

Abstract
Dysfunctions of genome caretaker genes contribute to genomic instability and tumor initiation. Because many of the caretaker genes are also essential for cell viability, permanent loss of function of these genes would prohibit further tumor progression. How essential caretaker genes contribute to tumorigenesis is not fully understood. Here, we report a “hit-and-run” mode of action for an essential caretaker gene in tumorigenesis. Using a BRCA2-interacting protein BCCIP as a platform, we found that a conditional BCCIP knockdown and concomitant p53 deletion caused rapid development of medulloblastomas, which bear a wide spectrum of alternations involving the Sonic hedgehog (Shh) pathway, consistent with a caretaker responsibility of BCCIP on genomic integrity. Surprisingly, the progressed tumors have spontaneously lost the transgenic BCCIP knockdown cassette and restored BCCIP expression. Thus, a transient down-regulation of BCCIP, but not necessarily a permanent mutation, is sufficient to initiate tumorigenesis. Once the malignant transformation has been accomplished and autonomous cancer growth has been established, BCCIP reverses its role from a tumor initiation suppressor to become a requisite for progression. This exemplifies a new type of tumor suppressor, which is distinct from the classical tumor suppressors that are often permanently abrogated during tumorigenesis. It has major implications on how a non-mutagenic or transient regulation of essential caretaker gene contributes to tumorigenesis. We further suggest that BCCIP represents a paradoxical class of modulators for tumorigenesis, as a Suppressor for Initiation but a Requisite for Progression (SIRP).
Keywords: DNA Repair, tumor suppressor, homologous recombination, genomic instability, BRCA2, BCCIP, Suppressor of Initiation and Requisite for Progression (SIRP), medulloblastoma
Introduction
Genomic instability is one of the enabling characters during tumorigenesis (1). It can be caused by dysregulations in DNA repair, compromised DNA replication fidelity, imprecise chromosome segregation during mitosis, improper cell cycle coordination, etc. (2). A tumor suppressor gene involved in the maintenance of genomic integrity is often referred to as a caretaker (3). Although defective caretakers may not immediately promote tumor growth, they cause stochastic genomic alterations that increase the risk of inactivating gatekeeper tumor suppressors and activations of growth-promoting oncogenes. The loss of gatekeeper functions and activation of pro-growth oncogenes then causes oncogenic transformation and imminent tumor growth and progression. Theoretically, once the defects of caretakers have caused the activation of autonomous pro-growth pathways and malignant transformation, the caretaker deficiency may no longer be a requisite for subsequent tumor progression. Thus caretaker defects may act in a “hit-and-run” manner to initiate tumorigenesis, and it is possible that only a transient dysfunction may be sufficient to trigger tumorigenesis. However, experimental evidence of such prediction is rare.
Many of the DNA repair genes are not only critical for the maintenance of genomic integrity, but also essential for cell viability and proliferation because of their fundamental involvement in DNA replication and mitosis. Therefore, a permanent inactivation of these genes may prohibit tumor progression despite its contribution to the initiation of tumorigenesis. There has been a conundrum on how these viability-essential caretaker genes modulate tumorigenesis. In this study, we constructed a conditional BCCIP knockdown transgenic mouse model. In this model, the expression of BCCIP is down-regulated by shRNA but can be restored when a spontaneous deletion of the knockdown cassette occurs due to growth pressure during tumor progression. We found that BCCIP modulates tumorigenesis in an un-conventional way: it serves as a tumor suppressor, but later is required for tumor progression.
BCCIP was originally identified as a BRCA2 and CDKN1A (Cip1/waf1/p2 1) interacting protein (4). Knockdown of BCCIP caused significant reduction of homologous recombination (5, 6), spontaneous chromatid aberrations including single chromatid breaks and sister chromatid union (7), cytokinesis failure (8), and cell cycle dysregulation (9-15). BCCIP is also an essential gene, and persistent deficiency of BCCIP leads to proliferation defects and mitosis failure (7, 8, 14). Down-regulation of BCCIP expression has been observed in some human cancers (16-18), but significant BCCIP mutations have not been reported in cancer tissues. In previous studies, we found that BCCIP deficiency caused proliferation arrest among neural progenitor cells, leading to severe neurogenesis defects (14). In this study, we showed that these neurogenesis defects can be rescued by a concomitant deletion of p53 gene. However, this leads to rapid formations of medulloblastoma with activation of the Sonic hedgehog (Shh) signaling pathway, confirming a caretaker tumor suppressor responsibility for BCCIP. Interestingly, reappearance of BCCIP protein expression was found in final stage medulloblastomas as a result of spontaneous deletion of the BCCIP shRNA expression transgene cassette. Our data suggest that BCCIP deficiency works in a “hit-and-run” manner to promote tumorigenesis. Thus, BCCIP has a paradoxical role in tumorigenesis, as a suppressor for the initiation and a requisite for the tumor progression once the oncogenic transformation has been accomplished, revealing an unconventional class of modulators for tumorigenesis.
Materials and Methods
Mouse strains
The names of moue lines and their corresponding genotypes used in this study are listed in Table 1. The FVB-LoxPshBCCIP+/+ mice were generated at the transgenic mice core facility of Rutgers Robert Wood Johnson Medical School as described previously (7). The glial fibrillary acidic protein (GFAP) promoter driven Cre recombinase (GFAP-Cre) transgenic mice (FVB-Tg(GFAP-Cre)25Mes/J) were obtained from the Jackson Laboratory (stock number: 004600 ). These mice were interbred to obtain BCCIP-CON (LoxPshBCCIP+/−;GFAP-Cre−/−) and BCCIP-CKD (LoxPshBCCIP+/−;GFAP-Cre+/− ) mice. To generate BCCIP-CKD;p53LoxP/LoxP (LoxPshBCCIP+/−; p53LoxP/LoxP;GFAP-Cre+/−) mice, the p53-floxed transgenic mice (B6.129P2-Trp53tm1Brn/J) were obtained from the Jackson Laboratory (stock number: 008462 ) and then interbred with BCCIP-CKD mice to generate BCCIP-CKD;Trp53LoxP/wt (LoxPshBCCIP+/−; p53LoxP/wt;GFAP-Cre+/−) mice, which were then intercrossed with p53-floxed transgenic mice to generate the BCCIP-CKD;p53LoxP/LoxP mice. During breeding, the GFAP-Cre transgene was routinely carried by the male to avoid germ-line BCCIP disruption due to low level of Cre expression in the ovaries. Mice used in p53 deficient study resulted from backcrossing mix FVB and B6.129P2. All routine mouse care and handling was approved by and performed according to the guidelines for the institutional animal care committee.
Table 1.
summary of the mouse genotypes, designation used in text, brain size, medulloblastoma incidence, and onset times.
| Genotype(s) | Designation in text | Brain size | Incidence of Medulloblastoma/total number of mice | Onset (days) for medullobl astoma |
|---|---|---|---|---|
| LoxPshBCCIP−/−; p53wt/wt;GFAP-Cre+/− | BCCIP-CON;p53wt/wt | Normal | 0/32 | N/A |
| LoxPshBCCIP−/−; p53LoxP/wt; GFAP-Cre+/− | BCCIP-CON;p53LoxP/wt | Normal | 0/31 | N/A |
| LoxPshBCCIP−/−; p53LoxP/LoxP;GFAP-Cre+/− | BCCIP-CON;p53LoxP/LoxP | Normal | 0/45 | N/A |
| LoxPshBCCIP+/−; p53wt/wt;GFAP-Cre+/− | BCCIP-CKD;p53wt/wt | Microcephaly | 0/35 | N/A |
| LoxPshBCCIP+/−; p53LoxP/wt;GFAP-Cre+/− | BCCIP-CKD;p53LoxP/wt | Reduced Microcephaly | 0/29 | N/A |
| LoxPshBCCIP+/−; p53LoxP/LoxP;GFAP-Cre+/− | BCCIP-CKD;p53LoxP/LoxP | Normal | 45/45 | 95±15 |
| LoxPshBCCIP+/−;GFAP-Cre−/−; p53wt/wt | BCCIP-CON | Normal | (N/A) | N/A |
| LoxPshBCCIP+/−;GFAP-Cre+/−; p53wt/wt | BCCIP-CKD (same as BCCIP-CKD;p53wt/wt) | Microcephaly | (N/A) | N/A |
| GFAP-Cre+/− | GFAP-Cre | Normal | (N/A) | N/A |
Western blotting analysis
Western blots were performed with procedures as described previously (7). Primary antibodies used were mBCCIP (7) , Cre (1:2000, 69050-3, Novagen), p53 (1:2000, sc-6243, Santa Cruz), PCNA (1:2500, sc-53407,Santa Cruz), PTEN (1:1000, #9559, cell signaling), and phospho-PTEN-Ser380 (1:1000, #9551, cell signaling)
Histological and immune-histochemical (IHC) analysis
Mice brains were fixed in 10% buffered formalin for 24-48 hours before paraffin embedding. All paraffin-embedded sections were cut at 5 μm. These sections were stained with hematoxylin and eosin (H&E) according to standard procedures. IHC analysis of tissue were performed by permeablizing with 0.1% Triton X-100 in PBS for 10 mins, quenching endogenous peroxides with 3% hydrogen peroxide for 10 mins, followed by blocking, primary and secondary antibody incubation. Immunoreactivity was visualized with 3,3’- diaminobenzidine (DAB) (D5637, Sigma). Positive staining appears as brown nuclear staining, whereas nuclei counterstained with hematoxylin appear as blue color. All cryosection immunofluorescence staining was performed after antigen retrieval by boiling in 0.01 M Citric acid buffer (pH 6.0). The following primary antibodies were used: calbindin D-28K (1:500, C9848, Sigma), NeuN (1:100, MAB377, Millipore), GFAP (1:400, ab360, Abcam), Ki67 (1:300, ab15580, Abcam) and synaptophysin (1:100, ab8049, Abcam).
PCR Genotyping, and Quantitative real-time RT-PCR (qPCR) analysis
The genotypes (see Table 1 for a list) were identified by PCR of tail snip DNA prepared by using proteinase K digestion and phenol-chloroform extraction. The primers used to screen the genotypes are summarized in Table S1. Total RNA was extracted from mouse tissues with the GeneElute Mammalian total RNA miniprep kit (Sigma-RTN70). The complementary DNA was generated with oligo(dT) primers using Omniscript RT kit (QIAGEN-205111). Real-time PCR was performed in duplicate with TaqMan PCR mixture (Applied Biosystems) in the 7000 ABI sequence detection system. The expression of genes was normalized to the housekeeping gene GAPDH. All qPCR primers were part of the pre-made Applied Biosystems TaqMan packages as summarized in Table S2.
cDNA sequencing of BCCIP, PTEN and Ptch1
The cDNA was synthesized as described previously (7). The paired primers used to amply the full-length coding regions of BCCIP and PTEN are listed in Table S3. Six overlapping fragments were amplified to cover the full-length coding region of mouse Ptch1 cDNA using the primer sets listed in Table S3, including those previously used by others (19). These primers were also used to sequence the amplified PCR products. For BCCIP and PTEN cDNA, two additional primers (shown in the last two rows of Table S3) were also used for sequencing.
Results
Rescue of neurogenesis defects by concurrent p53 deletion in BCCIP conditional knockdown mice
On the chromosome, the BCCIP gene overlaps with its neighboring genes (9). To avoid potential interference with genes flanking BCCIP by conventional knockout approaches, we constructed a conditional BCCIP knockdown transgenic mouse line designated LoxPshBCCIP (7). By crossing the LoxPshBCCIP+/+ mice with the GFAP-Cre transgenic mice, we found that BCCIP knockdown caused proliferation defects on embryonic neural progenitors (14). Because BCCIP deficiency caused accumulation of DNA damage in the proliferative progenitor cells and spontaneous p53 activation (7, 14), we asked whether the neurogenesis defects in BCCIP deficient mice can be rescued by concurrent p53 deletion. We crossed LoxPshBCCIP mice with the conditional p53 knockout mice, hereafter designated p53LoxP/LoxP (20), where exons 2-10 of the Trp53 gene are flanked by LoxP sites and can be conditionally deleted by expression of Cre-recombinase. After further crossing with GFAP-Cre mice (21), we generated mice with 6 genotypes as detailed in the Table 1.
As shown in Figure 1, the GFAP-Cre mediated p53 deletion indeed rescued the microcephaly in the BCCIP-CKD mice (Figs. 1A and 1B), and corrected the abnormal cerebral and cerebellar structures observed in BCCIP-CKD mice (Fig. 1C). In addition, p53 heterozygosity (BCCIP-CKD;p53LoxP/wt) partially improved cerebellar development, and slightly corrected the microcephaly (Fig. 1C). While the BCCIP-CKD mice displayed major motor coordination deficits (14), the BCCIP-CKD;p53LoxP/LoxP (LoxPshBCCIP+/−; p53LoxP/LoxP;GFAP-Cre+/−) mice were able to successfully complete the balance beam test. These observations suggest that p53 is required for the previously reported neurogenesis defects in BCCIP deficient mice, and concurrent p53 deletion rescues the neurodevelopmental deficits.
Fig. 1. p53 deficiency rescues microcephaly and neurological defects of BCCIP-CKD mice.

Panel A shows the average size of the brains (at age P21) from mice of 6 different genotypes (see Table 1 for details on genotypes). Panel B is a set of representative brains among the BCCIP-CON and BCCIP-CKD mice combined with wild type p53 (p53wt/wt), heterozygous p53 deletion (p53loxP/wt), or homozygous p53 deletion (p53LoxP/LoxP). Panel C is a set of representative H&E staining histology of cerebrum and cerebellum at age P21. Neural markers: NeuN, Calbindin (D-28K), and GFAP.
Development of medulloblastoma in conditional BCCIP knockdown (BCCIP-CKD) mice with concurrent p53 deletion
While the GFAP-Cre mediated p53 deletion rescued the neurogenesis defects of BCCIP-CKD mice, 100% of the BCCIP-CKD;p53LoxP/LoxP (LoxPshBCCIP+/−; p53LoxP/LoxP;GFAP-Cre+/−) mice became moribund and were diagnosed with medulloblastoma within the cerebellum with an average onset of 95 days of age (Fig. 2A and 2B, Table 1). However, medulloblastomas were not present in BCCIP-CKD; p53 wt/wt (LoxPshBCCIP+/−; p53wt/wt;GFAP-Cre+/−) or in the BCCIP-CON;p53LoxP/LoxP (LoxPshBCCIP−/−; p53LoxP/LoxP;GFAP-Cre+/−) mice, when observed throughout their lifespan. Necropsy and histology studies illustrated that the medulloblastoma foci are within the external granular layer of the cerebellum (Fig. 2B). The tumor tissues are immune-positive for synaptophysin, a marker for medulloblastomas (Fig. 2C). These medulloblastomas also showed high proliferative indices by Ki67 staining (Fig. 2D). The typical histological characteristics are similar to what is observed in human medulloblastomas. These findings show that medulloblastoma formation can be initiated in BCCIP deficient mice. In contrast to BCCIP-CKD;p53LoxP/LoxP (LoxPshBCCIP+/−; p53LoxP/LoxP;GFAP-Cre+/−) mice, we did not observe medulloblastoma in BCCIP-CKD;p53LoxP/wt (LoxPshBCCIP+/−; p53LoxP/wt;GFAP-Cre+/−) mice (Fig. 2), indicating that p53 complete loss of function is required for medulloblastoma formation in BCCIP-CKD mice (Table 1). Conditional p53 knockout alone did not induce medulloblastoma in the BCCIP-CON;p53LoxP/LoxP (LoxPshBCCIP−/−; p53LoxP/LoxP;GFAP-Cre+/−) mice. This is consistent with previous report (22).
Fig. 2. Medulloblastomas in conditional BCCIP knockdown & p53 knockout mice.

The LoxPshBCCIP conditional BCCIP knockdown (BCCIP-CKD) mice were crossed with conditional p53 knockout (p53LoxP/LoxP) mice, and then with the GFAP-Cre mice that expressed the Cre recombinase during embryogenesis. This resulted in mice with 6 different genotypes as shown in the insert of panel A and Table 1. These mice were observed for tumor formation for over 48 months. Panel A shows the survival curve of the mice. Medulloblastomas were formed in 100% of the BCCIP-CKD;p53LoxP/LoxP mice with an average onset of 95 days. None of the other mice developed medulloblastoma. Some of the BCCIP-CON;p53LoxP/LoxP mice were sacrificed due to growth of benign lipomas at older ages (see text for details). Panel B shows the gross morphology of medulloblastoma formed in the BCCIP & p53 deficient BCCIP-CKD;p53LoxP/LoxP mice, and H&E staining of the representative tumors. The top left inserts show the medulloblastoma (indicated by arrows) from two mouse brains. The top-right insert shows that the tumor foci are within the cerebellar external granule layer with a typical histology of medulloblastoma. The bottom inserts are H&E staining of the tissues at the magnifications as indicated.
Panels C and D are IHC staining of the tumor tissue for synaptophysin (a medulloblastoma marker) and Ki67 (a proliferation marker).
However at older ages, a few BCCIP-CON;p53LoxP/LoxP (LoxPshBCCIP−/−; p53LoxP/LoxP;GFAP-Cre+/−) and BCCIP-CON;p53LoxP/wt (LoxPshBCCIP−/−; p53wt/LoxP;GFAP-Cre+/−) mice developed non-central nervous system (CNS) tumors (including sarcomas, lipomas and lymphnoid tumors), and the knockdown of BCCIP accelerated non-CNS tumor formation in p53 heterozygous mice, when comparing the survival of BCCIP-CKD;p53LoxP/wt (LoxPshBCCIP+/−; p53LoxP/wt;GFAP-Cre+/−) with that of BCCIP- CON;p53LoxP/wt (LoxPshBCCIP−/−; p53LoxP/wt;GFAP-Cre+/−) (p=0.004, t-test). The formation of non-CNS tumors may be due to basal levels of GFAP promoter activity in tissues other than CNS (23, 24). Together, Figure 2 strongly suggests that BCCIP knockdown resulted in a high prevalence of medulloblastoma tumorigenesis, revealing a tumor-suppressing role of BCCIP.
The medulloblastomas formed in BCCIP deficient mice have inactivation of the Ptch1 gatekeeper tumor suppressor, and a wide range of alternations in the sonic hedgehog (Shh) pathway
Tumor suppressors can be classified as gatekeepers or caretakers (3), and they suppress tumorigenesis through distinct mechanisms. The gatekeeper tumor suppressors (such as RB, PTCH1, PTEN, etc.) often restrict cell proliferation, and a gatekeeper inactivation directly promotes tumor growth. On the other hand, the caretaker tumor suppressors do not directly control cell growth but maintain the genomic integrity. Caretaker deficiency causes stochastic genomic alterations that subsequently inactivate gatekeepers or activate oncogenes to cause tumor growth. Because BCCIP has been implicated in the maintenance of genomic integrity (5-8, 10), and BCCIP deficiency alone does not promote cell proliferation of neuronal progenitors in the external granule layer of the cerebellum (14), we speculated that compromised BCCIP may have caused secondary genetic alterations to inactivate certain gatekeeper genes thus enabling medulloblastoma tumorigenesis. The Shh growth-signaling pathway is one of the major regulators of granule neuron progenitor cells during neurodevelopment. Shh-signaling is normally suppressed by PTCH1, a gatekeeper gene that is often inactivated in human sporadic and hereditary medulloblastomas (25-27). In addition, Shh-mutated subtype tumors is thought to originate from the cerebellar external granule layer and has the tendency to be located away from the brainstem within the cerebellar hemispheres (28, 29). Coincidentally, we noticed the medulloblastomas that developed in BCCIP-CKD;p53LoxP/LoxP mice were mostly located in the external granule layer, away from the brainstem, and the embryonic external granule layer was the most BCCIP deficient affected region as reported previously (14). Therefore, we analyzed the Shh status among medulloblastomas formed in BCCIP-CKD;p53LoxP/LoxP using non-tumor cerebellar tissues from age-matched control mice. A critical negative regulator of the Shh pathway is the tumor suppressor Ptch1, which suppresses smoothened (Smo) dependent activation of Gli1 transcriptional activity (30-33). When Ptch1 function is impaired, the Shh pathway is activated to promote cell growth. Using qPCR with primers covering the region of exons 17-18 of mouse Ptch1, we measured the expression of Ptch1 in the tumors, and found a significant reduction of Ptch1 expression in 16 of 24 tested tumors (Fig. 4A). As expected, we were not able to amplify the full-length Ptch1 cDNA from the 16 cases. Among the 8 cases with apparent normal level of Ptch1 mRNA based on qPCR of exons 17-18, we were able to amplify the approximately normal sized Ptch1 cDNA from 6 samples. Therefore, only 6 out of the 24 cases have the expression of approximate full-length coding mRNA of Ptch1, and the other 18 cases likely had genetic alternations that prohibited the amplification of their cDNA. Furthermore, each of the amplified Ptch1 cDNAs contained an inactivation mutation (Table 2) that was verified in the genomic DNA of the same tumor. It is also striking that all mutations in the amplified cDNA were either deletions or insertions of multiple base pairs that resulted in truncation of Ptch1. We did not observe point mutations in these tumor samples (Table 2). These data suggest that Ptch1 is a critical target for inactivation due to the genomic instability in the context of medulloblastoma formation after BCCIP knockdown. Thus, the majority of the tumors have lost Ptch1 and the remaining cases have deletions/insertions with multiple bases. These observations are consistent with a role of BCCIP in DNA double strand break repair, including homologous recombination (5, 6).
Fig. 4. PTEN expression in in medulloblastomas.
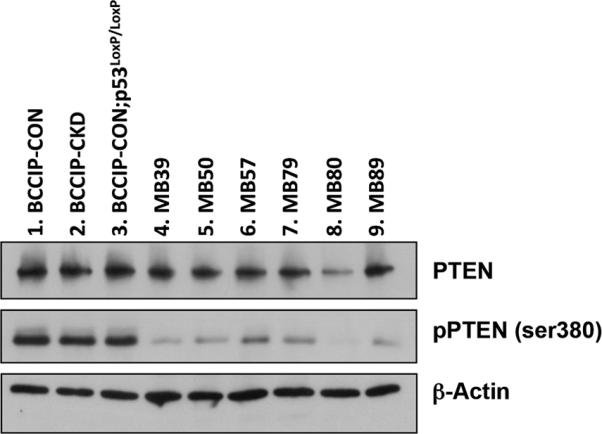
Western blots were performed to measure the levels of PTEN and pPTEN-ser380 in the medulloblastomas (lanes 4-9). As the controls, proteins were extracted from the cerebellums of BCCIP-CON (LoxPshBCCIP−/−; p53wt/wt; GFAP-Cre+/−), BCCIP-CKD (LoxPshBCCIP+/−; p53wt/wt; GFAP-Cre+/−), BCCIP-CON;p53LoxP/LoxP (LoxPshBCCIP−/−; p53LoxP/LoxP; GFAPCre+/−) mice of 3 month-old (Lanes1, 2 and 3).
Table 2.
Altered Sequence of Ptch1 cDNA in medulloblastomas
| Tumor ID | Ptch1 mutations | Altered sequences (parenthesized sequences are the deleted segments in tumor, underlined sequences are the duplicated segments) |
|---|---|---|
| MB20 | 14nt duplication in exon 10 | CTGGCTGGCGTCCTGTTGGTTGCGTCCTGTTGGTTGCGCTGTCAGTGG |
| MB36 | 14nt deletion in exon 16 | CAGACTGGCA(GCCGAGACAAGCCC)ATCGACATTA |
| MB59 | 4nt deletion in exon 10 | GTCCTGTTGG (TTGC) GCTGTCAGTG |
| MB73 | 14nt duplication in exon 10 | CTGGCTGGCGTCCTGTTGGTTGCGTCCTGTTGGTTGCGCTGTCAGTGG |
| MB79 | 7nt deletion in exon 18 | CCTACGAGAC(ACCTCAG)ACTTTGTGGA |
| MB89 | Deletion: 3nt of exon 11, Intron-11, and 123nt of exon 12. | ATTCCATTTG(AGG-Intron 11-ACAGGACTGGGGAGTGCCTCAAGCGCACCGGAGCCAGCGTGGCCCTCACCTCCATCAGCAATGTCACCGCCTTCTTCATGGCCGCATTGATCCCTATCCCTGCCCTGCGAGCGTTCTCCCTCC)AGGCTGCTGT |
In the Shh pathway, the critical downstream target of Ptch1 is Smo, which controls the expression of Gli1, Atoh1, N-Myc and D-cyclins. These downstream elements are known to be critical regulators of granule neuron progenitors (34-37), which are the origins of medulloblastoma. Along with the Ptch1 mutations and loss of Ptch1 expression, we observed high levels of Smo, Gli1, Atoh1, N-Myc, and D-cyclins in the medulloblastomas compared with controls (Table S4, and Fig. 3B). Together, these data suggest that the initial BCCIP knockdown had triggered the inactivation of the Ptch1 gatekeeper tumor suppressor, and caused widespread alterations leading to the activation of the Shh signaling pathway, which is known to promote medulloblastoma formation.
Fig. 3. Dysregulation of the sonic hedgehog (Shh) signaling pathway in medulloblastomas.

The qPCR analyses were used to measure the relative level of mRNA in medulloblastoma tissues derived from BCCIP-CKD;p53LoxP/LoxP mice. The mRNA levels of age-matched control (3-months old) cerebellums were also measured. Panel A shows the relative level of Ptch1 mRNA in 24 medulloblastoma samples. Control samples are (from left to right): BCCIP-CON (LoxPshBCCIP+/−; p53wt/wt;GFAP-Cre−/−)(first 2 control data points from the left, n=2), BCCIP-CKD (LoxPshBCCIP+/−; p53wt/wt;GFAP-Cre+/− )(control data points 3 and 4 from the left, n=2), GFAP-Cre (LoxPshBCCIP−/−; p53wt/wt;GFAP-Cre+/−) (n=2) and BCCIP-CON;p53LoxP/LoxP (LoxPshBCCIP−/−; p53LoxP/LoxP;GFAPCre+/− )(n=2). The average of Ptch1 mRNA level and its standard deviation were calculated from control samples. Based on the normal distribution, the 95% confidence limit (p<0.05) was set at 1.96 fold of the standard deviation, and the 99% confidence limit (p<0.01) was set at 2.58 fold of the standard deviation. The 95% and 99% confidence ranges are marked in the graph. A total of 16 cases displayed reduction of Ptch1 mRNA levels. Panel B shows the wide range of mRNA up-regulation of key components of the Shh pathway as indicated in the specific panels. A total of 12 MB samples were analyzed in Smo, Gli1, Atoch1, N-Myc and 16 MB samples were analyzed in Ccnd1 and Ccnd2.
PTEN status in medulloblastoma induced by BCCIP deficiency
Because the gatekeeper PTEN has been implicated in brain tumors including medulloblastoma (38)(39, 40). We also investigated whether there is a genetic changes on PTEN. However, we did not find evidence of PTEN mRNA reduction, nor mutations in PTEN coding region cDNA. Western blot revealed relatively normal expression level of PTEN protein within most of the tumor tissues (Fig. 4). Thus PTEN inactivation seems to be an unlikely cause of the tumors obtained from the BCCIP conditional knockdown mice. However, we observed a reduced PTEN Ser-380 phosphorylation compared to control tissues (Fig. 4). Although it had been suggested that C-terminal phosphorylation of PTEN may regulate its stability (41, 42), we did not observe an altered PTEN protein levels in the tumor tissues as shown in Figure 4. The significance of the reduced PTEN c-terminal phosphorylation in the tumor tissues remains to be determined.
Restored BCCIP expression in medulloblastomas
In our mouse model, BCCIP expression was conditionally knocked down by GFAP-mediated Cre expression during embryogenesis, which differs from the classical knockout approach. First, the knockout approach would completely abolish the expression of wild type BCCIP, the knockdown approach is unlikely to do so, leaving a reduced BCCIP expression. Second, the knockout approach permanently deletes the endogenous BCCIP allele and BCCIP expression cannot be restored during the later stages of tumor progression. Conversely, the knockdown approach leaves the endogenous BCCIP locus intact and there is a possibility to restore BCCIP expression. To exam BCCIP expression in the tumor tissues, we measured BCCIP protein levels in the medulloblastomas and of age-matched (3 month-old) control cerebellums (BCCIP-CON, BCCIP-CKD, GFAP-Cre and BCCIP-CON;p53LoxP/LoxP mice). To our surprise, a restored expression of BCCIP protein among all tumor samples was observed (Fig. 5A). We further compared BCCIP levels of the tumor regions (Cb-T) with the tumor-neighboring cerebellum region (Cb-NT) that may contain both tumor and normal cells, and the cortex non-tumor (Cx) region from the same animal. Again, the BCCIP expression detected in tumor tissues was significantly higher than non-tumor tissues among all measured 24 samples (see Fig. 5B for 3 representative cases). As expected the down-regulation of p53 expression was observed in the tumor tissues (Fig. 5B), and all tumor tissues displayed higher expression levels of PCNA, a proliferation marker (Fig. 5B). As expected, the normal tissue have detectable level of p53 (Figure 5B), due to the presence of GFAP-negative cells in the normal tissues. A small and reduced level of p53 can be detected in the tumor tissue (Fig. 5B, lanes 2, 5, and 8) are likely contributed by mixed normal cells in the tumor tissues.
Fig. 5. BCCIP expression in the medulloblastoma formed from the BCCIP-knockdown mice.

Proteins and DNA from tumor tissues (Cb-T), tumor-bordering cerebellum (Cb-NT), and non-tumor cortex (Cx) were subjected to Western blot and PCR genotype.
Panel A shows the BCCIP and Cre protein levels. It reveals an up-regulation of BCCIP in medulloblastoma tissues (Lanes 5-9) compared to the cerebellums from BCCIP-CON (LoxPshBCCIP+/−; p53wt/wt;GFAP-Cre−/−) (Lane 1), BCCIP-CKD (LoxPshBCCIP+/−; p53wt/wt;GFAP-Cre+/−) (Lane 2), GFAP-Cre (LoxPshBCCIP−/−; p53wt/wt;GFAP-Cre+/−) (Lane 3) and BCCIP-CON;p53LoxP/LoxP (LoxPshBCCIP−/−; p53LoxP/LoxP;GFAP-Cre+/− ) (Lane 4) mice.
Panel B shows the BCCIP, p53, and PCNA protein expression level in three representative mice, MB-20, 25, and 37. Lanes 2, 5 and 8 are the medulloblastoma tissues (Cb-T,); Lanes 3, 6 and 9 are tissues from tumor-bordering regions of the cerebellums (Cb-NT); and Lanes 1, 4 and 7 are the tissues of cerebrum tissues (Cx) of the same mice. Up-regulation of BCCIP protein and down-regulation of p53 levels were detected in medulloblastoma tissues (Cb-T, Lane 2, 5 and 8).
Panel C shows the relative mRNA levels of BCCIP in medulloblastoma using GAPDH as an internal control. Control samples are age-matched (3-month old) cerebellums of BCCIP-CON (LoxPshBCCIP+/−; p53wt/wt;GFAP-Cre−/−)(first 2 control data points from the left, n=2), BCCIP-CKD (LoxPshBCCIP+/−; p53wt/wt;GFAP-Cre+/− )(control data points 3 and 4 from the left, n=2), GFAP-Cre (LoxPshBCCIP−/−; p53wt/wt;GFAP-Cre+/− ) (n=2) and BCCIP-CON;p53LoxP/LoxP (LoxPshBCCIP−/−; p53LoxP/LoxP;GFAP-Cre+/− )(n=2). The average of BCCIP mRNA level and its standard deviation were calculated from control samples. Based on the normal distribution, the 95% confidence limit (p<0.05) was set at 1.96 fold of the standard deviation, and the 99% confidence limit (p<0.01) was set at 2.58 fold of the standard deviation. The 95% and 99% confidence ranges are marked in the panel. 16 medulloblastoma samples showed reduction of BCCIP mRNA levels.
Panel D shows the loss of the BCCIP shRNA expression cassette in the medulloblastomas. Shown are representative PCR genotyping results for the DNA extracted from medulloblastoma tissues (MB-T), tails (MB-Tail), non-tumor cortex (MB-Cx), and non-tumor cerebellums (MB-NT) from three mice. Panel 1 shows the detection of the split U6 promoter LoxPshBCCIP. The panel 2 detects the reconstituted U6-shBCCIP cassette after Cre-mediated recombination. The panel 3 detects the floxed p53 allele (and the wild type p53 allele, lower band). Panel 4 detects the p53 allele post Cre-mediated LoxP recombination. Panel 5 shows the presence of GFAP-Cre transgenes in the tissues. GAPDH was used as a PCR control (panel 6). The genotype structure and approximate primer locations for PCR genotype for panels 1-4 are shown on the right side of each panel. As shown here, all medulloblastoma tissues (MB-T) lost the BCCIP shRNA expression cassette, although they all carry the GFAPCre cassette and recombined p53 allele.
We used qPCR measurements to confirm elevated levels of BCCIP in medulloblastomas (Fig. 5C). These data confirmed that BCCIP expression was restored in the tumor tissues. Although the BCCIP-CKD mouse brains were reported to have lower level of BCCIP expression during embryogenesis at newborn age than non-knockdown mice (14), we noticed a minimum reduction of protein level in the whole brain at age of 3 months (compare lane 2 of Figure 5A with that of lane 1, and control data points 3 and 4 with the rest of the control data points in Figure 5C). This is due to the conditional nature of our GFAP-Cre system that only targets the GFAP-positive cells for BCCIP silencing. During embryogenesis and at newborn, GFAP is expressed in multi-purpose progenitor cells, and these cells constitute a major portion of brain tissue. Thus a clear BCCIP down regulation can be verified among embryo E15.5 neurospheres and newborn (P1) brain tissues as reported earlier (14). However, at the time of medulloblastoma development (3 months), GFAP is mainly expressed in astrocytes, which are no longer a major portion in the brain tissues. When the 3-month-old brain tissue is used to measure BCCIP level, it cannot reflect the BCCIP knockdown status in the GFAP-positive cell population.
To identify the cause of the restored BCCIP expression in the tumor tissues that originally possessed the BCCIP shRNA expression cassette, we analyzed the status of the BCCIP shRNA cassette among the genomic DNA from the tumor tissues. Interestingly, we found that all of the analyzed tumor samples had lost the transgenic BCCIP shRNA expression cassette LoxPshBCCIP, while they maintained the deleted p53 alleles and the GFAP-Cre cassette (see Fig. 5D for three representative cases). Therefore, it seems that the tumors have found a way to restore BCCIP expression by deleting the BCCIP shRNA expression cassette, likely by a growth pressure of the tumor cells. No BCCIP mutation was identified from the tumor tissues analyzed.
These surprising findings imply that expression of BCCIP is required for the continued growth or progression of medulloblastomas. The genomic instability generated by the initial and transient BCCIP down-regulation was sufficient to cause oncogenic transformation due to a stochastic activation of pro-growth Shh signaling (Figs. 3 and 4). Furthermore, the proliferative tumor cells have even higher level of BCCIP RNA and proteins than normal tissue, implying a role of BCCIP in promoting tumor progression, which is consistent with the essential function of BCCIP in cell growth.
Discussion
BCCIP represents a class of tumorigenesis modulators that function as Suppressor of Initiation but a Requisite for Progression (SIRP)
In this study, we have shown that conditional BCCIP knockdown contributes to the development of medulloblastomas. Based on the functions of BCCIP in DNA repair, cell cycle regulation, and mitosis (5-7, 10, 11), it is conceivable that BCCIP functions as a caretaker tumor suppressor gene. Reduced BCCIP function is expected to cause genomic instability that can inactivate gatekeeper tumor suppressors, which is consistent with the observed alterations in the gatekeeper pathways (i.e. Shh) and the high penetrance of medulloblastomas in the BCCIP-CKD;p53LoxP/LoxP mice (Table 1, Fig. 2). These data strongly suggest that BCCIP has a tumor suppressor responsibility. However, it was paradoxical that BCCIP expression was restored and expressed at even higher levels in the fully developed tumors (Fig. 5), which implies a possible tumor promoter role for BCCIP. As a consolidated model, we propose that the initial BCCIP deficiency (caused by BCCIP shRNA expression) elicited stochastic genomic instability that eventually compromised the function of gatekeepers in the Shh pathway. Once the pro-growth pathways were activated, they are autonomous in supporting cell growth, which caused oncogenic transformation of the neural progenitor cells. At this stage, BCCIP deficiency is no longer needed to trigger transformation and becomes an obstacle for the progression of the transformed cells, due to BCCIP's functions for DNA replication, DNA repair, and mitosis. In the context of tumor progression, the sustained rapid proliferation favors those cells with spontaneous deletion of the BCCIP knockdown cassette. Thus, during tumor progression, when gatekeeper functions of Ptch1 have been inactivated, BCCIP become essential for a sustained growth of the tumor. This mode of action underscores the paradoxical roles of BCCIP in tumorigenesis. We postulate that BCCIP represents a unique class of modulators of tumorigenesis that are Suppressors of Initiation but Requisite for Progression (SIRP).
We further suggest that BCCIP exhibits a typical mode of action for essential caretaker genes in tumorigenesis. On one hand, a transient or subtle down regulation of these genes is sufficient to trigger genomic instability, leading to oncogenic transformations from autonomous pro-proliferation signaling networks. On the other hand, the sustained growth of transformed cells demands the availability of the same genes due to their essential roles in cell division and proliferation. Unlike the classical tumor suppressors that are often found mutated in the tumor tissue, the SIRP types of genes regulate tumorigenesis in an unconventional way. It only takes a transient or subtle dysfunction to trigger the oncogenic transformations, and their deficiencies work in a “hit-and-run” manner to initiate tumorigenesis. Thus, inactivation mutations in the SIRP genes may be rare in fully developed tumors, and a transient and epigenetic down-regulation of SIRP genes may be more prevalent in conferring tumorigenesis. This scenario may help explaining how some epigenetically regulated tumor suppressors play a role in tumorigenesis. It further raises several critical questions when considering the roles of DNA repair genes in tumorigenesis. First, do essential DNA repair genes typically function as conventional tumor suppressors? Secondly, are there any other DNA repair genes possess SIRP roles in tumorigenesis? Lastly, what environmental factors play a role in the transient dysregulation of the SIRP genes to promote tumorigenesis?
Conditional knockdown as an ideal approach to identify genes with paradoxical functions at different stages of tumorigenesis
As a gene involved in genomic integrity, BCCIP is unlikely the only gene with the paradoxical SIRP functions. The conventional knockout approach is unlikely to reveal the SIRP genes with multiple and often opposing roles at different stages of tumorigenesis. As discussed in a previous report (7), we initially opted to use the conditional knockdown approach to minimize the possibility of interference by BCCIP deletion on its overlapping genes. This approach leaves the endogenous BCCIP locus intact. When BCCIP is indeed critical for tumor progression, the growth pressure of transformed cells would favor cells with a spontaneous deletion of the BCCIP knockdown cassette. In addition, this approach down-regulates, but does not mutate or abolish BCCIP and has advantages over the conventional knockout approach as it can faithfully reproduce situations where gene down-regulation, but not mutation, contributes to tumorigenesis. On the contrary a conventional knockout strategy would permanently delete the gene(s) of interest. The absence of essential functions of the gene(s), would prevent tumor progression, thus development of advanced tumors would be unlikely. We were fortunate to observe such a unique SIRP role of BCCIP in tumorigenesis because we opted to use a conditional knockdown approach.
Distinct features of the BCCIP knockdown medulloblastomas mouse model
Although p53 deletion alone in the GFAP-Cre or Nestin-Cre based conditional knockout mice is not sufficient to cause medulloblastoma in many DNA repair deficient mouse models (this study, and (22)), p53 deficiency combined with DSB repair defects (including NHEJ and HR) and DNA damage signaling can induce medulloblastoma (43). It has been shown that Nestin-Cre mediated conditional knockout of BRCA2, and NBS1 also caused medulloblastomas (44, 45). In these mouse models, the Nestin promoter becomes active at around embryonic day 11 (E11), primarily in the central and peripheral nervous system during embryogenesis (46). In our study, we crossed FVB-LoxPshBCCIP+/+ mice with FVB-Tg(GFAP-Cre) transgenic mice that express Cre recombinase under the control of the human GFAP promoter. Although only glial cells are immune-reactive for GFAP in adult brain, embryonic GFAP-promoter activity is not restricted to glial progenitor cells. Much like Nestin-Cre, GFAP-promoter is active in multi-potent stem cells including neural progenitor cells during embryogenesis with a peak activity at around embryonic day 13.5 (E13.5) (21). However, there is a difference between the two transgenic mouse strains. Nestin-Cre mice express Cre recombinase in common neural progenitors mostly during embryogenesis, while hGFAP-Cre transgenic mice express Cre recombinase in both embryonic common progenitor cells as well as in adult glial cells (21).
The conditional homozygous BRCA2 deletion in Brca2Nestin-cre mice resulted in neural development defects and medulloblastoma when p53 was also deleted (44). As a BRCA2 interacting protein, BCCIP has been shown to play a role in homologous recombination, cell cycle regulation, and chromosome stability (5, 6, 8). Although certain features, such as viable mice with no tumor formation in BCCIP or BRCA2 deficiency alone are similar, additional characteristics of the BCCIP-CKD mice make it distinguishable from the BRCA2 knockout mice. First, homozygous p53 deletion almost completely rescued microcephaly in BCCIP-CKD mice (Fig.1), but it only partially rescued the microcephaly in BRCA2 knockout mice (44). Secondly, when both copies of the p53 gene were deleted, 100% of the BCCIP-CKD mice developed medulloblastoma, but only 83% of the BRCA2 knockout mice developed tumors with a similar onset of about 3 months (44). Lastly, none of the BCCIP-CKD mice with p53 heterozygosity developed tumors, yet 72% of the BRCA2 conditional knockout mice with p53 heterozygosity developed medulloblastoma with a concurrent loss of the second copy of the p53 in the tumor cells (44). This is a drop from 83% (p53 homozygous) to 72% (p53 heterozygous) in BRCA2 knockout mice, comparing to 100% (p53 homozygous) to 0% (p53 heterozygous) for BCCIP knockdown mice. It implies that BCCIP deficiency induced medulloblastoma is more dependent on complete loss of p53 function than BRCA2 knockout. However, it was noted that the BRCA2 study used the constitutive p53 deficient mice rather than a conditional p53 LoxP mice (44).
Nbs1 is another gene involved in DNA damage response. The Nbs1Nestin-Cre conditional knockout mice had severe neural degeneration, ataxia, and microcephaly, to a similar extent as our BCCIP-CKD mice (45). In addition, p53 deletion remarkably rescued the microcephaly and neural degeneration phenotype of NBS1Nestin-Cre knockout mice as with BCCIP-CKD mice. However, unlike in BCCIP-CKD mice, p53 deletion did not promote tumor formation in NBS1Nestin-cre mice (45). These distinct features suggest that BCCIP's roles in neurodevelopment and medulloblastoma formation may be independent of NBS1 and BRCA2. However, it is possible that NBS1 may be another essential gene with SIRP function, and un-reversible NBS1 deficiency (in the knockout mouse model) may have had prevented the transformed progenitor cells from a sustained progression into medulloblastomas.
Activation of the Shh pathway in medulloblastoma in BCCIP deficient mice
Medulloblastoma is the most common childhood malignant brain tumor. This type of tumor originates from granule neural progenitors in the cerebellum (47). Based on gene expression profiling, different subtypes of human medulloblastomas are characterized by alterations of multiple cell growth signaling pathways, including the Shh, Wnt, and the Notch signaling (27, 48-50). More than 25% of sporadic human medulloblastomas have mutations in key components of the Shh signaling pathway, such as Ptch1 and Smo (33, 48). Interestingly, 100% of the medulloblastomas developed in the BCCIP-CKD; p53LoxP/LoxP mice had abnormalities in at least one, and often multiple, components of the Shh pathway (Table S4, Fig. 3). A striking feature of Ptch1 mutations from our BCCIP knockdown mice is that majority of the tumor have lost the Ptch1 gene (18/24), and the remaining mutations (6/24) involve large deletions or duplication of normal sequences in the Ptch1 gene but do not involve single base pair mutations. This feature indicate that BCCIP deficiency induced Ptch1 mutations are likely due to DNA repair mechanisms involved in large re-arrangements of DNA fragments, which is consistent with BCCIP's role in DNA double strand break repair and replication slippage.
PTEN status in medulloblastoma
Reduced PTEN expression was shown in human medulloblastomas (39). In addition, frequent loss of heterozygosity of chromosome 10q, where PTEN is located (10q23.31) was observed in human medulloblastomas (40). Although reduced expression levels of PTEN was observed in a small fraction of tumors (1 out 9 as shown in Figure 4), and we did not detect mutations in the Pten ORF region (Fig. 4). Interestingly, we observed a reduced PTEN phosphorylation at Ser380 in the tumor tissues (Fig. 4). The significance of this observation remains to be investigated.
In summary, our study showed that, due to BCCIP's roles in the maintenance of genomic integrity, conditional BCCIP deficiency synergizes with loss of p53 to promote medulloblastoma. This suggests a tumor suppressor role for BCCIP. However, BCCIP is also required for tumor progression. These observations revealed a paradoxical class of modulators of tumorigenesis that act as suppressors of initiation but are requisites for progression (SIRP). We further propose that only a transient down-regulation, but not necessarily a permanent mutation, of a SIRP is sufficient to confer tumorigenesis. This concept has major implications on how dynamic regulation of essential caretaker genes, such as by epigenetic mechanisms of regulation, can contribute to tumorigenesis.
Supplementary Material
Acknowledgements
This research was supported by NIH R01CA156706 to ZS, and by the Transgenic & Knockout Mouse and the Histopathology & Imaging shared resources of Rutgers Cancer Institute of New Jersey (P30CA072720). We thank Dr. W. Hu (Rutgers Cancer Institute of New Jersey) for suggestions on qPCR analysis.
Footnotes
The authors disclose no potential conflicts of interest.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Shen Z. Genomic instability and cancer: an introduction. Journal of molecular cell biology. 2011;3(1):1–3. doi: 10.1093/jmcb/mjq057. [DOI] [PubMed] [Google Scholar]
- 3.Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386(6627):761, 763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- 4.Liu J, Yuan Y, Huan J, Shen Z. Inhibition of breast and brain cancer cell growth by BCCIPalpha, an evolutionarily conserved nuclear protein that interacts with BRCA2. Oncogene. 2001;20(3):336–345. doi: 10.1038/sj.onc.1204098. [DOI] [PubMed] [Google Scholar]
- 5.Lu H, et al. The BRCA2-interacting protein BCCIP functions in RAD51 and BRCA2 focus formation and homologous recombinational repair. Mol Cell Biol. 2005;25(5):1949–1957. doi: 10.1128/MCB.25.5.1949-1957.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu H, Yue J, Meng X, Nickoloff JA, Shen Z. BCCIP regulates homologous recombination by distinct domains and suppresses spontaneous DNA damage. Nucleic Acids Res. 2007;35(21):7160–7170. doi: 10.1093/nar/gkm732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu H, et al. Essential Roles of BCCIP in Mouse Embryonic Development and Structural Stability of Chromosomes. PLoS Genet. 2011;7(9):e1002291. doi: 10.1371/journal.pgen.1002291. doi:10.1371/journal.pgen.1002291 Pages: 1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meng X, Fan J, Shen Z. Roles of BCCIP in chromosome stability and cytokinesis. Oncogene. 2007;26(43):6253–6260. doi: 10.1038/sj.onc.1210460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meng X, Liu J, Shen Z. Genomic structure of the human BCCIP gene and its expression in cancer. Gene. 2003;302(1-2):139–146. doi: 10.1016/s0378-1119(02)01098-3. [DOI] [PubMed] [Google Scholar]
- 10.Meng X, Liu J, Shen Z. Inhibition of G1 to S cell cycle progression by BCCIP beta. Cell Cycle. 2004;3(3):343–348. [PubMed] [Google Scholar]
- 11.Meng X, Lu H, Shen Z. BCCIP functions through p53 to regulate the expression of p21Waf1/Cip1. Cell Cycle. 2004;3(11):1457–1462. doi: 10.4161/cc.3.11.1213. [DOI] [PubMed] [Google Scholar]
- 12.Meng X, Yue J, Liu Z, Shen Z. Abrogation of the transactivation activity of p53 by BCCIP down-regulation. J Biol Chem. 2007;282(3):1570–1576. doi: 10.1074/jbc.M607520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan J, Wray J, Meng X, Shen Z. BCCIP is required for the nuclear localization of the p21 protein. Cell Cycle. 2009;8(18):3019–3024. [PMC free article] [PubMed] [Google Scholar]
- 14.Huang YY, Lu H, Liu S, Droz-Rosario R, Shen Z. Requirement of mouse BCCIP for neural development and progenitor proliferation. PLoS One. 2012;7(1):e30638. doi: 10.1371/journal.pone.0030638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mao N, Zhou Q, Kojic M, Perez-Martin J, Holloman WK. Ortholog of BRCA2-interacting protein BCCIP controls morphogenetic responses during DNA replication stress in Ustilago maydis. DNA Repair (Amst) 2007;6(11):1651–1660. doi: 10.1016/j.dnarep.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J, Lu H, Ohgaki H, Merlo A, Shen Z. Alterations of BCCIP, a BRCA2 interacting protein, in astrocytomas. BMC Cancer. 2009;9:268. doi: 10.1186/1471-2407-9-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roversi G, et al. Identification of novel genomic markers related to progression to glioblastoma through genomic profiling of 25 primary glioma cell lines. Oncogene. 2006;25(10):1571–1583. doi: 10.1038/sj.onc.1209177. [DOI] [PubMed] [Google Scholar]
- 18.Rewari A, et al. BCCIP as a prognostic marker for radiotherapy of laryngeal cancer. Radiother Oncol. 2009;90(2):183–188. doi: 10.1016/j.radonc.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee Y, et al. Patched2 modulates tumorigenesis in patched1 heterozygous mice. Cancer Res. 2006;66(14):6964–6971. doi: 10.1158/0008-5472.CAN-06-0505. [DOI] [PubMed] [Google Scholar]
- 20.Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14(8):994–1004. [PMC free article] [PubMed] [Google Scholar]
- 21.Zhuo L, et al. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31(2):85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- 22.Frappart PO, et al. Recurrent genomic alterations characterize medulloblastoma arising from DNA double-strand break repair deficiency. Proc Natl Acad Sci U S A. 2009;106(6):1880–1885. doi: 10.1073/pnas.0806882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci. 1994;14(3 Pt 1):1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brenner M. Structure and transcriptional regulation of the GFAP gene. Brain Pathol. 1994;4(3):245–257. doi: 10.1111/j.1750-3639.1994.tb00840.x. [DOI] [PubMed] [Google Scholar]
- 25.Wechsler-Reya R, Scott MP. The developmental biology of brain tumors. Annu Rev Neurosci. 2001;24:385–428. doi: 10.1146/annurev.neuro.24.1.385. [DOI] [PubMed] [Google Scholar]
- 26.Zindy F, et al. Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 2007;67(6):2676–2684. doi: 10.1158/0008-5472.CAN-06-3418. [DOI] [PubMed] [Google Scholar]
- 27.Gilbertson RJ, Ellison DW. The origins of medulloblastoma subtypes. Annu Rev Pathol. 2008;3:341–365. doi: 10.1146/annurev.pathmechdis.3.121806.151518. [DOI] [PubMed] [Google Scholar]
- 28.Marino S. Medulloblastoma: developmental mechanisms out of control. Trends Mol Med. 2005;11(1):17–22. doi: 10.1016/j.molmed.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 29.Gibson P, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468(7327):1095–1099. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stecca B, Ruiz i Altaba A. Brain as a paradigm of organ growth: Hedgehog-Gli signaling in neural stem cells and brain tumors. J Neurobiol. 2005;64(4):476–490. doi: 10.1002/neu.20160. [DOI] [PubMed] [Google Scholar]
- 31.Dellovade T, Romer JT, Curran T, Rubin LL. The hedgehog pathway and neurological disorders. Annu Rev Neurosci. 2006;29:539–563. doi: 10.1146/annurev.neuro.29.051605.112858. [DOI] [PubMed] [Google Scholar]
- 32.Ruiz i Altaba A, Palma V, Dahmane N. Hedgehog-Gli signalling and the growth of the brain. Nat Rev Neurosci. 2002;3(1):24–33. doi: 10.1038/nrn704. [DOI] [PubMed] [Google Scholar]
- 33.Zurawel RH, et al. Analysis of PTCH/SMO/SHH pathway genes in medulloblastoma. Genes Chromosomes Cancer. 2000;27(1):44–51. doi: 10.1002/(sici)1098-2264(200001)27:1<44::aid-gcc6>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 34.Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130(1):15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- 35.Huard JM, Forster CC, Carter ML, Sicinski P, Ross ME. Cerebellar histogenesis is disturbed in mice lacking cyclin D2. Development. 1999;126(9):1927–1935. doi: 10.1242/dev.126.9.1927. [DOI] [PubMed] [Google Scholar]
- 36.Pogoriler J, Millen K, Utset M, Du W. Loss of cyclin D1 impairs cerebellar development and suppresses medulloblastoma formation. Development. 2006;133(19):3929–3937. doi: 10.1242/dev.02556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee Y, et al. A molecular fingerprint for medulloblastoma. Cancer Res. 2003;63(17):5428–5437. [PubMed] [Google Scholar]
- 38.Castellino RC, et al. Heterozygosity for Pten promotes tumorigenesis in a mouse model of medulloblastoma. PLoS One. 2010;5(5):e10849. doi: 10.1371/journal.pone.0010849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartmann W, et al. Phosphatidylinositol 3'-kinase/AKT signaling is activated in medulloblastoma cell proliferation and is associated with reduced expression of PTEN. Clin Cancer Res. 2006;12(10):3019–3027. doi: 10.1158/1078-0432.CCR-05-2187. [DOI] [PubMed] [Google Scholar]
- 40.Griffin CA, Hawkins AL, Packer RJ, Rorke LB, Emanuel BS. Chromosome abnormalities in pediatric brain tumors. Cancer Res. 1988;48(1):175–180. [PubMed] [Google Scholar]
- 41.Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20(14):5010–5018. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Z, et al. Reduced expression of PTEN and increased PTEN phosphorylation at residue Ser380 in gastric cancer tissues: a novel mechanism of PTEN inactivation. Clinics and research in hepatology and gastroenterology. 2013;37(1):72–79. doi: 10.1016/j.clinre.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Frappart PO, McKinnon PJ. Mouse models of DNA double-strand break repair and neurological disease. DNA Repair (Amst) 2008;7(7):1051–1060. doi: 10.1016/j.dnarep.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frappart PO, Lee Y, Lamont J, McKinnon PJ. BRCA2 is required for neurogenesis and suppression of medulloblastoma. EMBO J. 2007;26(11):2732–2742. doi: 10.1038/sj.emboj.7601703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frappart PO, et al. An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nat Med. 2005;11(5):538–544. doi: 10.1038/nm1228. [DOI] [PubMed] [Google Scholar]
- 46.Tronche F, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23(1):99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- 47.Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer. 2010;10(5):319–331. doi: 10.1038/nrc2818. [DOI] [PubMed] [Google Scholar]
- 48.Monje M, Beachy PA, Fisher PG. Hedgehogs, flies, Wnts and MYCs: the time has come for many things in medulloblastoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29(11):1395–1398. doi: 10.1200/JCO.2010.34.0547. [DOI] [PubMed] [Google Scholar]
- 49.Northcott PA, et al. Medulloblastoma comprises four distinct molecular variants. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29(11):1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho YJ, et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29(11):1424–1430. doi: 10.1200/JCO.2010.28.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.