move the field forward. Ann N Y Acad Sci. 2010;1199:69–76. doi: 10.1111/j.1749-6632.2009.05359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Barone E, Cenini G, Di Domenico F, Martin S, Sultana R, Mancuso C, et al. Long-term high-dose atorvastatin decreases brain oxidative and nitrosative stress in a preclinical model of Alzheimer disease: a novel mechanism of action. Pharmacol Res. 2011;63(3):172–180. doi: 10.1016/j.phrs.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Butterfield DA, Barone E, Mancuso C. Cholesterol-independent neuroprotective and neurotoxic activities of statins: perspectives for statin use in Alzheimer disease and other age-related neurodegenerative disorders. Pharmacol Res. 2011;64(3):180–186. doi: 10.1016/j.phrs.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tokui T, Nakai D, Nakagomi R, Yawo H, Abe T, Sugiyama Y. Pravastatin, an HMG-CoA reductase inhibitor, is transported by rat organic anion transporting polypeptide, oatp2. Pharm Res. 1999;16(6):904–908. doi: 10.1023/a:1018838405987. [DOI] [PubMed] [Google Scholar]
- 128.Yang L, Shah K, Wang H, Karamyan VT, Abbruscato TJ. Characterization of neuroprotective effects of biphalin, an opioid receptor agonist, in a model of focal brain ischemia. J Pharmacol Exp Ther. 2011;339(2):499–508. doi: 10.1124/jpet.111.184127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yang L, Wang H, Shah K, Karamyan VT, Abbruscato TJ. Opioid receptor agonists reduce brain edema in stroke. Brain Res. 2011;1383:307–316. doi: 10.1016/j.brainres.2011.01.083. [DOI] [PubMed] [Google Scholar]
- 130.Gao B, Hagenbuch B, Kullak-Ublick GA, Benke D, Aguzzi A, Meier PJ. Organic anion-transporting polypeptides mediate transport of opioid peptides across blood-brain barrier. J Pharmacol Exp Ther. 2000;294(1):73–79. [PubMed] [Google Scholar]
- 131.Sugiyama D, Kusuhara H, Taniguchi H, Ishikawa S, Nozaki Y, Aburatani H, et al. Functional characterization of rat brain-specific organic anion transporter (Oatp14) at the blood-brain barrier: high affinity transporter for thyroxine. J Biol Chem. 2003;278(44):43489–43495. doi: 10.1074/jbc.M306933200. [DOI] [PubMed] [Google Scholar]
- 132.Taogoshi T, Nomura A, Murakami T, Nagai J, Takano M. Transport of prostaglandin E1 across the blood-brain barrier in rats. J Pharm Pharmacol. 2005;57(1):61–66. doi: 10.1211/0022357055173. [DOI] [PubMed] [Google Scholar]
- 133.Chu C, Li JY, Boado RJ, Pardridge WM. Blood-brain barrier genomics and cloning of a novel organic anion transporter. J Cereb Blood Flow Metab. 2008;28(2):291–301. doi: 10.1038/sj.jcbfm.9600538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325(6101):253–257. doi: 10.1038/325253a0. [DOI] [PubMed] [Google Scholar]
- 135.Tao-Cheng JH, Nagy Z, Brightman MW. Tight junctions of brain endothelium in vitro are enhanced by astroglia. J Neurosci. 1987;7(10):3293–3299. doi: 10.1523/JNEUROSCI.07-10-03293.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Neuhaus J, Risau W, Wolburg H. Induction of blood-brain barrier characteristics in bovine brain endothelial cells by rat astroglial cells in transfilter coculture. Ann N Y Acad Sci. 1991;633:578–580. doi: 10.1111/j.1749-6632.1991.tb15667.x. [DOI] [PubMed] [Google Scholar]
- 137.Hayashi Y, Nomura M, Yamagishi S, Harada S, Yamashita J, Yamamoto H. Induction of various blood-brain barrier properties in non-neural endothelial cells by close apposition to co-cultured astrocytes. Glia. 1997;19(1):13–26. [PubMed] [Google Scholar]
- 138.Willis CL, Leach L, Clarke GJ, Nolan CC, Ray DE. Reversible disruption of tight junction complexes in the rat blood-brain barrier, following transitory focal astrocyte loss. Glia. 2004;48(1):1–13. doi: 10.1002/glia.20049. [DOI] [PubMed] [Google Scholar]
- 139.Willis CL, Nolan CC, Reith SN, Lister T, Prior MJ, Guerin CJ, et al. Focal astrocyte loss is followed by microvascular damage, with subsequent repair of the blood-brain barrier in the apparent absence of direct astrocytic contact. Glia. 2004;45(4):325–337. doi: 10.1002/glia.10333. [DOI] [PubMed] [Google Scholar]
- 140.Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16(1):1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 141.Goldberg M, De Pitta M, Volman V, Berry H, Ben Jacob E. Nonlinear gap junctions enable long-distance propagation of pulsating calcium waves in astrocyte networks. PLoS Comput Biol. 2010;6(8) doi: 10.1371/journal.pcbi.1000909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pelligrino DA, Vetri F, Xu HL. Purinergic mechanisms in gliovascular coupling. Semin Cell Dev Biol. 2011;22(2):229–236. doi: 10.1016/j.semcdb.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 144.Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia. 2010;58(9):1094–1103. doi: 10.1002/glia.20990. [DOI] [PubMed] [Google Scholar]
- 145.Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem. 2006;98(3):641–653. doi: 10.1111/j.1471-4159.2006.03913.x. [DOI] [PubMed] [Google Scholar]
- 146.Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32(1):1–14. [PubMed] [Google Scholar]
- 147.Kimelberg HK, Goderie SK, Higman S, Pang S, Waniewski RA. Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J Neurosci. 1990;10(5):1583–1591. doi: 10.1523/JNEUROSCI.10-05-01583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ronaldson PT, Bendayan M, Gingras D, Piquette-Miller M, Bendayan R. Cellular localization and functional expression of P-glycoprotein in rat astrocyte cultures. J Neurochem. 2004;89(3):788–800. doi: 10.1111/j.1471-4159.2004.02417.x. [DOI] [PubMed] [Google Scholar]
- 149.Ronaldson PT, Bendayan R. HIV-1 viral envelope glycoprotein gp120 triggers an inflammatory response in cultured rat astrocytes and regulates the functional expression of P-glycoprotein. Mol Pharmacol. 2006;70(3):1087–1098. doi: 10.1124/mol.106.025973. [DOI] [PubMed] [Google Scholar]
- 150.Nies AT, Jedlitschky G, Konig J, Herold-Mende C, Steiner HH, Schmitt HP, et al. Expression and immunolocalization of the multidrug resistance proteins, MRP1-MRP6 (ABCC1-ABCC6), in human brain. Neuroscience. 2004;129(2):349–360. doi: 10.1016/j.neuroscience.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 151.Hirrlinger J, Moeller H, Kirchhoff F, Dringen R. Expression of multidrug resistance proteins (Mrps) in astrocytes of the mouse brain: a single cell RT-PCR study. Neurochem Res. 2005;30(10):1237–1244. doi: 10.1007/s11064-005-8795-y. [DOI] [PubMed] [Google Scholar]
- 152.Minich T, Riemer J, Schulz JB, Wielinga P, Wijnholds J, Dringen R. The multidrug resistance protein 1 (Mrp1), but not Mrp5, mediates export of glutathione and glutathione disulfide from brain astrocytes. J Neurochem. 2006;97(2):373–384. doi: 10.1111/j.1471-4159.2006.03737.x. [DOI] [PubMed] [Google Scholar]
- 153.Ronaldson PT, Ashraf T, Bendayan R. Regulation of multidrug resistance protein 1 by tumor necrosis factor alpha in cultured glial cells: involvement of nuclear factor-kappaB and c-Jun N-terminal kinase signaling pathways. Mol Pharmacol. 2010;77(4):644–659. doi: 10.1124/mol.109.059410. [DOI] [PubMed] [Google Scholar]
- 154.Bronger H, Konig J, Kopplow K, Steiner HH, Ahmadi R, Herold-Mende C, et al. ABCC drug efflux pumps and organic anion uptake transporters in human gliomas and the blood-tumor barrier. Cancer Res. 2005;65(24):11419–11428. doi: 10.1158/0008-5472.CAN-05-1271. [DOI] [PubMed] [Google Scholar]
- 155.del Rio-Hortega P Microglia. Cytology and Cellular Pathology of the Nervous System. In: Penfield W, editor. Hoecher. 1932. pp. 481–584. [Google Scholar]
- 156.Raivich G, Bohatschek M, Kloss CU, Werner A, Jones LL, Kreutzberg GW. Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Brain Res Rev. 1999;30(1):77–105. doi: 10.1016/s0165-0173(99)00007-7. [DOI] [PubMed] [Google Scholar]
- 157.Dickson DW, Mattiace LA, Kure K, Hutchins K, Lyman WD, Brosnan CF. Microglia in human disease, with an emphasis on acquired immune deficiency syndrome. Lab Invest. 1991;64(2):135–156. [PubMed] [Google Scholar]
- 158.Speth C, Dierich MP, Sopper S. HIV-infection of the central nervous system: the tightrope walk of innate immunity. Mol Immunol. 2005;42(2):213–228. doi: 10.1016/j.molimm.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 159.Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61(11):1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 160.Rivest S. Molecular insights on the cerebral innate immune system. Brain Behav Immun. 2003;17(1):13–19. doi: 10.1016/s0889-1591(02)00055-7. [DOI] [PubMed] [Google Scholar]
- 161.Deierborg T, Roybon L, Inacio AR, Pesic J, Brundin P. Brain injury activates microglia that induce neural stem cell proliferation ex vivo and promote differentiation of neurosphere-derived cells into neurons and oligodendrocytes. Neuroscience. 2010;171(4):1386–1396. doi: 10.1016/j.neuroscience.2010.09.045. [DOI] [PubMed] [Google Scholar]
- 162.Faustino JV, Wang X, Johnson CE, Klibanov A, Derugin N, Wendland MF, et al. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci. 2011;31(36):12992–13001. doi: 10.1523/JNEUROSCI.2102-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wei Z, Chigurupati S, Arumugam TV, Jo DG, Li H, Chan SL. Notch activation enhances the microglia-mediated inflammatory response associated with focal cerebral ischemia. Stroke. 2011;42(9):2589–2594. doi: 10.1161/STROKEAHA.111.614834. [DOI] [PubMed] [Google Scholar]
- 164.Aloisi F. Immune function of microglia. Glia. 2001;36(2):165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- 165.Eder C. Ion channels in microglia (brain macrophages) Am J Physiol. 1998;275(2 Pt 1):C327–C342. doi: 10.1152/ajpcell.1998.275.2.C327. [DOI] [PubMed] [Google Scholar]
- 166.Faff L, Ohlemeyer C, Kettenmann H. Intracellular pH regulation in cultured microglial cells from mouse brain. J Neurosci Res. 1996;46(3):294–304. doi: 10.1002/(SICI)1097-4547(19961101)46:3<294::AID-JNR2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 167.Klee R, Heinemann U, Eder C. Voltage-gated proton currents in microglia of distinct morphology and functional state. Neuroscience. 1999;91(4):1415–1424. doi: 10.1016/s0306-4522(98)00710-6. [DOI] [PubMed] [Google Scholar]
- 168.Newell EW, Schlichter LC. Integration of K+ and Cl- currents regulate steady-state and dynamic membrane potentials in cultured rat microglia. J Physiol. 2005;567(Pt 3):869–890. doi: 10.1113/jphysiol.2005.092056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Eder C. Regulation of microglial behavior by ion channel activity. J Neurosci Res. 2005;81(3):314–321. doi: 10.1002/jnr.20476. [DOI] [PubMed] [Google Scholar]
- 170.Noda M, Nakanishi H, Nabekura J, Akaike N. AMPA-kainate subtypes of glutamate receptor in rat cerebral microglia. J Neurosci. 2000;20(1):251–258. doi: 10.1523/JNEUROSCI.20-01-00251.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Maher F. Immunolocalization of GLUT1 and GLUT3 glucose transporters in primary cultured neurons and glia. J Neurosci Res. 1995;42(4):459–469. doi: 10.1002/jnr.490420404. [DOI] [PubMed] [Google Scholar]
- 172.Lee G, Schlichter L, Bendayan M, Bendayan R. Functional expression of P-glycoprotein in rat brain microglia. J Pharmacol Exp Ther. 2001;299(1):204–212. [PubMed] [Google Scholar]
- 173.Ronaldson PT, Lee G, Dallas S, Bendayan R. Involvement of P-glycoprotein in the transport of saquinavir and indinavir in rat brain microvessel endothelial and microglia cell lines. Pharm Res. 2004;21(5):811–818. doi: 10.1023/b:pham.0000026433.27773.47. [DOI] [PubMed] [Google Scholar]
- 174.Dallas S, Zhu X, Baruchel S, Schlichter L, Bendayan R. Functional expression of the multidrug resistance protein 1 in microglia. J Pharmacol Exp Ther. 2003;307(1):282–290. doi: 10.1124/jpet.103.054304. [DOI] [PubMed] [Google Scholar]
- 175.Dallas S, Ronaldson PT, Bendayan M, Bendayan R. Multidrug resistance protein 1-mediated transport of saquinavir by microglia. Neuroreport. 2004;15(7):1183–1186. doi: 10.1097/00001756-200405190-00020. [DOI] [PubMed] [Google Scholar]
- 176.Dore-Duffy P, Cleary K. Morphology and properties of pericytes. Methods Mol Biol. 2011;686:49–68. doi: 10.1007/978-1-60761-938-3_2. [DOI] [PubMed] [Google Scholar]
- 177.Hori S, Ohtsuki S, Hosoya K, Nakashima E, Terasaki T. A pericyte-derived angiopoietin-1 multimeric complex induces occludin gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. J Neurochem. 2004;89(2):503–513. doi: 10.1111/j.1471-4159.2004.02343.x. [DOI] [PubMed] [Google Scholar]
- 178.Al Ahmad A, Taboada CB, Gassmann M, Ogunshola OO. Astrocytes and pericytes differentially modulate blood-brain barrier characteristics during development and hypoxic insult. J Cereb Blood Flow Metab. 2011;31(2):693–705. doi: 10.1038/jcbfm.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gonul E, Duz B, Kahraman S, Kayali H, Kubar A, Timurkaynak E. Early pericyte response to brain hypoxia in cats: an ultrastructural study. Microvasc Res. 2002;64(1):116–119. doi: 10.1006/mvre.2002.2413. [DOI] [PubMed] [Google Scholar]
- 180.Dore-Duffy P, Owen C, Balabanov R, Murphy S, Beaumont T, Rafols JA. Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc Res. 2000;60(1):55–69. doi: 10.1006/mvre.2000.2244. [DOI] [PubMed] [Google Scholar]
- 181.Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468(7323):557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 182.Berezowski V, Landry C, Dehouck MP, Cecchelli R, Fenart L. Contribution of glial cells and pericytes to the mRNA profiles of P-glycoprotein and multidrug resistance-associated proteins in an in vitro model of the blood-brain barrier. Brain Res. 2004;1018(1):1–9. doi: 10.1016/j.brainres.2004.05.092. [DOI] [PubMed] [Google Scholar]
- 183.Ben Menachem E, Johansson BB, Svensson TH. Increased vulnerability of the blood-brain barrier to acute hypertension following depletion of brain noradrenaline. J Neural Transm. 1982;53(2–3):159–167. doi: 10.1007/BF01243407. [DOI] [PubMed] [Google Scholar]
- 184.Cohen Z, Molinatti G, Hamel E. Astroglial and vascular interactions of noradrenaline terminals in the rat cerebral cortex. J Cereb Blood Flow Metab. 1997;17(8):894–904. doi: 10.1097/00004647-199708000-00008. [DOI] [PubMed] [Google Scholar]
- 185.Cohen Z, Bonvento G, Lacombe P, Hamel E. Serotonin in the regulation of brain microcirculation. Prog Neurobiol. 1996;50(4):335–362. doi: 10.1016/s0301-0082(96)00033-0. [DOI] [PubMed] [Google Scholar]
- 186.Vaucher E, Hamel E. Cholinergic basal forebrain neurons project to cortical microvessels in the rat: electron microscopic study with anterogradely transported Phaseolus vulgaris leucoagglutinin and choline acetyltransferase immunocytochemistry. J Neurosci. 1995;15(11):7427–7441. doi: 10.1523/JNEUROSCI.15-11-07427.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tong XK, Hamel E. Regional cholinergic denervation of cortical microvessels and nitric oxide synthase-containing neurons in Alzheimer’s disease. Neuroscience. 1999;92(1):163–175. doi: 10.1016/s0306-4522(98)00750-7. [DOI] [PubMed] [Google Scholar]
- 188.Vaucher E, Tong XK, Cholet N, Lantin S, Hamel E. GABA neurons provide a rich input to microvessels but not nitric oxide neurons in the rat cerebral cortex: a means for direct regulation of local cerebral blood flow. J Comp Neurol. 2000;421(2):161–171. [PubMed] [Google Scholar]
- 189.Buxton RB, Frank LR. A model for the coupling between cerebral blood flow and oxygen metabolism during neural stimulation. J Cereb Blood Flow Metab. 1997;17(1):64–72. doi: 10.1097/00004647-199701000-00009. [DOI] [PubMed] [Google Scholar]
- 190.Paemeleire K. The cellular basis of neurovascular metabolic coupling. Acta Neurol Belg. 2002;102(4):153–157. [PubMed] [Google Scholar]
- 191.Cucullo L, Couraud PO, Weksler B, Romero IA, Hossain M, Rapp E, et al. Immortalized human brain endothelial cells and flow-based vascular modeling: a marriage of convenience for rational neurovascular studies. J Cereb Blood Flow Metab. 2008;28(2):312–328. doi: 10.1038/sj.jcbfm.9600525. [DOI] [PubMed] [Google Scholar]
- 192.Vital SA, Terao S, Nagai M, Granger DN. Mechanisms underlying the cerebral microvascular responses to angiotensin II-induced hypertension. Microcirculation. 2010;17(8):641–649. doi: 10.1111/j.1549-8719.2010.00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Shen Q, Du F, Huang S, Duong TQ. Spatiotemporal characteristics of postischemic hyperperfusion with respect to changes in T1, T2, diffusion, angiography, and blood-brain barrier permeability. J Cereb Blood Flow Metab. 2011;31(10):2076–2085. doi: 10.1038/jcbfm.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sood RR, Taheri S, Candelario-Jalil E, Estrada EY, Rosenberg GA. Early beneficial effect of matrix metalloproteinase inhibition on blood-brain barrier permeability as measured by magnetic resonance imaging countered by impaired long-term recovery after stroke in rat brain. J Cereb Blood Flow Metab. 2008;28(2):431–438. doi: 10.1038/sj.jcbfm.9600534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Del Zoppo GJ. The neurovascular unit, matrix proteases, and innate inflammation. Ann N Y Acad Sci. 2010;1207:46–49. doi: 10.1111/j.1749-6632.2010.05760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hynes RO, Lander AD. Contact and adhesive specificities in the associations, migrations, and targeting of cells and axons. Cell. 1992;68(2):303–322. doi: 10.1016/0092-8674(92)90472-o. [DOI] [PubMed] [Google Scholar]
- 197.Tilling T, Engelbertz C, Decker S, Korte D, Huwel S, Galla HJ. Expression and adhesive properties of basement membrane proteins in cerebral capillary endothelial cell cultures. Cell Tissue Res. 2002;310(1):19–29. doi: 10.1007/s00441-002-0604-1. [DOI] [PubMed] [Google Scholar]
- 198.Tilling T, Korte D, Hoheisel D, Galla HJ. Basement membrane proteins influence brain capillary endothelial barrier function in vitro. J Neurochem. 1998;71(3):1151–1157. doi: 10.1046/j.1471-4159.1998.71031151.x. [DOI] [PubMed] [Google Scholar]
- 199.Savettieri G, Di L I, Catania C, Licata L, Pitarresi GL, D’Agostino S, et al. Neurons and ECM regulate occludin localization in brain endothelial cells. Neuroreport. 2000;11(5):1081–1084. doi: 10.1097/00001756-200004070-00035. [DOI] [PubMed] [Google Scholar]
- 200.Baldwin K, Orr S, Briand M, Piazza C, Veydt A, McCoy S. Acute ischemic stroke update. Pharmacotherapy. 2010;30(5):493–514. doi: 10.1592/phco.30.5.493. [DOI] [PubMed] [Google Scholar]
- 201.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, et al. Heart disease and stroke statistics–2011 update: a report from the American Heart Association. Circulation. 2011;123(4):e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Barreto G, White RE, Ouyang Y, Xu L, Giffard RG. Astrocytes: targets for neuroprotection in stroke. Cent Nerv Syst Agents Med Chem. 2011;11(2):164–173. doi: 10.2174/187152411796011303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4(5):399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 204.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67(2):181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Liu S, Levine SR, Winn HR. Targeting ischemic penumbra: part I – from pathophysiology to therapeutic strategy. J Exp Stroke Transl Med. 2010;3(1):47–55. doi: 10.6030/1939-067x-3.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Adibhatla RM, Hatcher JF. Tissue plasminogen activator (tPA) and matrix metalloproteinases in the pathogenesis of stroke: therapeutic strategies. CNS Neurol Disord Drug Targets. 2008;7(3):243–253. doi: 10.2174/187152708784936608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Adibhatla RM, Hatcher JF, Dempsey RJ. Lipids and lipidomics in brain injury and diseases. AAPS J. 2006;8(2):E314–E321. doi: 10.1007/BF02854902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Adibhatla RM, Hatcher JF, Larsen EC, Chen X, Sun D, Tsao FH. CDP-choline significantly restores phosphatidylcholine levels by differentially affecting phospholipase A2 and CTP: phosphocholine cytidylyltransferase after stroke. J Biol Chem. 2006;281(10):6718–6725. doi: 10.1074/jbc.M512112200. [DOI] [PubMed] [Google Scholar]
- 209.Candelario-Jalil E. Injury and repair mechanisms in ischemic stroke: considerations for the development of novel neurotherapeutics. Curr Opin Investig Drugs. 2009;10(7):644–654. [PubMed] [Google Scholar]
- 210.Jahan R, Vinuela F. Treatment of acute ischemic stroke: intravenous and endovascular therapies. Expert Rev Cardiovasc Ther. 2009;7(4):375–387. doi: 10.1586/erc.09.13. [DOI] [PubMed] [Google Scholar]
- 211.Kalaria RN, Ballard C. Stroke and cognition. Curr Atheroscler Rep. 2001;3(4):334–339. doi: 10.1007/s11883-001-0028-5. [DOI] [PubMed] [Google Scholar]
- 212.Lobysheva NV, Tonshin AA, Selin AA, Yaguzhinsky LS, Nartsissov YR. Diversity of neurodegenerative processes in the model of brain cortex tissue ischemia. Neurochem Int. 2009;54(5–6):322–329. doi: 10.1016/j.neuint.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 213.Wong CH, Crack PJ. Modulation of neuro-inflammation and vascular response by oxidative stress following cerebral ischemia-reperfusion injury. Curr Med Chem. 2008;15(1):1–14. doi: 10.2174/092986708783330665. [DOI] [PubMed] [Google Scholar]
- 214.Schild L, Reiser G. Oxidative stress is involved in the permeabilization of the inner membrane of brain mitochondria exposed to hypoxia/reoxygenation and low micromolar Ca2+ FEBS J. 2005;272(14):3593–3601. doi: 10.1111/j.1742-4658.2005.04781.x. [DOI] [PubMed] [Google Scholar]
- 215.Witt KA, Mark KS, Huber J, Davis TP. Hypoxia-inducible factor and nuclear factor kappa-B activation in blood-brain barrier endothelium under hypoxic/reoxygenation stress. J Neurochem. 2005;92(1):203–214. doi: 10.1111/j.1471-4159.2004.02871.x. [DOI] [PubMed] [Google Scholar]
- 216.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10(11):1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- 217.Yang Y, Rosenberg GA. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011;42(11):3323–3328. doi: 10.1161/STROKEAHA.110.608257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zhao LR, Navalitloha Y, Singhal S, Mehta J, Piao CS, Guo WP, et al. Hematopoietic growth factors pass through the blood-brain barrier in intact rats. Exp Neurol. 2007;204(2):569–573. doi: 10.1016/j.expneurol.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Svedin P, Hagberg H, Savman K, Zhu C, Mallard C. Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia-ischemia. J Neurosci. 2007;27(7):1511–1518. doi: 10.1523/JNEUROSCI.4391-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Nagel S, Su Y, Horstmann S, Heiland S, Gardner H, Koziol J, et al. Minocycline and hypothermia for reperfusion injury after focal cerebral ischemia in the rat: effects on BBB breakdown and MMP expression in the acute and subacute phase. Brain Res. 2008;1188:198–206. doi: 10.1016/j.brainres.2007.10.052. [DOI] [PubMed] [Google Scholar]
- 221.Wang G, Guo Q, Hossain M, Fazio V, Zeynalov E, Janigro D, et al. Bone marrow-derived cells are the major source of MMP-9 contributing to blood-brain barrier dysfunction and infarct formation after ischemic stroke in mice. Brain Res. 2009;1294:183–192. doi: 10.1016/j.brainres.2009.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Bauer AT, Burgers HF, Rabie T, Marti HH. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J Cereb Blood Flow Metab. 2010;30(4):837–848. doi: 10.1038/jcbfm.2009.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Kumari R, Willing LB, Patel SD, Baskerville KA, Simpson IA. Increased cerebral matrix metalloprotease-9 activity is associated with compromised recovery in the diabetic db/db mouse following a stroke. J Neurochem. 2011;119(5):1029–1040. doi: 10.1111/j.1471-4159.2011.07487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Brouns R, Wauters A, De Surgeloose D, Marien P, De Deyn PP. Biochemical markers for blood-brain barrier dysfunction in acute ischemic stroke correlate with evolution and outcome. Eur Neurol. 2011;65(1):23–31. doi: 10.1159/000321965. [DOI] [PubMed] [Google Scholar]
- 225.Gu Y, Zheng G, Xu M, Li Y, Chen X, Zhu W, et al. Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J Neurochem. 2012;120(1):147–156. doi: 10.1111/j.1471-4159.2011.07542.x. [DOI] [PubMed] [Google Scholar]
- 226.Bhattacharjee AK, Nagashima T, Kondoh T, Tamaki N. Quantification of early blood-brain barrier disruption by in situ brain perfusion technique. Brain Res Brain Res Protoc. 2001;8(2):126–131. doi: 10.1016/s1385-299x(01)00094-0. [DOI] [PubMed] [Google Scholar]
- 227.Pfefferkorn T, Rosenberg GA. Closure of the blood-brain barrier by matrix metalloproteinase inhibition reduces rtPA-mediated mortality in cerebral ischemia with delayed reperfusion. Stroke. 2003;34(8):2025–2030. doi: 10.1161/01.STR.0000083051.93319.28. [DOI] [PubMed] [Google Scholar]
- 228.Nagaraja TN, Keenan KA, Fenstermacher JD, Knight RA. Acute leakage patterns of fluorescent plasma flow markers after transient focal cerebral ischemia suggest large openings in blood-brain barrier. Microcirculation. 2008;15(1):1–14. doi: 10.1080/10739680701409811. [DOI] [PubMed] [Google Scholar]
- 229.Strbian D, Durukan A, Tatlisumak T. Rodent models of hemorrhagic stroke. Curr Pharm Des. 2008;14(4):352–358. doi: 10.2174/138161208783497723. [DOI] [PubMed] [Google Scholar]
- 230.Fischer S, Gerriets T, Wessels C, Walberer M, Kostin S, Stolz E, et al. Extracellular RNA mediates endothelial-cell permeability via vascular endothelial growth factor. Blood. 2007;110(7):2457–2465. doi: 10.1182/blood-2006-08-040691. [DOI] [PubMed] [Google Scholar]
- 231.Yeh WL, Lu DY, Lin CJ, Liou HC, Fu WM. Inhibition of hypoxia-induced increase of blood-brain barrier permeability by YC-1 through the antagonism of HIF-1alpha accumulation and VEGF expression. Mol Pharmacol. 2007;72(2):440–449. doi: 10.1124/mol.107.036418. [DOI] [PubMed] [Google Scholar]
- 232.Lee SW, Kim WJ, Jun HO, Choi YK, Kim KW. Angiopoietin-1 reduces vascular endothelial growth factor-induced brain endothelial permeability via upregulation of ZO-2. Int J Mol Med. 2009;23(2):279–284. [PubMed] [Google Scholar]
- 233.Yamauchi A, Dohgu S, Nishioku T, Shuto H, Naito M, Tsuruo T, et al. An inhibitory role of nitric oxide in the dynamic regulation of the blood-brain barrier function. Cell Mol Neurobiol. 2007;27(3):263–270. doi: 10.1007/s10571-007-9139-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Heo JH, Han SW, Lee SK. Free radicals as triggers of brain edema formation after stroke. Free Radic Biol Med. 2005;39(1):51–70. doi: 10.1016/j.freeradbiomed.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 235.Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32(2):200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 236.Pillai DR, Dittmar MS, Baldaranov D, Heidemann RM, Henning EC, Schuierer G, et al. Cerebral ischemia-reperfusion injury in rats–a 3 T MRI study on biphasic blood-brain barrier opening and the dynamics of edema formation. J Cereb Blood Flow Metab. 2009;29(11):1846–1855. doi: 10.1038/jcbfm.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Wallace BK, Jelks KA, O’Donnell ME. Ischemia-induced stimulation of cerebral microvascular endothelial cell Na-K-Cl cotransport involves p38 and JNK MAP kinases. Am J Physiol Cell Physiol. 2011 doi: 10.1152/ajpcell.00261.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Lam TI, Wise PM, O’Donnell ME. Cerebral microvascular endothelial cell Na/H exchange: evidence for the presence of NHE1 and NHE2 isoforms and regulation by arginine vasopressin. Am J Physiol Cell Physiol. 2009;297(2):C278–C289. doi: 10.1152/ajpcell.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Vemula S, Roder KE, Yang T, Bhat GJ, Thekkumkara TJ, Abbruscato TJ. A functional role for sodium-dependent glucose transport across the blood-brain barrier during oxygen glucose deprivation. J Pharmacol Exp Ther. 2009;328(2):487–495. doi: 10.1124/jpet.108.146589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kim GW, Lewen A, Copin J, Watson BD, Chan PH. The cytosolic antioxidant, copper/zinc superoxide dismutase, attenuates blood-brain barrier disruption and oxidative cellular injury after photothrombotic cortical ischemia in mice. Neuroscience. 2001;105(4):1007–1018. doi: 10.1016/s0306-4522(01)00237-8. [DOI] [PubMed] [Google Scholar]
- 241.Strasser A, Stanimirovic D, Kawai N, McCarron RM, Spatz M. Hypoxia modulates free radical formation in brain microvascular endothelium. Acta Neurochir Suppl. 1997;70:8–11. doi: 10.1007/978-3-7091-6837-0_3. [DOI] [PubMed] [Google Scholar]
- 242.Nito C, Kamada H, Endo H, Niizuma K, Myer DJ, Chan PH. Role of the p38 mitogen-activated protein kinase/cytosolic phospholipase A2 signaling pathway in blood-brain barrier disruption after focal cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. 2008;28(10):1686–1696. doi: 10.1038/jcbfm.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Salvemini D, Doyle TM, Cuzzocrea S. Superoxide, peroxynitrite and oxidative/nitrative stress in inflammation. Biochem Soc Trans. 2006;34(Pt 5):965–970. doi: 10.1042/BST0340965. [DOI] [PubMed] [Google Scholar]
- 244.Schreibelt G, Kooij G, Reijerkerk A, van Doorn R, Gringhuis SI, van der PS, et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007;21(13):3666–3676. doi: 10.1096/fj.07-8329com. [DOI] [PubMed] [Google Scholar]
- 245.Wang X, Barone FC, Aiyar NV, Feuerstein GZ. Interleukin-1 receptor and receptor antagonist gene expression after focal stroke in rats. Stroke. 1997;28(1):155–161. doi: 10.1161/01.str.28.1.155. [DOI] [PubMed] [Google Scholar]
- 246.Petty MA, Lo EH. Junctional complexes of the blood-brain barrier: permeability changes in neuroinflammation. Prog Neurobiol. 2002;68(5):311–323. doi: 10.1016/s0301-0082(02)00128-4. [DOI] [PubMed] [Google Scholar]
- 247.McCarron RM, Wang L, Racke MK, McFarlin DE, Spatz M. Cytokine-regulated adhesion between encephalitogenic T lymphocytes and cerebrovascular endothelial cells. J Neuroimmunol. 1993;43(1–2):23–30. doi: 10.1016/0165-5728(93)90071-6. [DOI] [PubMed] [Google Scholar]
- 248.Stins MF, Gilles F, Kim KS. Selective expression of adhesion molecules on human brain microvascular endothelial cells. J Neuroimmunol. 1997;76(1–2):81–90. doi: 10.1016/s0165-5728(97)00036-2. [DOI] [PubMed] [Google Scholar]
- 249.Staykova M, Maxwell L, Willenborg D. Kinetics and polarization of the membrane expression of cytokine-induced ICAM-1 on rat brain endothelial cells. J Neuropathol Exp Neurol. 2000;59(2):120–128. doi: 10.1093/jnen/59.2.120. [DOI] [PubMed] [Google Scholar]
- 250.Poller B, Drewe J, Krahenbuhl S, Huwyler J, Gutmann H. Regulation of BCRP (ABCG2) and P-glycoprotein (ABCB1) by cytokines in a model of the human blood-brain barrier. Cell Mol Neurobiol. 2010;30(1):63–70. doi: 10.1007/s10571-009-9431-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Yu C, Argyropoulos G, Zhang Y, Kastin AJ, Hsuchou H, Pan W. Neuroinflammation activates Mdr1b efflux transport through NFkappaB: promoter analysis in BBB endothelia. Cell Physiol Biochem. 2008;22(5–6):745–756. doi: 10.1159/000185558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Ahishali B, Kaya M, Kalayci R, Uzun H, Bilgic B, Arican N, et al. Effects of lipopolysaccharide on the blood-brain barrier permeability in prolonged nitric oxide blockade-induced hypertensive rats. Int J Neurosci. 2005;115(2):151–168. doi: 10.1080/00207450590519030. [DOI] [PubMed] [Google Scholar]
- 253.Didier N, Romero IA, Creminon C, Wijkhuisen A, Grassi J, Mabondzo A. Secretion of interleukin-1beta by astrocytes mediates endothelin-1 and tumour necrosis factor-alpha effects on human brain microvascular endothelial cell permeability. J Neurochem. 2003;86(1):246–254. doi: 10.1046/j.1471-4159.2003.01829.x. [DOI] [PubMed] [Google Scholar]
- 254.Haseloff RF, Blasig IE, Bauer HC, Bauer H. In search of the astrocytic factor(s) modulating blood-brain barrier functions in brain capillary endothelial cells in vitro. Cell Mol Neurobiol. 2005;25(1):25–39. doi: 10.1007/s10571-004-1375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Wolburg H, Neuhaus J, Kniesel U, Krauss B, Schmid EM, Ocalan M, et al. Modulation of tight junction structure in blood-brain barrier endothelial cells. Effects of tissue culture, second messengers and cocultured astrocytes. J Cell Sci. 1994;107(Pt 5):1347–1357. doi: 10.1242/jcs.107.5.1347. [DOI] [PubMed] [Google Scholar]
- 256.Benarroch EE. Astrocyte-neuron interactions: implications for epilepsy. Neurology. 2009;73(16):1323–1327. doi: 10.1212/WNL.0b013e3181bd432d. [DOI] [PubMed] [Google Scholar]
- 257.Sievers J, Schmidtmayer J, Parwaresch R. Blood monocytes and spleen macrophages differentiate into microglia-like cells when cultured on astrocytes. Ann Anat. 1994;176(1):45–51. doi: 10.1016/s0940-9602(11)80414-0. [DOI] [PubMed] [Google Scholar]
- 258.Tanaka J, Maeda N. Microglial ramification requires nondiffusible factors derived from astrocytes. Exp Neurol. 1996;137(2):367–375. doi: 10.1006/exnr.1996.0038. [DOI] [PubMed] [Google Scholar]
- 259.Eder C, Klee R, Heinemann U. Distinct soluble astrocytic factors induce expression of outward K+ currents and ramification of brain macrophages. Neurosci Lett. 1997;226(3):147–150. doi: 10.1016/s0304-3940(97)00281-4. [DOI] [PubMed] [Google Scholar]
- 260.Woodman SE, Benveniste EN, Nath A, Berman JW. Human immunodeficiency virus type 1 TAT protein induces adhesion molecule expression in astrocytes. J Neurovirol. 1999;5(6):678–684. doi: 10.3109/13550289909021296. [DOI] [PubMed] [Google Scholar]
- 261.Toneatto S, Finco O, van der PH, Abrignani S, Annunziata P. Evidence of blood-brain barrier alteration and activation in HIV-1 gp120 transgenic mice. AIDS. 1999;13(17):2343–2348. doi: 10.1097/00002030-199912030-00005. [DOI] [PubMed] [Google Scholar]
- 262.Cowell RM, Xu H, Galasso JM, Silverstein FS. Hypoxic-ischemic injury induces macrophage inflammatory protein-1alpha expression in immature rat brain. Stroke. 2002;33(3):795–801. doi: 10.1161/hs0302.103740. [DOI] [PubMed] [Google Scholar]
- 263.Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Effects of the chemokine CCL2 on blood-brain barrier permeability during ischemia-reperfusion injury. J Cereb Blood Flow Metab. 2006;26(6):797–810. doi: 10.1038/sj.jcbfm.9600229. [DOI] [PubMed] [Google Scholar]
- 264.Strecker JK, Minnerup J, Gess B, Ringelstein EB, Schabitz WR, Schilling M. Monocyte chemoattractant protein-1-deficiency impairs the expression of IL-6, IL-1beta and G-CSF after transient focal ischemia in mice. PLoS One. 2011;6(10):e25863. doi: 10.1371/journal.pone.0025863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Lee SR, Guo SZ, Scannevin RH, Magliaro BC, Rhodes KJ, Wang X, et al. Induction of matrix metalloproteinase, cytokines and chemokines in rat cortical astrocytes exposed to plasminogen activators. Neurosci Lett. 2007;417(1):1–5. doi: 10.1016/j.neulet.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 266.Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36(2):180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- 267.John GR, Lee SC, Brosnan CF. Cytokines: powerful regulators of glial cell activation. Neuroscientist. 2003;9(1):10–22. doi: 10.1177/1073858402239587. [DOI] [PubMed] [Google Scholar]
- 268.Tuttolomondo A, Di Raimondo D, di Sciacca R, Pinto A, Licata G. Inflammatory cytokines in acute ischemic stroke. Curr Pharm Des. 2008;14(33):3574–3589. doi: 10.2174/138161208786848739. [DOI] [PubMed] [Google Scholar]
- 269.Buckwalter MS, Wyss-Coray T. Modelling neuroinflammatory phenotypes in vivo. J Neuroinflammation. 2004;1(1):10. doi: 10.1186/1742-2094-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Doyle KP, Cekanaviciute E, Mamer LE, Buckwalter MS. TGFbeta signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J Neuroinflammation. 2010;7:62. doi: 10.1186/1742-2094-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Orzylowska O, Oderfeld-Nowak B, Zaremba M, Januszewski S, Mossakowski M. Prolonged and concomitant induction of astroglial immunoreactivity of interleukin-1beta and interleukin-6 in the rat hippocampus after transient global ischemia. Neurosci Lett. 1999;263(1):72–76. doi: 10.1016/s0304-3940(99)00043-9. [DOI] [PubMed] [Google Scholar]
- 272.Lau LT, Yu AC. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J Neurotrauma. 2001;18(3):351–359. doi: 10.1089/08977150151071035. [DOI] [PubMed] [Google Scholar]
- 273.Mander P, Borutaite V, Moncada S, Brown GC. Nitric oxide from inflammatory-activated glia synergizes with hypoxia to induce neuronal death. J Neurosci Res. 2005;79(1–2):208–215. doi: 10.1002/jnr.20285. [DOI] [PubMed] [Google Scholar]
- 274.Love S. Oxidative stress in brain ischemia. Brain Pathol. 1999;9(1):119–131. doi: 10.1111/j.1750-3639.1999.tb00214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Gursoy-Ozdemir Y, Can A, Dalkara T. Reperfusion-induced oxidative/nitrative injury to neurovascular unit after focal cerebral ischemia. Stroke. 2004;35(6):1449–1453. doi: 10.1161/01.STR.0000126044.83777.f4. [DOI] [PubMed] [Google Scholar]
- 276.Yasuda Y, Tateishi N, Shimoda T, Satoh S, Ogitani E, Fujita S. Relationship between S100beta and GFAP expression in astrocytes during infarction and glial scar formation after mild transient ischemia. Brain Res. 2004;1021(1):20–31. doi: 10.1016/j.brainres.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 277.Jiang J, Wang W, Sun YJ, Hu M, Li F, Zhu DY. Neuroprotective effect of curcumin on focal cerebral ischemic rats by preventing blood-brain barrier damage. Eur J Pharmacol. 2007;561(1–3):54–62. doi: 10.1016/j.ejphar.2006.12.028. [DOI] [PubMed] [Google Scholar]
- 278.Dallasta LM, Pisarov LA, Esplen JE, Werley JV, Moses AV, Nelson JA, et al. Blood-brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am J Pathol. 1999;155(6):1915–1927. doi: 10.1016/S0002-9440(10)65511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia potentiate damage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke. 2006;37(4):1087–1093. doi: 10.1161/01.STR.0000206281.77178.ac. [DOI] [PubMed] [Google Scholar]
- 280.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, et al. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2(7):788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 281.Dziewulska D, Mossakowski MJ. Cellular expression of tumor necrosis factor a and its receptors in human ischemic stroke. Clin Neuropathol. 2003;22(1):35–40. [PubMed] [Google Scholar]
- 282.Clausen BH, Lambertsen KL, Babcock AA, Holm TH, Dagnaes-Hansen F, Finsen B. Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J Neuroinflammation. 2008;5:46. doi: 10.1186/1742-2094-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008;28(9):2221–2230. doi: 10.1523/JNEUROSCI.5643-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Akundi RS, Candelario-Jalil E, Hess S, Hull M, Lieb K, Gebicke-Haerter PJ, et al. Signal transduction pathways regulating cyclooxygenase-2 in lipopolysaccharide-activated primary rat microglia. Glia. 2005;51(3):199–208. doi: 10.1002/glia.20198. [DOI] [PubMed] [Google Scholar]
- 285.Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998;18(5):1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Piao CS, Kim JB, Han PL, Lee JK. Administration of the p38 MAPK inhibitor SB203580 affords brain protection with a wide therapeutic window against focal ischemic insult. J Neurosci Res. 2003;73(4):537–544. doi: 10.1002/jnr.10671. [DOI] [PubMed] [Google Scholar]
- 287.Fleegal MA, Hom S, Borg LK, Davis TP. Activation of PKC modulates blood-brain barrier endothelial cell permeability changes induced by hypoxia and posthypoxic reoxygenation. Am J Physiol Heart Circ Physiol. 2005;289(5):H2012–H2019. doi: 10.1152/ajpheart.00495.2005. [DOI] [PubMed] [Google Scholar]
- 288.Saito K, Takeshita K, Ueda J, Ozawa T. Two reaction sites of a spin label, TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl), with hydroxyl radical. J Pharm Sci. 2003;92(2):275–280. doi: 10.1002/jps.10304. [DOI] [PubMed] [Google Scholar]
- 289.Tsuhako MH, Augusto O, Linares E, Chadi G, Giorgio S, Pereira CA. Tempol ameliorates murine viral encephalomyelitis by preserving the blood-brain barrier, reducing viral load, and lessening inflammation. Free Radic Biol Med. 2010;48(5):704–712. doi: 10.1016/j.freeradbiomed.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Zhelev Z, Bakalova R, Aoki I, Matsumoto K, Gadjeva V, Anzai K, et al. Nitroxyl radicals for labeling of conventional therapeutics and noninvasive magnetic resonance imaging of their permeability for blood-brain barrier: relationship between structure, blood clearance, and MRI signal dynamic in the brain. Mol Pharm. 2009;6(2):504–512. doi: 10.1021/mp800175k. [DOI] [PubMed] [Google Scholar]
- 291.Cuzzocrea S, McDonald MC, Mazzon E, Filipe HM, Costantino G, Caputi AP, et al. Beneficial effects of tempol, a membrane-permeable radical scavenger, in a rodent model of splanchnic artery occlusion and reperfusion. Shock. 2000;14(2):150–156. doi: 10.1097/00024382-200014020-00013. [DOI] [PubMed] [Google Scholar]
- 292.Rak R, Chao DL, Pluta RM, Mitchell JB, Oldfield EH, Watson JC. Neuroprotection by the stable nitroxide Tempol during reperfusion in a rat model of transient focal ischemia. J Neurosurg. 2000;92(4):646–651. doi: 10.3171/jns.2000.92.4.0646. [DOI] [PubMed] [Google Scholar]
- 293.Kwon TH, Chao DL, Malloy K, Sun D, Alessandri B, Bullock MR. Tempol, a novel stable nitroxide, reduces brain damage and free radical production, after acute subdural hematoma in the rat. J Neurotrauma. 2003;20(4):337–345. doi: 10.1089/089771503765172291. [DOI] [PubMed] [Google Scholar]
- 294.Deng-Bryant Y, Singh IN, Carrico KM, Hall ED. Neuroprotective effects of tempol, a catalytic scavenger of peroxynitrite-derived free radicals, in a mouse traumatic brain injury model. J Cereb Blood Flow Metab. 2008;28(6):1114–1126. doi: 10.1038/jcbfm.2008.10. [DOI] [PubMed] [Google Scholar]
- 295.van Vliet EA, Redeker S, Aronica E, Edelbroek PM, Gorter JA. Expression of multidrug transporters MRP1, MRP2, and BCRP shortly after status epilepticus, during the latent period, and in chronic epileptic rats. Epilepsia. 2005;46(10):1569–1580. doi: 10.1111/j.1528-1167.2005.00250.x. [DOI] [PubMed] [Google Scholar]
- 296.Hayashi K, Pu H, Andras IE, Eum SY, Yamauchi A, Hennig B, et al. HIV-TAT protein upregulates expression of multidrug resistance protein 1 in the blood-brain barrier. J Cereb Blood Flow Metab. 2006;26(8):1052–1065. doi: 10.1038/sj.jcbfm.9600254. [DOI] [PubMed] [Google Scholar]
- 297.Hayashi K, Pu H, Tian J, Andras IE, Lee YW, Hennig B, et al. HIV-Tat protein induces P-glycoprotein expression in brain microvascular endothelial cells. J Neurochem. 2005;93(5):1231–1241. doi: 10.1111/j.1471-4159.2005.03114.x. [DOI] [PubMed] [Google Scholar]
- 298.Seelbach MJ, Brooks TA, Egleton RD, Davis TP. Peripheral inflammatory hyperalgesia modulates morphine delivery to the brain: a role for P-glycoprotein. J Neurochem. 2007;102(5):1677–1690. doi: 10.1111/j.1471-4159.2007.04644.x. [DOI] [PubMed] [Google Scholar]
- 299.Murozono M, Matsumoto S, Okada S, Nagaoka D, Isshiki A, Watanabe Y. Reduction of brain infarction induced by a transient brain ischemia in mdr1a knockout mice. Neurochem Res. 2009;34(9):1555–1561. doi: 10.1007/s11064-009-9943-6. [DOI] [PubMed] [Google Scholar]
- 300.Murozono M, Matsumoto S, Matsumoto E, Isshiki A, Watanabe Y. Neuroprotective and neurotoxic effects of cyclosporine A on transient focal ischemia in mdr1a knockout mice. Eur J Pharmacol. 2004;498(1–3):115–118. doi: 10.1016/j.ejphar.2004.06.068. [DOI] [PubMed] [Google Scholar]
- 301.Choo EF, Kurnik D, Muszkat M, Ohkubo T, Shay SD, Higginbotham JN, et al. Differential in vivo sensitivity to inhibition of P-glycoprotein located in lymphocytes, testes, and the blood-brain barrier. J Pharmacol Exp Ther. 2006;317(3):1012–1018. doi: 10.1124/jpet.105.099648. [DOI] [PubMed] [Google Scholar]
- 302.Kaye SB. Reversal of drug resistance in ovarian cancer: where do we go from here? J Clin Oncol. 2008;26(16):2616–2618. doi: 10.1200/JCO.2008.16.2123. [DOI] [PubMed] [Google Scholar]
- 303.Kannan P, John C, Zoghbi SS, Halldin C, Gottesman MM, Innis RB, et al. Imaging the function of P-glycoprotein with radiotracers: pharmacokinetics and in vivo applications. Clin Pharmacol Ther. 2009;86(4):368–377. doi: 10.1038/clpt.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Kassan M, Montero MJ, Sevilla MA. In vitro antioxidant activity of pravastatin provides vascular protection. Eur J Pharmacol. 2010;630(1–3):107–111. doi: 10.1016/j.ejphar.2009.12.037. [DOI] [PubMed] [Google Scholar]
- 305.Noe B, Hagenbuch B, Stieger B, Meier PJ. Isolation of a multispecific organic anion and cardiac glycoside transporter from rat brain. Proc Natl Acad Sci U S A. 1997;94(19):10346–10350. doi: 10.1073/pnas.94.19.10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Egleton RD, Davis TP. Transport of the delta-opioid receptor agonist [D-penicillamine2,5] enkephalin across the blood-brain barrier involves transcytosis1. J Pharm Sci. 1999;88(4):392–397. doi: 10.1021/js980410+. [DOI] [PubMed] [Google Scholar]
- 307.Dagenais C, Graff CL, Pollack GM. Variable modulation of opioid brain uptake by P-glycoprotein in mice. Biochem Pharmacol. 2004;67(2):269–276. doi: 10.1016/j.bcp.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 308.Sharom FJ, Yu X, Lu P, Liu R, Chu JW, Szabo K, et al. Interaction of the P-glycoprotein multidrug transporter (MDR1) with high affinity peptide chemosensitizers in isolated membranes, reconstituted systems, and intact cells. Biochem Pharmacol. 1999;58(4):571–586. doi: 10.1016/s0006-2952(99)00139-2. [DOI] [PubMed] [Google Scholar]
- 309.Pepper MS. Transforming growth factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 1997;8(1):21–43. doi: 10.1016/s1359-6101(96)00048-2. [DOI] [PubMed] [Google Scholar]
- 310.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 311.Lebrin F, Deckers M, Bertolino P, ten Dijke P. TGF-beta receptor function in the endothelium. Cardiovasc Res. 2005;65(3):599–608. doi: 10.1016/j.cardiores.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 312.Wu X, Ma J, Han JD, Wang N, Chen YG. Distinct regulation of gene expression in human endothelial cells by TGF-beta and its receptors. Microvasc Res. 2006;71(1):12–19. doi: 10.1016/j.mvr.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 313.Seki T, Yun J, Oh SP. Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circ Res. 2003;93(7):682–689. doi: 10.1161/01.RES.0000095246.40391.3B. [DOI] [PubMed] [Google Scholar]
- 314.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002;21(7):1743–1753. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Watabe T, Nishihara A, Mishima K, Yamashita J, Shimizu K, Miyazawa K, et al. TGF-beta receptor kinase inhibitor enhances growth and integrity of embryonic stem cell-derived endothelial cells. J Cell Biol. 2003;163(6):1303–1311. doi: 10.1083/jcb.200305147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Ishihara H, Kubota H, Lindberg RL, Leppert D, Gloor SM, Errede M, et al. Endothelial cell barrier impairment induced by glioblastomas and transforming growth factor beta2 involves matrix metalloproteinases and tight junction proteins. J Neuropathol Exp Neurol. 2008;67(5):435–448. doi: 10.1097/NEN.0b013e31816fd622. [DOI] [PubMed] [Google Scholar]
- 317.Dohgu S, Yamauchi A, Takata F, Naito M, Tsuruo T, Higuchi S, et al. Transforming growth factor-beta1 upregulates the tight junction and P-glycoprotein of brain microvascular endothelial cells. Cell Mol Neurobiol. 2004;24(3):491–497. doi: 10.1023/B:CEMN.0000022776.47302.ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53(2):245–282. [PubMed] [Google Scholar]
- 319.Junge CE, Lee CJ, Hubbard KB, Zhang Z, Olson JJ, Hepler JR, et al. Protease-activated receptor-1 in human brain: localization and functional expression in astrocytes. Exp Neurol. 2004;188(1):94–103. doi: 10.1016/j.expneurol.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 320.Bushell TJ, Plevin R, Cobb S, Irving AJ. Characterization of proteinase-activated receptor 2 signalling and expression in rat hippocampal neurons and astrocytes. Neuropharmacology. 2006;50(6):714–725. doi: 10.1016/j.neuropharm.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 321.Noorbakhsh F, Tsutsui S, Vergnolle N, Boven LA, Shariat N, Vodjgani M, et al. Proteinase-activated receptor 2 modulates neuroinflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J Exp Med. 2006;203(2):425–435. doi: 10.1084/jem.20052148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.McCoy KL, Traynelis SF, Hepler JR. PAR1 and PAR2 couple to overlapping and distinct sets of G proteins and linked signaling pathways to differentially regulate cell physiology. Mol Pharmacol. 2010;77(6):1005–1015. doi: 10.1124/mol.109.062018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Pompili E, Fabrizi C, Nori SL, Panetta B, Geloso MC, Corvino V, et al. Protease-activated receptor-1 expression in rat microglia after trimethyltin treatment. J Histochem Cytochem. 2011;59(3):302–311. doi: 10.1369/0022155410397996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Domotor E, Bartha K, Machovich R, Adam-Vizi V. Protease-activated receptor-2 (PAR-2) in brain microvascular endothelium and its regulation by plasmin and elastase. J Neurochem. 2002;80(5):746–754. doi: 10.1046/j.0022-3042.2002.00759.x. [DOI] [PubMed] [Google Scholar]
- 325.Kim YV, Di Cello F, Hillaire CS, Kim KS. Differential Ca2+ signaling by thrombin and protease-activated receptor-1-activating peptide in human brain microvascular endothelial cells. Am J Physiol Cell Physiol. 2004;286(1):C31–C42. doi: 10.1152/ajpcell.00157.2003. [DOI] [PubMed] [Google Scholar]
- 326.Yamamoto C, Sugato M, Fujiwara Y, Kaji T. Selective promotion of plasminogen activator inhibitor-1 secretion by activation of proteinase-activated receptor-1 in cultured human brain microvascular pericytes: comparison with endothelial cells. Biol Pharm Bull. 2005;28(2):208–211. doi: 10.1248/bpb.28.208. [DOI] [PubMed] [Google Scholar]
- 327.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109(8):3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 328.Guo H, Singh I, Wang Y, Deane R, Barrett T, Fernandez JA, et al. Neuroprotective activities of activated protein C mutant with reduced anticoagulant activity. Eur J Neurosci. 2009;29(6):1119–1130. doi: 10.1111/j.1460-9568.2009.06664.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 329.Fabrizi C, Pompili E, Panetta B, Nori SL, Fumagalli L. Protease-activated receptor-1 regulates cytokine production and induces the suppressor of cytokine signaling-3 in microglia. Int J Mol Med. 2009;24(3):367–371. doi: 10.3892/ijmm_00000241. [DOI] [PubMed] [Google Scholar]
- 330.Thiyagarajan M, Fernandez JA, Lane SM, Griffin JH, Zlokovic BV. Activated protein C promotes neovascularization and neurogenesis in postischemic brain via protease-activated receptor 1. J Neurosci. 2008;28(48):12788–12797. doi: 10.1523/JNEUROSCI.3485-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Nagai M, Terao S, Yilmaz G, Yilmaz CE, Esmon CT, Watanabe E, et al. Roles of inflammation and the activated protein C pathway in the brain edema associated with cerebral venous sinus thrombosis. Stroke. 2010;41(1):147–152. doi: 10.1161/STROKEAHA.109.562983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Zlokovic BV, Griffin JH. Cytoprotective protein C pathways and implications for stroke and neurological disorders. Trends Neurosci. 2011;34(4):198–209. doi: 10.1016/j.tins.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Deane R, LaRue B, Sagare AP, Castellino FJ, Zhong Z, Zlokovic BV. Endothelial protein C receptor-assisted transport of activated protein C across the mouse blood-brain barrier. J Cereb Blood Flow Metab. 2009;29(1):25–33. doi: 10.1038/jcbfm.2008.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Mendioroz M, Fernandez-Cadenas I, Alvarez-Sabin J, Rosell A, Quiroga D, Cuadrado E, et al. Endogenous activated protein C predicts hemorrhagic transformation and mortality after tissue plasminogen activator treatment in stroke patients. Cerebrovasc Dis. 2009;28(2):143–150. doi: 10.1159/000225907. [DOI] [PubMed] [Google Scholar]
- 335.Kelso EB, Ferrell WR, Lockhart JC, Elias-Jones I, Hembrough T, Dunning L, et al. Expression and proinflammatory role of proteinase-activated receptor 2 in rheumatoid synovium: ex vivo studies using a novel proteinase-activated receptor 2 antagonist. Arthritis Rheum. 2007;56(3):765–771. doi: 10.1002/art.22423. [DOI] [PubMed] [Google Scholar]
- 336.Tindell AG, Kelso EB, Ferrell WR, Lockhart JC, Walsh DA, Dunning L, et al. Correlation of protease-activated receptor-2 expression and synovitis in rheumatoid and osteoarthritis. Rheumatol Int. 2011 doi: 10.1007/s00296-011-2102-9. [DOI] [PubMed] [Google Scholar]
- 337.Fagan SC, Cronic LE, Hess DC. Minocycline Development for Acute Ischemic Stroke. Transl Stroke Res. 2011;2(2):202–208. doi: 10.1007/s12975-011-0072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006;83(5):711–730. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Downes CE, Crack PJ. Neural injury following stroke: are Toll-like receptors the link between the immune system and the CNS? Br J Pharmacol. 2010;160(8):1872–1888. doi: 10.1111/j.1476-5381.2010.00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173(6):3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 341.Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175(7):4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- 342.Bowman CC, Rasley A, Tranguch SL, Marriott I. Cultured astrocytes express toll-like receptors for bacterial products. Glia. 2003;43(3):281–291. doi: 10.1002/glia.10256. [DOI] [PubMed] [Google Scholar]
- 343.Farina C, Krumbholz M, Giese T, Hartmann G, Aloisi F, Meinl E. Preferential expression and function of Toll-like receptor 3 in human astrocytes. J Neuroimmunol. 2005;159(1–2):12–19. doi: 10.1016/j.jneuroim.2004.09.009. [DOI] [PubMed] [Google Scholar]