

Abstract
Background
Azathioprine (AZA), a pro-drug metabolized to the active metabolites 6-thioguanine nucleotides (6TGN), is a steroid-sparing therapy for Crohn’s disease (CD). This trial investigated whether AZA therapy is optimized by individualized dosing based on thiopurine methyltransferase (TPMT) activity and 6TGN concentrations.
Methods
This multicenter, double-blind, randomized controlled trial compared the efficacy and safety of weight-based vs. individualized AZA dosing in inducing and maintaining remission in adults and children with steroid-treated CD. The primary outcome was clinical remission (CR) at 16 weeks. In the weight-based arm, subjects received 2.5 mg/kg/d. In the individualized dosing arm, the initial AZA dose was 1.0 mg/kg/d (if intermediate TPMT) or 2.5 mg/kg/d (if normal TPMT). Starting at week 5, the dose was adjusted to target 6TGN concentrations of 250–400 pmol/8x108 red blood cells (RBC), or to a maximal dose of 4 mg/kg/d.
Results
After randomizing 50 subjects, the trial was stopped prematurely due to insufficient enrollment. In intention-to-treat analysis, CR rates at week 16 were 40% in the individualized arm vs. 16% in the weight-based arm (p=0.11). In per-protocol (PP) analysis, week 16 CR rates were 60% in the individualized arm and 25% in the weight-based arm (p=0.12). At week 16, median 6TGN concentrations in PP remitters and non-remitters were 216 and 149 pmol/8x108 RBC respectively (p=0.07).
Conclusions
Despite trends favoring individualized over weight-based AZA dosing, there were no statistically significant differences in efficacy, likely due to low statistical power and inability to achieve the target 6TGN concentrations in the individualized arm.
Keywords: Azathioprine, thiopurine methyltransferase, dosing, Crohn’s disease
INTRODUCTION
The thiopurines, mercaptopurine (MP) and its pro-drug, azathioprine (AZA), are steroid-sparing and maintenance therapies in patients with moderate to severe Crohn’s disease (CD)1–3. Unfortunately approximately 40–50% of CD patients fail to respond, or cannot tolerate treatment.
Differences in response and toxicity may reflect inter-individual variations in thiopurine metabolism. AZA is an inactive pro-drug metabolized by competing pathways. One pathway leads to the formation of 6-thioguanine nucleotides (6TGN) that, in some retrospective studies, have been correlated with clinical response4. In a competing pathway, the enzyme thiopurine methyltransferase (TPMT) catalyzes the formation of 6-methyl-mercaptopurine ribonucleotides (6MMPR) that have been correlated with hepatotoxicity4. TPMT activity is largely determined genetically. Alleles conferring high (TPMTH) and low TPMT activity (TPMTL) are inherited in autosomal, co-dominant fashion.
Approximately 89% of Caucasians carry only TPMTH alleles (TPMTH/TPMTH) and have normal TPMT activity (>23.6 EU/mL Red Blood Cell (RBC)); 11% are heterozygous (TPMTH/TPMTL) and have intermediate activity (6.7–23.5 EU/mL RBC); and 0.3% are homozygous for the same TPMTL allele (TPMTL/TPMTL), or are heterozygous with two different TPMTL alleles (TPMTL/TPMTL’; compound heterozygotes) and have low or undetectable activity (<6.7 EU/mL RBC) 4.
Current, evidence-based, therapeutic guidelines recommended weight-based dosing (MP 1.0–1.5 mg/kg/day and AZA 2.0–3.5 mg/kg/day) that ignores the variation of TPMT activity2, 3. In patients who are homozygotes for low activity, severe, potentially fatal, myelosuppression occurs within weeks of initiating therapy5. Thiopurine therapy should be avoided in such patients. For heterozygotes with intermediate activity, the risk of myelosuppression correlates with excessive production of the 6TGN metabolites at standard AZA/MP doses. Such patients should be treated with lower than usual AZA/MP doses. Conversely, some patients fail to respond because of insufficient production of the 6TGN metabolites due to an excessive TPMT activity, or other, unknown mechanisms. These patients may benefit from higher than the standard doses4.
Measuring TPMT activity has been recommended by the FDA and the American Gastroenterological Association before initiating weight-based dosing6, 7. Further dose adjustments may be guided by metabolite concentrations, aiming at improving response and minimizing toxicity 4. The aims of this randomized trial were to compare the efficacy and safety of standard, weight-based AZA dosing versus individualized AZA dosing in steroid-dependent, steroid-refractory and steroid-naive Crohn’s disease patients. In the weight-based arm, AZA was dosed based at 2.5 mg/kg/day, with dosage adjustments limited to toxicity, such as leukopenia or abnormal liver chemistries. In the individualized dosing arm, subjects received an initial AZA dose based on their baseline TPMT activity. Subsequent dose adjustments were made targeting 6TGN concentrations within a therapeutic window.
MATERIALS AND METHODS
Patients
This multicenter, randomized, controlled study was conducted in twelve adult and pediatric centers in the United States and Canada between July 2005 and Dec 2007. Adults (18≤ age ≤70) and children (10≤ age<18) with established CD and with intermediate or normal TPMT enzyme activity (≥6.7 EU/ml RBC; Prometheus Labs, San Diego, CA) were eligible for the study.
The initial eligibility criteria were steroid-dependent CD or steroid-refractory CD. Steroid-dependent CD was defined as inactive CD (Crohn’s Disease Activity Index (CDAI) <150 for adults; modified CDAI (mCDAI) < 150 for children8) while receiving prednisone 10–40 mg/day or budesonide 3–9 mg/day for at least 12 weeks prior to screening; and inability to taper prednisone below 10 mg/day or budesonide below 3 mg/day without experiencing a flare within the previous 6 months. Steroid-refractory CD was defined as moderately active CD (CDAI or mCDAI 200 – 450) despite treatment with prednisone ≥ 20 mg/day (if weighing ≥ 40 kg) or 0.5 mg/kg/day (if weighing <40 kg), or budesonide ≤ 9 mg/day for the previous 4 weeks prior to the screening evaluation. To increase enrollment, eligibility was extended after April 2006 to subjects with steroid-naïve CD. These subjects had moderately active CD (CDAI or mCDAI 200 – 450) despite aminosalicylates and/or antibiotics, or were on no therapy. All steroid-naïve patients were treated for active disease with either 40 mg/day of prednisone or 9mg/day of budesonide prior to randomization to one of the two AZA dosing arms. Previous exposure to steroids was allowed, as long as subjects had not been treated with steroids for 4 weeks prior to screening.
Prior treatment with MP or AZA was an initial exclusion criterion. After April 2006, in order to increase enrollment, we allowed subjects who had previously been treated with AZA or MP and had at least a 6-month washout period before screening. Oral and/or rectal mesalamine, rectal steroids, and antibiotics used primarily to treat CD, were discontinued at the screening visit.
Additional exclusion criteria were: 1) Low TPMT activity (<6.7 U/mL RBC) at the screening visit. 2) History of a blood transfusion within 3 months before screening (transfusions interfere with the determination of TPMT activity). 3) History of prior allergic reaction to AZA or MP (pancreatitis, fever, arthritis or septic shock). 4) History of leukopenia (white blood count (WBC) < 3,500/mm3, absolute neutrophil count (ANC) < 2,000/mm3), thrombocytopenia (platelet count < 150,000) or hepatitis (ALT/AST > 2 x ULN; alkaline phosphatase or gamma GT > 1.5 x ULN). 5) Prior treatment with natalizumab. 6) Treatment with immunosuppressants (methotrexate, cyclosporin, mycophenolate, tacrolimus, thalidomide) or biologic therapies (other than natalizumab) in the 3 months before screening. 7) Treatment with any of the following medications in the 2 weeks before screening: allopurinol (inhibits xanthine oxidase and increases the risk of leukopenia), trimethoprim-sulfamethoxazole (inhibits folate metabolism and may increase the risk of leukopenia), non-steroidal anti-inflammatory agents (NSAIDs) or aspirin > 81 mg/day (both may cause flares of CD), cholestyramine or other drugs interfering with enterohepatic circulation (may influence AZA bioavailability), furosemide and thiazide diuretics (inhibit TPMT activity and may increase the risk of leukopenia), and fish-oil preparations (may influence disease activity).
Study Design
Eligible subjects were randomized in a 1:1 ratio to weight-based dosing or individualized dosing. Random allocation sequences were generated by the study statistician using SAS® Software version 8, Cary, NC. Permuted blocks of size 2 and 4 were randomly generated within sites, stratified by adult versus pediatric status. Random assignments were generated by the Data Management Center prior to initiation of the study. The site coordinators enrolled participants. The sites requested randomization of subjects via a web site. An automated SAS program was used to retrieve the next treatment assignment for that stratum (site, adult vs. pediatric) from the pre-computed randomization sequence. The treatment assignment was faxed by secure fax to the central study pharmacist, who dispensed and shipped drug to the site coordinator on blinded cards.
Azathioprine dosing in the two arms is depicted in Figure 1. In the induction phase of the study (weeks #0–28), subjects in the weight-based dosing arm received AZA at a daily dose of 1.0 mg/kg/day for weeks #0 and #1, and at a daily dose of 2.5 mg/kg/day for weeks #2–28. For subjects in the individualized dosing arm, the starting induction dose was determined by the baseline TPMT activity. Subjects with intermediate and normal TPMT activity began AZA at 1.0 mg/kg/day and 2.5 mg/kg/day respectively. Starting at week #5, the Data Management Center (DMC) adjusted the dose to achieve a RBC 6TGN concentration of 250–400 pmol/8 x 108 RBCs, or to a maximum daily dose of 4mg/kg body weight, whichever was achieved first. Doses were changed in increments of 12.5 mg if the dose was less than 50 mg/day, and increments of 25 mg if the dose was ≥50 mg/day. Subjects in clinical remission at week 28 entered into the maintenance phase of the study (weeks 29–52). Dosing continued in the same manner as in the induction phase.
FIGURE 1.

Azathioprine dosing in the two arms
For either group, in the event of myelosuppression or hepatotoxicity, the DMC made dosage adjustments according to a pre-specified algorithm. Daily doses were rounded upward or downward to the nearest multiple of 12.5 mg if the total dose was less than 50 mg/day. If the total dose was 50 mg or higher, then it was rounded to the nearest 25 mg increment.
Steroid-naïve subjects entered the study with moderately active disease (CDAI or mCDAI of 200–450), requiring prednisone or budesonide. At the screening visit, subjects were treated at the discretion of the investigator with either 40 mg/day of prednisone, or 9mg/day of budesonide. In all subjects, prednisone or budesonide were tapered by week #13 according to a pre-specified steroid taper.
Prometheus Laboratories, Inc. performed the TPMT, 6TGN and 6MMPR assays and transmitted the results to the DMC. Results of the routine laboratory assessments were entered on a secure DMC website. Using defined protocols, a computer program determined whether it was necessary to make a dose adjustment.
Subjects, investigators and study coordinators were blinded to treatment assignment, baseline TPMT activity, and metabolite concentrations. Azathioprine (50 mg) and placebo (in tablets identical in size, shape and color to the AZA tablets) were provided by Prometheus Laboratories. Subjects received the study drug and placebo in “blister-packed” cards. Each week’s blister pack contained 3 blisters per day, each blister labeled either A, B or C. Subjects were instructed which columns to take, in order to receive the correct dose, while also allowing for a dose change within the same blister pack. Dose adjustments were made by changing the combination of columns in the blister pack. To maintain blinding, blister packs contained both active AZA and placebo in order to allow both the weight-based dosing and the individualized dosing blister packs to look identical, and have the ability to change the columns and achieve the proper dose.
Safety and Efficacy Evaluations
The primary outcome measure was clinical remission at week#16. For the steroid-dependent subjects, clinical remission was defined as complete withdrawal of corticosteroids, and Crohn’s Disease Activity Index (CDAI) score <150 in adults, or modified CDAI (mCDAI) score <150 in children. For the steroid-refractory and the steroid-naïve subjects, clinical remission was defined as CDAI score <150 (or mCDAI <150 in children), and a reduction of at least 70 points from the baseline score (CDAI or mCDAI), and complete withdrawal of corticosteroids.
Subjects were evaluated at weeks -2 (screening visit), 0 (baseline visit), and 2, 4, 8, 12, 16, 20, 24, 28, 36, 44 and 52. Measures of disease activity (CDAI in adults, mCDAI in children), measures of quality of life (IBDQ in adults, IMPACT in children), adverse events, and concomitant medications were recorded at each visit. TPMT activity was measured at the screening visit and on week #28. Complete blood counts and liver chemistries were performed at each visit. AZA metabolites (6TGN and 6MMPR) were measured at weeks 4, 8, 12, 16, 20, 24, 28, 36, and 44.
Additional clinical evaluations and laboratory tests were required after dose adjustments in the individualized-dose group. To maintain blinding, subjects in the weight-based dose group were selected by a computer-generated program to receive a “sham” dose adjustment, which required the same additional evaluations and tests as the individualized-dose group. In both arms, additional safety visits were required when cytotoxicity or hepatotoxicity occurred.
Unscheduled visits were required for any worsening symptoms. Subjects whose condition worsened (increase in the CDAI/mCDAI > 100 points above baseline, or CDAI/mCDAI > 450, or at the discretion of the investigator) received rescue corticosteroids: The prednisone dose was increased to 40 mg/d for one week, then decreased by 5 mg per week to a dose of 20 mg/d, and subsequently decreased by 2.5 mg per week until it is discontinued. For subjects receiving budesonide, the dose was increased to 9 mg/day and tapered by 3 mg every 4 weeks. Subjects who did not respond after one week at 40 mg/d of prednisone or 9 mg/d of budesonide were withdrawn as treatment failures. Subjects who remained on steroids at week #28 were considered treatment failures.
In the maintenance phase of the study, subjects who developed an increase in CDAI of >100 points from the 28 week score, had scores greater than 200 for two consecutive visits, or according required treatment with corticosteroids, infliximab or any other treatment for active CD, were considered treatment failures.
Subjects who developed any of the following events were also considered treatment failures and were withdrawn from the study: severe CD requiring parenteral steroids, hospitalization or surgery; worsening perianal disease that made continued participation in the study ill advised; myelosuppression or hepatotoxicity that did not resolve after the drug had been discontinued for 3 weeks; pancreatitis, severe allergic reaction, opportunistic infection, malignancy, intolerable adverse effects, or any disease and/or laboratory abnormalities that, in the investigators’ judgment, made continued participation in the study ill advised.
Statistical Analysis
The primary outcome measure was the proportion of subjects achieving clinical remission at week #16. We assumed that a) 80% of subjects enrolled would be steroid-naïve and 20% steroid-dependent or steroid-refractory; b) the dropout rates for the individualized and weight-based arms would be 15 and 20% respectively; c) the per protocol remission rates for the individualized and weight-based arms in the steroid-naïve group would be 85% and 65% respectively; and d) the per protocol remission rates for individualized and weight-based arms in the steroid-dependent and steroid-refractory groups would be 75% and 55% respectively. Under these assumptions, we expected remission rates of 66.4% and 47.25% for the individualized and weight-based arms respectively. With an alpha level of 0.05, the number needed in each group to achieve 80% power was 113.
Secondary outcome measures were: 1) Proportion of subjects maintaining clinical remission at week #28; 2) Proportion of subjects maintaining clinical remission at week #52; 3) Frequencies of adverse events (AE); 4) Corticosteroid use; 5) Adult and pediatric health-related quality of life (HRQOL) index scores (Inflammatory Bowel Disease Questionnaire (IBDQ) in adults and IMPACT in children); and 6) Metabolite concentrations, TPMT activity, serum transaminases, and cell counts.
The primary analysis was based on the intention-to-treat (ITT) principle. All randomized subjects who received at least one dose of the study drug were included in the intent-to-treat analysis. A secondary, per-protocol analysis (PP) was performed in which pre-defined non-compliers, protocol violators and voluntary withdrawals were excluded. Subgroup analyses were planned by age (pediatric and adult) and by disease category (steroid-dependent, steroid-refractory and steroid-naïve).
Baseline characteristics measured on a nominal or ordinal scale were compared using a Fisher’s exact test or Wilcoxon rank-sum test as appropriate. Continuous variables (after appropriate transformation to normalize the data, if required) were compared using the unpaired t-test.
For the ITT analysis the proportion of subjects in remission at week #16 was defined as the number of subjects in remission at week #16 compared to the total number of subjects at inception. For the PP analysis, we excluded all subjects who were non-compliers with the protocol. Frequencies of AEs were compared using chi square. Mean disease activity indices (CDAI, mCDAI), mean corticosteroid dose, mean quality of life indices (IBDQ, IMPACT), 6TGN and 6MMPR concentrations, cell counts (white blood cells (WBC) and absolute neutrophil count (ANC)), and serum transaminases (alanine aminotransferase (ALT) and aspartate aminotransferase (AST)) were compared using mixed effect models (or weighted least squares estimates as appropriate) to estimate a treatment effect. Baseline TPMT activity and TPMT activity during therapy at week #28 were compared using the Mann-Whitney rank-sum test. All tests of significance were two-tailed at the 0.05 level.
Another pre-planned analysis was an examination of metabolites at early time points (concentrations, concentration ratios, and changes in concentrations after dose adjustments) as predictors of remission in univariate and multiple logistic regression analyses.
We used SAS Software (Cary, NC), version 9, for the analyses.
Roles of Steering Committee, Executive Committee, Data and Safety Monitoring Board and Funding Organizations
The study was designed by the P.I. (SBH) and the Executive Committee (TD, MD, RS and ES) and was funded by the National Institutes of Health. Prometheus Laboratories, Inc. supplied the AZA and placebo tablets and provided partial support for the TPMT, 6TGN and 6MMPR assays. Prometheus Laboratories did not have any role in study design, data analysis, data interpretation, or writing of the manuscript. The Executive committee oversaw all major decisions regarding study structure and scientific design. The committee met quarterly to assess enrollment and to address any issues regarding recruitment and trial operations. The committee addressed any questions related to eligibility, protocol violations or study inquiries. A five-member Data and Safety Monitoring Board DSMB acted in an advisory capacity to the National Institute of Diabetes, Digestive and Kidney Diseases (NIDDK) to monitor patient safety and evaluate the efficacy of the intervention.
ETHICAL CONSIDERATIONS
The institutional review board or ethics committee at each site approved the protocols. All subjects gave written informed consent. The study was registered at www.clinicaltrials.gov (NCT00113503).
RESULTS
Subject characteristics
Sixty-nine subjects (53 adults and 16 children) were screened. We did not capture reasons for screen failure. Fifty subjects (45 adults and 5 children) were randomized to the weight-based (n=25) and individualized dosing (n=25). Patient characteristics are listed in Table 1. Patient flow is depicted in Figure 2.
TABLE 1.
Patient Characteristics
| Weight-based dose (n=25) | Individualized dose (n=25) | P | |
|---|---|---|---|
| Male | 10 (40%) | 12 (48%) | 0.78 |
| Mean age (years) | 36.5±16.1 | 34.1±13.1 | 0.57 |
| Mean weight (kg) | 70.1±20.5 | 73.2±19.8 | 0.59 |
| White | 24 (96%) | 23 (92%) | 1.0 |
| Adults/children | 22/3 | 23/2 | 1.0 |
| Disease duration (months) | 70±114 | 114±127 | 0.20 |
| Disease location | 0.68 | ||
| Ileal | 9 (36%) | 11 (44%) | |
| Colonic | 4 (16%) | 2 (8%) | |
| Ileocolic | 12 (48%) | 12 (48%) | |
| Steroid status | 0.49 | ||
| Steroid-dependent | 12 (48%) | 12 (48%) | |
| Steroid-refractory | 9 (36%) | 6 (24%) | |
| Steroid-naive | 4 (16%) | 7 (28%) | |
| Baseline TPMT (EU/mL RBC) | 28.9±7.6 | 28.6±9.2 | 0.90 |
| High/Intermediate TPMT | 21/4 | 19/6 | 0.73 |
| Current smoking | 2 (8%) | 3 (12%) | 1.0 |
FIGURE 2.

Flow of patients
Efficacy
The trial began enrolling subjects in July 2005. Due to inadequate enrollment, the principal investigator (SBH), with the advice and consent of the NIDDK sponsors, stopped enrollment after two and a half years, in December 2007. Only 5 children were enrolled into the study, so separate data are not presented for the pediatric population. The primary outcome measure of clinical remission at week #16 was achieved by 40% (10 of 25) of subjects randomized to the individualized dosing arm, compared to 16% (4 of 25) of subjects in the weight-based dosing arm (p=0.11) (Table 2). Individualized dosing was associated with an absolute increase in the probability of the primary outcome of 24% (95% CI, 0.01% – 47.99%; p=0.05), or a relative increase of 250% (95% CI, 90.29% – 692.21%; p=0.11).
TABLE 2.
Clinical remission rates at week 16 in the intention-to-treat and per-protocol analyses.
| Intention to Treat | Per protocol | |
|---|---|---|
| Weight-based dosing | 4/25 (16%) * | 3/12 (25%) ** |
| Individualized dosing | 10/25 (40%) | 9/15 (60%) |
P=0.11
P=0.12
In the per protocol (PP) analysis, remission rates at week 16 were 60% (9/15) and 25% (3/12) in the individualized and weight-based dosing groups respectively (p=0.12). There were no differences in the ITT remission rates at week #28 (3/25 (12%) vs. 8/25 (32%) in the weight-based and individualized dosing arms respectively; p=0.17) and week #52 (2/25 in both arms) (Figure 2). There were no differences between the two groups in CDAI, IBDQ, or steroid use during the course of the trial (Supplemental Material).
In a pre-specified analysis, steroid status was the only patient characteristic associated with the primary outcome of remission at week #16. Remission rates were 37.1% (13/35) for steroid-dependent and -naïve patients, vs. 6.7% (1/15) for steroid-refractory patients (p=0.04; Fisher’s exact test). The small number of subjects precluded the planned analyses of metabolites at early time points as predictors of remission.
Azathioprine dosing
In the PP analysis, there were no differences between the two study arms in the mean AZA doses at week 16 (Table 3). However, these comparisons conceal differences according to baseline TPMT. Among normal TPMT metabolizers, mean AZA doses were 2.3 and 3.4 mg/kg/day in the weight-based and individualized dosing arms respectively (p<0.0001). Among intermediate TPMT metabolizers, mean doses were 2.4 and 1.5 mg/kg/day in the weight-based and individualized dosing arms respectively (p=0.02). As might have been expected from the study design, mean AZA dose in the weight-based arm was similar to the initial dose (2.5 mg/kg/day) in both normal (2.3 mg/kg/day) and intermediate TPMT metabolizers (2.4 mg/kg/day). In the individualized dosing arm, mean AZA doses at week #16 were higher than the starting doses for both the normal TPMT metabolizers (3.4 vs. 2.5 mg/kg/day; p=0.15) and the intermediate TPMT metabolizers (1.5 vs. 1.0 mg/kg/day; p=0.004) (Table 3).
TABLE 3.
AZA doses and metabolite concentrations in the per protocol subjects at week #16, analyzed by baseline TPMT activity
| INDIVIDUALIZED DOSE | WEIGHT-BASED DOSE | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||
| N | Mean (SD) | Median (range) | N | Mean (SD) | Median (range) | Means p value | Medians M-W p | ||
| Dose | Overall | 15 | 2.7 (1.06) | 3.1 (0.6–4.0) | 12 | 2.3 (0.41) | 2.4 (1.6–2.7) | 0.23 | 0.16 |
| Normal TPMT | 9 | 3.4 (0.35) | 3.4 (2.8–4.0) | 10 | 2.3 (0.45) | 2.4 (1.6–2.7) | <0.0001 | 0.0003 | |
| Intermediate TPMT | 6 | 1.5 (0.65) | 1.8 (0.6–2.1) | 2 | 2.4 (0.01) | 2.4 (2.4–2.4) | 0.02 | 0.07 | |
| TGN | Overall | 14* | 216 (102) | 199 (81–413) | 12 | 230.0 (242) | 156 (81–972) | 0.86 | 0.40 |
| Normal TPMT | 8* | 198 (70) | 197 (81–286) | 10 | 163 (71) | 150 (81–297) | 0.32 | 0.36 | |
| Intermediate TPMT | 6 | 242 (137) | 212 (88–413) | 2 | 563 (578) | 563 (154–972) | 0.58 | 0.64 | |
| MMPR | Overall | 14* | 4872 (4437) | 2873 (782–14631) | 12 | 3407 (3877) | 2076 (782–15152) | 0.38 | 0.37 |
| Normal TPMT | 8* | 7035 (4798) | 6362 (2193–14631) | 10 | 3861 (4119) | 2292 (1230–15152) | 0.16 | 0.08 | |
| Intermediate TPMT | 6 | 1987 (1224) | 1977 (782–3494) | 2 | 1138 (503) | 1138 (782–1493) | 0.22 | 0.64 | |
Metabolite data at week 16 were missing for one subject with normal TPMT activity in the individualized dosing arm.
Pharmacokinetics
Mean 6TGN concentrations (230 vs. 215 pmol/8 x 108 RBCs in the weight-based and individualized arms; p=0.85) and 6MMPR concentrations (3407 vs. 5228 pmol/8 x 108 RBCs; p=0.26) were similar in the two treatment arms at week #16 (Table 3). Again however, these comparisons conceal differences according to baseline TPMT activity in each arm. Metabolite concentrations through the course of the trial in individual subjects within each treatment arm and stratified by baseline TPMT activity are depicted in scatter plot format in Figures #3A–3D. Surprisingly, mean 6TGN concentrations at week#16 among normal TPMT metabolizers in the individualized arm (198 pmol/8 x 108 RBCs) were below the “therapeutic” target of 250 pmol/8 x 108 RBCs, and not significantly higher than the corresponding concentrations of the normal TPMT metabolizers of the weight-based arm (163 pmol/8 x 108 RBCs). Median 6TGN concentrations for the two groups were 197 (81–286) and 150 (81–297) pmol/8 x 108 RBCs respectively (NS). It must be remembered that these (non-significant) differences in 6TGN concentrations occurred on a backdrop of different final doses at week 16 (median doses of 3.4 vs. 2.3 mg/kg/day in the individualized and weight-based dosing groups respectively (p<0.0001)).
FIGURE 3.

Scatter plot, mean values and 95% confidence intervals for the 6TGN and 6MMPR concentrations in individual subjects by TPMT activity in the individualized dosing arm (
 ) and in the weight-based arm (Δ) (per protocol analysis). A: 6TGN concentrations in normal metabolizers. B: 6MMPR concentrations in normal metabolizers. C: 6TGN concentrations in intermediate metabolizes. D: 6MMPR concentrations in intermediate metabolizes.
) and in the weight-based arm (Δ) (per protocol analysis). A: 6TGN concentrations in normal metabolizers. B: 6MMPR concentrations in normal metabolizers. C: 6TGN concentrations in intermediate metabolizes. D: 6MMPR concentrations in intermediate metabolizes.
As may have been predicted, intermediate TPMT metabolizers in the individualized arm achieved higher 6TGN concentrations than normal TPMT metabolizers in the same arm (despite a much lower AZA dose: 1.5 vs. 3.4 mg/kg/day): mean 6TGN 242 vs. 198 pmol/8 x 108 RBCs (p=0.067). In the weight-based arm, intermediate and normal TPMT metabolizers were similarly dosed, but intermediate TPMT metabolizers attained much higher 6TGN concentrations: mean 6TGN 563 vs. 163 pmol/8 x 108 RBCs (p=0.0001).
At week #16, median (range) 6TGN concentrations in PP remitters (n=12) and non-remitters (n=14; data for the fifteenth non-remitter were missing) were 216 (127–413) and 149 (81–972) pmol/8 x 108 RBCs respectively (p=0.07) (Figure 4). Mean 6TGN concentrations were 229 and 217 pmol/8 x 108 RBCs respectively (p=0.90).
FIGURE 4.
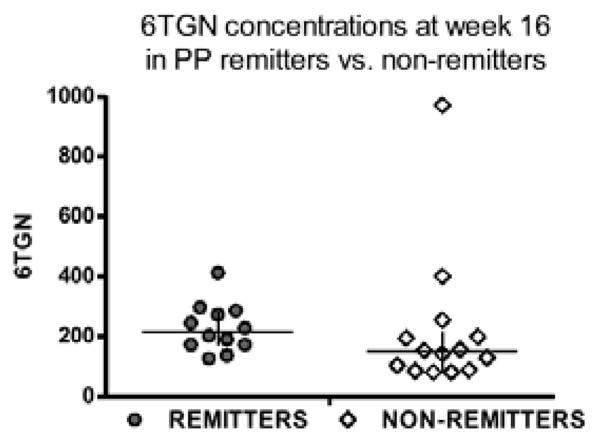
Scatter plot, median values and interquartile ranges of the 6TGN concentrations at week 16 in the per-protocol remitters (n=12) and non-remitters (n=14). Median 6TGN concentrations were 216 and 149 pmol/8 x 108 RBCs in remitters and non-remitters respectively (p=0.07).
[Note: There were 15 non-remitters at week 16, but 6TGN data were missing for one subject with normal TPMT activity in the individualized dosing arm. At week 12, this subject had TGN and 6MMP concentrations of 198 and 2633 pmol/8 x 108 RBCs respectively.]
Median (range) TPMT activity at baseline and at week #28 was 25.1 (5.5–43.4) and 26.3 (8.1–36.6) EU/mL RBC respectively (Mann-Whitney rank-sum test, p=0.87).
Safety
There was a high number of early withdrawals (Figure 2), including AZA hypersensitivity reactions (n=8), pancreatitis (n=1), erythema multifome (n=1) and other adverse events (n=2). Twenty-three patients withdrew before week 16: 15 within the first 4 weeks, and another 8 between weeks 4 and 16. Almost all withdrawals before week 16 were normal TMPT metabolizers. In the weight-based dosing arm, there were 21 normal (84%) and 4 intermediate (16%) TMPT metabolizers at the beginning of the trial. At week #16, 10 normal (83%) and 2 intermediate (17%) TMPT metabolizers remained in the trial. In the individualized dosing arm, there were 19 normal (76%) and 6 intermediate (24%) TMPT metabolizers at the beginning of the trial. At week #16, there were 9 (60%) and 6 (40%) respectively. There were no differences in the frequencies of severe myelosuppression or severe hepatotoxicity. Additionally, there were no differences between the two arms in the mean values of ALT, AST, WBC, and ANC. White blood counts were inversely correlated with 6TGN concentrations (r= −0.29, p<0.0001).
DISCUSSION
Standard, weight-based dosing of AZA in CD disregards the genetic variability of TPMT activity, which determines the relative production of the 6TGN and 6MMPR metabolites. Furthermore, standard dosing disregards other factors that may determine the absolute and relative concentrations of these metabolites. We hypothesized that individualized dosing based on baseline TPMT activity, and subsequently adjusted according to the 6TGN concentrations, might be more effective and safer than standard, weight-based AZA dosing in inducing remission of CD. Despite a trend favoring individualized dosing, we found no statistically significant difference in efficacy. The primary outcome of clinical remission at week #16 was achieved by 40% (10 of 25) in the individualized and 16% (4 of 25) in the weight-based dosing arms (p=0.11). In the PP analysis, remission rates at week 16 were 60% (9/15) and 25% (3/12) in the individualized and weight-based dosing arms respectively (p=0.12). Although there were no differences in adverse events between the arms, the safety of individualized dosing will need to be assessed in larger studies. Our findings should be viewed in the contexts of the inadequate enrollment of subjects, under-dosing in normal TPMT metabolizers, and high frequency of early withdrawals, mostly due to AZA-related adverse events.
Despite failing to meet the primary endpoint, our study provides some interesting lessons. First, there was a trend towards higher median 6TGN concentrations among PP remitters vs. non-remitters: 216 (127–413) vs. 149 (81–972) pmol/8 x 108 RBCs respectively (p=0.07). These findings, although based on the PP analysis and not statistically significant, suggest that higher 6TGN concentrations might be associated with improved efficacy and effectiveness.
As studies examining the correlation of 6-TGN concentrations with thiopurine effectiveness have yielded conflicting results, the concept of a therapeutic 6TGN window in CD has been controversial. A meta-analysis found that patients with 6-TGN concentrations higher than 230–260 pmol/8 x 108 were more likely to be in remission (62%) than those with concentrations below the threshold value (36%) (pooled odds ratio, 3.3; 95% confidence interval, 1.7–6.3; P < 0.001)9.
The potential importance of 6TGN (and 6MMPR) measurements has been bolstered by more recent studies that have assessed the effectiveness of AZA dose escalation 10–12 or AZA-allopurinol combination therapy 13, 14. Dubinsky et al. assessed the effects of 6-MP dose escalation on 6TGN concentrations in 51 IBD patients failing therapy 10. Responders to dose escalation achieved large increases in 6TGN levels (from 183 [39–298] to 306 [168–853] pmol/8 x 108 RBCs; P = 0.03), whereas there were small increases in the 6TGN levels of the non-responders (136 [50–378] to 155 [90–707]; P = 0.046). The absolute change in 6TGN levels from baseline to follow-up was significantly higher among the responders vs. non-responders (median: 122 [ −2 to +627] vs. 26 [ −177 to +503]; P = 0.0003). Both at baseline and upon dose escalation, responders were characterized by preferential 6TGN production and non-responders by preferential 6MMPR production. In a prospective study from New Zealand, 52 patients received AZA or 6-MP for 9 months dosed to target 6TGN concentrations of 235–450 pmol/8 x 108 RBCs. The percentage change in the short IBD score correlated significantly with 6TGN concentrations (r = 0.37, P = 0.01) 11. An Australian study assessed the utility of metabolite measurements in directing therapy in patients failing AZA12. Of the14 patients with low 6TGN and non-elevated 6MMPR concentrations that had AZA dose increases, 12 improved, 1 developed leucopenia, and 1 failed 12.
Data on the pharmacokinetics and effectiveness of AZA-allopurinol combination therapy also lend support to the concept of a therapeutic 6TGN window. Sparrow et al. treated 20 thiopurine non-responders who were preferential 6MMPR metabolizers with combination allopurinol 100 mg po daily and AZA or MP at 25–50% of the original dose 13. 6TGN concentrations increased from 191 ± 17 to 400 ± 37 pmol/8 x 108 RBCs (P < 0.001), while 6MMPR concentrations decreased from 10,605 ± 1278 to 2001± 437 pmol/8 x 108 RBCs (P < 0.001). Combination therapy was safe and was associated with decreased disease activity, steroid sparing and normalization of transaminases. In CD patients (n=12), the mean partial Harvey Bradshaw Index decreased from 4.9 ± 1.0 to 1.5 ± 0.3 (P = 0.001). In patients with ulcerative (n=6) or indeterminate colitis (n=2), the mean Mayo Score decreased from 4.1 ± 0.7 to 2.9 ± 0.7 (P =0.13) 13. The same group extended their findings in a report on the long term effectiveness and safety of AZA-combination therapy in 25 patients with Crohn’s disease and ulcerative colitis 14. Within the first month of therapy 6TGN metabolites increased from 186.5 ± 17.4 to 352.8 ± 37.8 pmol/8×108 (p=0.0001). Over the same period 6MMPR concentrations decreased from 11,966 ± 1697 to 2004 ± 536 pmol/8×108 (p<0.0001). The mean daily dosage of prednisone decreased from 19.8 ± 3.8 mg to 5.3 ± 2.7 mg (p=0.03). Thirteen patients had a minimum of one year follow-up, 9 of whom had continued on therapy for at least 2 years. All 13 remained in clinical remission at the last follow-up visit. No patients have had evidence of sustained thrombocytopenia or abnormal liver enzymes 14. Allopurinol thus shifts thiopurine metabolism in non-responding, preferential 6MMPR metabolizers towards 6TGN. In these open label studies, an increase in 6TGN concentrations from approximately 190 to 350–400 pmol/8×108 was associated with improved clinical response. Subsequent open-label studies from other institutions have also shown clinical benefits from combination AZA-allopurinol therapy15, 16.
In summary, accumulating evidence suggests that 1) higher 6TGN concentrations correlate with thiopurine effectiveness and 2) the relative abundance of the 6TGN and 6MMPN metabolites partly determines therapeutic success. An open label, randomized trial found no difference between 6TGN–adjusted and weight-based AZA dosing in patients with active CD (CDAI 150–450)17. The open label design, dose adjustment based on 6TGN concentrations only (i.e. not accounting for the baseline TPMT activity) and finally the nonattainment of target 6TGN concentrations in the adjusted arm are significant limitations of the trial.
A second important lesson from our study concerns the appropriate initial AZA dose and subsequent dose changes. Normal TPMT metabolizers in the individualized arm received incremental dose changes from a starting dose of 2.5 mg/kg/day, but their 6TGN concentrations at week#16 (mean and median values of 198 mol/8x108 RBCs) were well below the target window (250–400 mol/8x108 RBCs). As might have been expected, intermediate TPMT metabolizers in the individualized arm achieved concentrations much closer to the target (mean 242 and median 212 mol/8x108 RBCs). Our original study design specified dosage changes in 50 mg increments, but the regulatory agencies requested more conservative changes of 25 mg increments.
Assuming a higher starting dose and/or higher dose changes, the final doses in normal TPMT metabolizers would have been higher than 3.4 mg/kg/day (the mean dose at week 16 in our study) and more than two-fold higher than the dose in the intermediate TPMT metabolizers. Our results are congruent with those of the aforementioned study from New Zealand 11. In that study, IBD patients starting AZA or MP were genotyped for TPMT and were followed for 9 months. The thiopurine dose was individualized to target TGN concentrations of 235–450 pmol/8 x 108 RBCs. The mean initial dose (as AZA equivalents) was similar (approximately 1 mg/kg/d) for the two TPMT genotypes, but after 9 months the dose was 50% lower in the heterozygous group (0.9 vs. 1.8 mg/kg/day, P < 0.0001). Despite dose adjustment, median 6TGN concentrations still were two-fold higher in that group at the end of the follow-up period (505 vs. 273 pmol/8 x 108 RBCs, P = 0.02). This difference was three-fold when the concentration was adjusted for dose (578 vs. 183 pmol/8 x 108 per mg/kg/d, P =0.0007). The results were similar if TPMT phenotype was used instead of genotype 11. On the basis of that study and ours, we conclude that intermediate and normal TMPT metabolizers should be dosed differently. Intermediate TPMT metabolizers should be dosed at 1–1.5 mg/kg/day. Normal TPMT metabolizers may be treated with doses exceeding 3.5 mg/kg/day.
Thiopurine intolerance is a significant contributor to lack of efficacy. We observed a very high rate (46%; 23/50) of early drug discontinuation (Figure 2), mostly due to adverse events, including AZA hypersensitivity reactions (n=8), pancreatitis (n=1), erythema multifome (n=1), pneumonia/sepsis (n=1), severe myelosuppression (n=1) and other adverse events (n=3). Ansari et al. found that co-administration of allopurinol may reduce certain types of thiopurine intolerance (nausea, myalgia and fatigue)18. Interestingly, we did not observe these types of intolerance. Our subjects experienced other adverse events that may or may not have been abrogated by low dose AZA-allopurinol therapy.
Why then did our trial fail to meet its primary endpoint? There are several reasons: 1) Statistical power was low due to inadequate enrollment and premature termination of the trial; 2) There was a high frequency of early withdrawals, mostly due to AZA-related adverse events; and 3) As discussed, the starting dose and dose changes in the normal TPMT metabolizers were too conservative.
In summary, although we found a trend favoring individualized over weight-based AZA dosing, there were no statistically significant differences in efficacy, likely due to low statistical power and inability to achieve the target 6TGN concentrations in the individualized dosing arm. Clinicians should be aware that standard, weight based dosing (2.5 mg/kg/day) often produces subtherapeutic 6TGN concentrations among normal TPMT metabolizers. Conversely, intermediate TPMT metabolizers should be treated with lower doses (1.5 kg/kg/day). Prospective studies using higher doses or combination therapy with allopurinol are needed to address the hypothesis that 6TGN-guided dosing leads to higher efficacy in normal TPMT metabolizers.
Supplementary Material
Acknowledgments
Financial Support
Authors’ declaration of personal interests:
Themistocles Dassopoulos, M.D., has served as consultant for Prometheus.
Marla C. Dubinsky, M.D., has served as consultant for Prometheus.
Jennifer L. Bentsen, B.A.: No personal interests.
Christopher F. Martin, M.S.P.H.: No personal interests.
Joseph A. Galanko, Ph.D.: No personal interests.
Ernest Seidman, M.D. has served as consultant for Prometheus.
Robert S. Sandler, M.D., M.P.H. No personal interests.
Stephen B. Hanauer, M.D.: No personal interests.
Declaration of funding interests:
This work was supported by NIH grants U01 DK60083 (SBH) and P30DK34987 (RSS and JAG); the Meyerhoff IBD center at Johns Hopkins University, Baltimore, MD (TD); and the Crohn’s and Colitis Foundation of America (RSS). Prometheus Laboratories, Inc. supplied the azathioprine and placebo tablets and provided partial support for the enzyme and metabolite assays.
We acknowledge the investigators and staff of the participating centeres: University of Chicago Hospitals, Chicago, IL (Adults: Stephen B. Hanauer; Children: Barbara S. Kirschner); Johns Hopkins Hospital, Baltimore, MD (Themistocles Dassopoulos); Hospital for Sick Children, Toronto (Ann M. Griffiths); University of North Carolina, Chapel Hill, NC (Kim L. Isaacs); Atlanta Gastroenterology Associates, Atlanta, GA (Douglas C. Wolf); Mount Sinai Hospital, New York, NY (Maria T. Abreu); University of Pittsburgh Medical Center, Pittsburgh, PA (Miguel D. Regueiro); London Health Sciences Centre, London, Ontario (Brian G. Feagan); University of Alberta, Edmonton, Alberta (Richard N. Fedorak); Mayo Clinic, Rochester, MN (William J. Sandborn); Medical College of Wisconsin, Milwaukee, WI (David G. Binion)
Footnotes
Guarantor of the article
Themistocles Dassopoulos
All authors approved the final version of the manuscript.
Specific author contributions
Themistocles Dassopoulos: Study concept and design, acquisition of data, analysis and interpretation of data, drafting of the manuscript, critical revision of the manuscript for important intellectual content and statistical analysis; Marla C. Dubinsky: Study concept and design, acquisition of data, and critical revision of the manuscript for important intellectual content; Jennifer L. Bentsen: Acquisition of data; critical revision of the manuscript for important intellectual content and study supervision; Christopher F. Martin: Study design, acquisition of data, analysis and interpretation of data, critical revision of the manuscript for important intellectual content, statistical analysis and technical support; Joseph A. Galanko: Study design, acquisition of data, analysis and interpretation of data, critical revision of the manuscript for important intellectual content, statistical analysis and technical support; Robert S. Sandler: Study design, acquisition of data, analysis and interpretation of data, critical revision of the manuscript for important intellectual content, statistical analysis, technical support and study supervision; Stephen B. Hanauer: Study concept and design, acquisition of data, analysis and interpretation of data, critical revision of the manuscript for important intellectual content, obtained funding and study supervision.
References
- 1.Prefontaine E, Sutherland LR, Macdonald JK, Cepoiu M. Azathioprine or 6-mercaptopurine for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev. 2009;(1):CD000067. doi: 10.1002/14651858.CD000067.pub2. [DOI] [PubMed] [Google Scholar]
- 2.Dignass A, Van Assche G, Lindsay JO, et al. The second European evidence-based Consensus on the diagnosis and management of Crohn’s disease: Current management. J Crohns Colitis. 2010;4(1):28–62. doi: 10.1016/j.crohns.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Talley NJ, Abreu MT, Achkar JP, et al. An evidence-based systematic review on medical therapies for inflammatory bowel disease. Am J Gastroenterol. 2011;106 (Suppl 1):S2–25. doi: 10.1038/ajg.2011.58. [DOI] [PubMed] [Google Scholar]
- 4.Ha C, Dassopoulos T. Thiopurine therapy in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol. 2010;4(5):575–88. doi: 10.1586/egh.10.59. [DOI] [PubMed] [Google Scholar]
- 5.Colombel JF, Ferrari N, Debuysere H, et al. Genotypic analysis of thiopurine S-methyltransferase in patients with Crohn’s disease and severe myelosuppression during azathioprine therapy. Gastroenterology. 2000;118(6):1025–30. doi: 10.1016/s0016-5085(00)70354-4. [DOI] [PubMed] [Google Scholar]
- 6.Imuran ® (azathioprine) product information. Available at: www.accessdata.fda.gov/drugsatfda_docs/label/2011/016324s034s035lbl.pdf.
- 7.Lichtenstein GR, Abreu MT, Cohen R, Tremaine W. American Gastroenterological Association Institute technical review on corticosteroids, immunomodulators, and infliximab in inflammatory bowel disease. Gastroenterology. 2006;130(3):940–87. doi: 10.1053/j.gastro.2006.01.048. [DOI] [PubMed] [Google Scholar]
- 8.Belli DC, Seidman E, Bouthillier L, et al. Chronic intermittent elemental diet improves growth failure in children with Crohn’s disease. Gastroenterology. 1988;94(3):603–10. doi: 10.1016/0016-5085(88)90230-2. [DOI] [PubMed] [Google Scholar]
- 9.Osterman MT, Kundu R, Lichtenstein GR, Lewis JD. Association of 6-thioguanine nucleotide levels and inflammatory bowel disease activity: a meta-analysis. Gastroenterology. 2006;130(4):1047–53. doi: 10.1053/j.gastro.2006.01.046. [DOI] [PubMed] [Google Scholar]
- 10.Dubinsky MC, Yang H, Hassard PV, et al. 6-MP metabolite profiles provide a biochemical explanation for 6-MP resistance in patients with inflammatory bowel disease. Gastroenterology. 2002;122(4):904–15. doi: 10.1053/gast.2002.32420. [DOI] [PubMed] [Google Scholar]
- 11.Gardiner SJ, Gearry RB, Begg EJ, Zhang M, Barclay ML. Thiopurine dose in intermediate and normal metabolizers of thiopurine methyltransferase may differ three-fold. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2008;6(6):654–60. doi: 10.1016/j.cgh.2008.02.032. [DOI] [PubMed] [Google Scholar]
- 12.Haines ML, Ajlouni Y, Irving PM, et al. Clinical usefulness of therapeutic drug monitoring of thiopurines in patients with inadequately controlled inflammatory bowel disease. Inflammatory bowel diseases. 2011;17(6):1301–7. doi: 10.1002/ibd.21458. [DOI] [PubMed] [Google Scholar]
- 13.Sparrow MP, Hande SA, Friedman S, Cao D, Hanauer SB. Effect of allopurinol on clinical outcomes in inflammatory bowel disease nonresponders to azathioprine or 6-mercaptopurine. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2007;5(2):209–14. doi: 10.1016/j.cgh.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 14.Leung Y, Sparrow MP, Schwartz M, Hanauer SB. Long term efficacy and safety of allopurinol and azathioprine or 6-mercaptopurine in patients with inflammatory bowel disease. J Crohns Colitis. 2009;3(3):162–7. doi: 10.1016/j.crohns.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 15.Govani SM, Higgins PD. Combination of thiopurines and allopurinol: adverse events and clinical benefit in IBD. J Crohns Colitis. 2010;4(4):444–9. doi: 10.1016/j.crohns.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith MA, Blaker P, Marinaki AM, Anderson SH, Irving PM, Sanderson JD. Optimising outcome on thiopurines in inflammatory bowel disease by co-prescription of allopurinol. J Crohns Colitis. 2012;6(9):905–12. doi: 10.1016/j.crohns.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 17.Reinshagen M, Schutz E, Armstrong VW, et al. 6-thioguanine nucleotide-adapted azathioprine therapy does not lead to higher remission rates than standard therapy in chronic active crohn disease: results from a randomized, controlled, open trial. Clinical chemistry. 2007;53(7):1306–14. doi: 10.1373/clinchem.2007.086215. [DOI] [PubMed] [Google Scholar]
- 18.Ansari A, Patel N, Sanderson J, O’Donohue J, Duley JA, Florin TH. Low-dose azathioprine or mercaptopurine in combination with allopurinol can bypass many adverse drug reactions in patients with inflammatory bowel disease. Alimentary Pharmacology & Therapeutics. 2010;31(6):640–7. doi: 10.1111/j.1365-2036.2009.04221.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.