

SUMMARY
Nuclear export of unspliced and singly spliced viral mRNA is a critical step in the HIV life cycle. The structural basis by which the virus selects its own mRNA among more abundant host cellular RNAs for export has been a mystery for more than 25 years. Here, we describe an unusual topological structure that the virus uses to recognize its own mRNA. The viral Rev response element (RRE) adopts an “A”-like structure in which the two legs constitute two tracks of binding sites for the viral Rev protein and position the two primary known Rev-binding sites ~55 Å apart, matching the distance between the two RNA-binding motifs in the Rev dimer. Both the legs of the “A” and the separation between them are required for optimal RRE function. This structure accounts for the specificity of Rev for the RRE and thus the specific recognition of the viral RNA.
INTRODUCTION
Virtually all mRNAs in mammalian cells are spliced before leaving the nucleus. However, retroviral replication requires that some viral RNAs be exported while retaining some or all of their introns, since these RNAs will serve as mRNAs for the gag, gag-pol, and env gene products and as genomes to be encap-sidated in progeny virions. HIV-1 fulfills this requirement by encoding the Rev protein: this protein binds to an element within the env-coding region of the viral RNA, called the Rev response element (RRE). The Rev-RRE complex then engages Crm1 (Xpol1) and RanGTP to form a host export complex enabling the translocation through the nuclear pore complex. The secondary structure of the HIV-1 RRE includes a series of stems; stems I, II, III/IV, and V are arranged around a central four-way junction, with stem-loop II split into a proximal stem (IIA) and two distal stem-loops (IIB and IIC) around a three-way junction (Legiewicz et al., 2008). There is a high-affinity site for Rev in stem-loop IIB (Battiste et al., 1996; Cook et al., 1991; Heaphy et al., 1990; Heaphy et al., 1991; Kjems et al., 1991; Lusvarghi et al., 2013; Malim et al., 1989; Malim et al., 1990; Pond et al., 2009) and a second Rev-binding site (IA) in stem I (Daugherty et al., 2008). Initial occupancy of the IIB site (Cook et al., 1991; Tiley et al., 1992) is considered a prerequisite to Rev oligomerization (Daly et al., 1989; Daly et al., 1993; Malim and Cullen, 1991; Mann et al., 1994), in which as many as 10 to 12 Rev molecules may bind to a single RRE (Mann et al., 1994), and this assembly is necessary for Rev-mediated nuclear export of RRE-containing viral transcripts (Hoffmann et al., 2012).
Recent structures of Rev-Rev dimers reveal a hydrophobic core that drives Rev-Rev dimer formation and an arrangement in which two 17 residue arginine-rich motifs (ARMs) in the two monomers point away from each other, with a separation distance of ~55 Å and an angle of 120°–140° (Daugherty et al., 2010b; DiMattia et al., 2010). Oligomerization is essential for Rev function, as demonstrated by analyses of Rev mutants that are deficient in dimerization (Malim and Cullen, 1991; Zapp et al., 1991); indeed, the C-terminal dimerization domain of the protein can be replaced by unrelated dimerization domains without loss of function (Hoffmann et al., 2012). Formation of the multimeric Rev-RRE complex is driven in part by hydrophobic Rev-Rev interactions (Cook et al., 1991; Daugherty et al., 2008; Daugherty et al., 2010b; DiMattia et al., 2010; Mann et al., 1994; Pond et al., 2009), in addition to the binding of multiple Rev proteins to less well defined binding sites on the RRE. The cooperative binding has an affinity ~500 times greater than that of the tightest single Rev-IIB interaction (Daugherty et al., 2010a; Daugherty et al., 2008), and the multiple Rev-binding events may be driven and dictated by overall topological constraints (Daugherty et al., 2010a). While significant progress has been made in the past two decades in understanding the Rev-RRE interaction, little is known about the role of the RRE architecture in the specificity and cooperativity of Rev-RRE interactions. Since the Rev-RRE interaction lacks high sequence specificity, with only preferential binding to purine-rich major grooves, and Rev could potentially interact with other RNA targets with various affinities (Bayer et al., 2005; Landt et al., 2005; McColl et al., 1999; Mishra et al., 2009; Wang et al., 2010; Xu and Ellington, 1996), the basis for selection of HIV-1 mRNA for nucleocytoplasmic transport remains a mystery (Hammarskjold and Rekosh, 2011). The key to resolving this mystery is the three-dimensional structure of the RRE.
Despite recent remarkable progress in both X-ray crystallography and solution NMR spectroscopy, the RRE overall topology and three-dimensional structure remain unknown. Therefore, alternative strategies are warranted. Small angle X-ray scattering (SAXS) is a solution-based method, does not require crystallization, and has almost no size limitation. Meaningful protein structural models of large systems have been constructed from high-resolution domain structures and SAXS data using rigid body refinement (Koch et al., 2003). Thus, almost in parallel, heuristic topological structural models of RNAs can be constructed when accurate information about the secondary structure, relative accessibility of individual residues in an RNA and other structural information are available (Funari et al., 2000; Hajdin et al., 2010; Lipfert et al., 2008). In particular, construction of RNA structural models based on SAXS-derived envelopes is feasible, because the main structural RNA elements are duplexes that exist almost exclusively in A-form conformation with a root-mean-square-deviation (RMSD) of backbone heavy atoms within 1 ± 0.5 Å over all known structures in structural databases (Wang et al., 2009). The possible resolution of such an envelope is approximately defined by 2π/q, where q is the momentum transfer. Thus, in principle, scattering data recorded for q ranging up to 0.3 Å−1 is sufficient to identify an A-form duplex that has a diameter of 20–25 Å, q ranging up to 0.8 Å−1 can clearly delineate the major groove of an RNA (X.F. and Y-X.W., unpublished data), and high-resolution and quality SAXS/WAXS data with q up to 2.3 Å−1 can even reveal fine structural features such as the spacing between phosphate groups in DNA at a ~2 Å resolution (Zuo et al., 2006). Thus, SAXS/WAXS is a bona fide tool for structure determination and characterization. When used in combination with biochemical and functional studies, as we illustrate in this study, it can address important biological questions and becomes very useful especially when high-resolution structures of RNA are unattainable using conventional methods.
RESULTS
A Unique Global Topological Structure of the RRE RNA
The genomic location of the RRE and the secondary structure of the RRE fragment used in this study are illustrated in Figure 1A. This construct is identical to one whose secondary structure was mapped using SHAPE technology (Legiewicz et al., 2008); the RRE in genomic RNA from virus particles also has the same general secondary structure as that studied here (Watts et al., 2009; Wilkinson et al., 2008). We analyzed the three-dimensional structure of RRE RNA by SAXS. The experimental SAXS curve, with scattering intensity I(q) plotted versus momentum transfer q, together with the pair distance distribution function (PDDF) and the Kratky and Porod-Debye plots of the RRE, are shown in Figures 1B–1E, respectively. The Guinier region of the scattering curve (inset in Figure 1B) is linear, indicating that RRE is mono-disperse and homogeneous in solution (Jacrot, 1976). The scattering curve in the high-q region has fine features typical of nucleic acids, such as P1 and P2 peaks (Figure 1B) (Zuo et al., 2006), albeit with attenuated peak intensity compared to those of a simple duplex. This is likely due to the presence of nonduplex structure elements as well as dynamic conformational averaging within RRE molecules. The PDDF shows that the RRE has two main distance populations, one at ~25 Å (the shoulder at the left side of the curve in Figure 1C) and one at 50–60 Å. The second derivative of the PDDF (the inset in Figure 1C) indicates that the most populated distances in the RRE are ~10, ~25, and ~55 Å. The first two are related to the dimensions across an A-form RNA duplex, whose diameter is about 25 Å, while the latter suggests the presence of a structural feature in which two major structural segments are separated by 50–60 Å. The dimensionless Kratky (Rambo and Tainer, 2013) and Porod-Debye plots (Figures 1D and 1E), plotted as (qRg)2I(q)/I0 versus qRg and I(q)·q4 versus q4 (Rambo and Tainer, 2011), respectively, suggest that the RRE RNA is extended and open, rather than bundled double helices such as in the adenine riboswitch RNA (Serganov et al., 2004) (Figures 1D and 1E). These structural features are direct observables, not subject to possible bias due to limitation of software, and are therefore important in guiding the interpretation presented below.
Figure 1. The Schematic Location of the RRE in the HIV-1 Genomic RNA, the RRE Secondary Structure, SAXS Analysis, and the Molecular Envelope.

(A) The genomic location and secondary structure of the RRE. The green and blue ovals in the secondary structure highlight the two known primary binding sites for Rev.
(B) Scattering intensity in arbitrary units versus momentum transfer q in Å−1. Note that the SAXS/WAXS data were recorded up to q = 2.3 Å−1, corresponding to a convoluted spatial resolution of ~2.8 Å. The fine features of P1 and P2 in the scattering curve arise from helical interstrand pair distance correlation and reflect scattering interference among electrons within major and minor groves (Zuo et al., 2006). The inset in (B) shows the Guinier region of the scattering curve with a linear fit line.
(C) Pair distance distribution function (PDDF) with the inset showing the absolute value of the second derivative of the PDDF. The PDDF profile was calculated using GNOM (qmax = 0.30). The second derivative of the PDDF (inset in C) gives approximate peak positions of populated pair distances in the RRE.
(D) Dimensionless Kratky plot of the RRE (dotted) and adenine riboswitch (access code: 1Y26) (solid).
(E) Porod-Debye plot of the RRE (dotted) and adenine riboswitch (solid). Plots (D) and (E) indicate that the RRE structure is more extended and open than the adenine riboswitch.
(F) The molecular envelope of the RRE RNA drawn in mesh. A scale bar of 30 Å indicates the relative dimension of the RRE envelope. The spatial resolution of the envelope is ~21 Å. See also Figure S1, Table S2, and Movie S1.
The three-dimensional structure of the RRE was determined from the SAXS data using a two-step protocol (depicted in Movie S1 available online). The resulting average envelope is shown in Figure 1F. The RRE adopts an “A”-like topology, with one leg longer than the other. This open and extended structure appears to represent the native structure at physiological Mg2+ concentration. Even at higher Mg2+ concentrations, the overall topology remains similar (Figure S1). The major portions of the two legs, i.e., the two main structural segments, are separated by 50–60 Å, in agreement with the PDDF (Figure 1C). Such an extended and open structure may permit the binding by multiple copies of Rev.
Locations of the RRE RNA Domains
We identified the locations of the individual RRE domains using a SAXS-aided divide-and-conquer approach (Figures 2 and S2A–S2F). Briefly, the construct composed of domains II, III, and IV (referred to from now on as “domains II-III-IV”), has an overall shape and size that exclusively matches the short side of the “A” (magenta construct in Figures 2 and S2A–S2D and Movie S2), whereas the construct consisting of domains I and V (called “domains I-V” below) (cyan construct, Figure 2 and Movie S2), with its unique, long stem I, matches the long side of the “A” in both shape and length. The ~25 Å envelope thickness of the magenta construct is similar to that of a duplex, indicating that domains II and III, which are linked by a single residue, A104 (Figure 1A), along with IV may be coaxially stacked with a possible kink. The notion that domains II, III, and IV may be coaxially stacked was further supported by SAXS analysis of two more subcon-structs, II-III-IV-X and II-III-IV-C (Figures S2B–S2D). The sequences of subconstructs II-III-IV-X and II-III-IV-C (Figure S2B) are identical to that of subconstruct II-III-IV except for a 7 bp insertion at region IIB in sub-construct II-III-IV-X and an addition of 5 bp at the junction between domain II and domains III-IV in subconstruct II-III-IV-C (Figures S2B–S2D and Table S1). The subconstructs of II-III-IV-X and II-III-IV-C share very similar global shape with that of subconstruct II-III-IV (Figures S2B–S2D), indicating that the possible coaxial stacking between domain II and III-IV prevails in all three subconstructs. Lastly, the elongated and bent envelope of the cyan construct (right side of Figure 2) suggests that domains I and V may be arranged via either a coaxial stacking or helix-loop-helix interaction, the type of structure that has generally been observed for four-way junctions (Laing and Schlick, 2009).
Figure 2. Identification of the RRE Domain Locations.
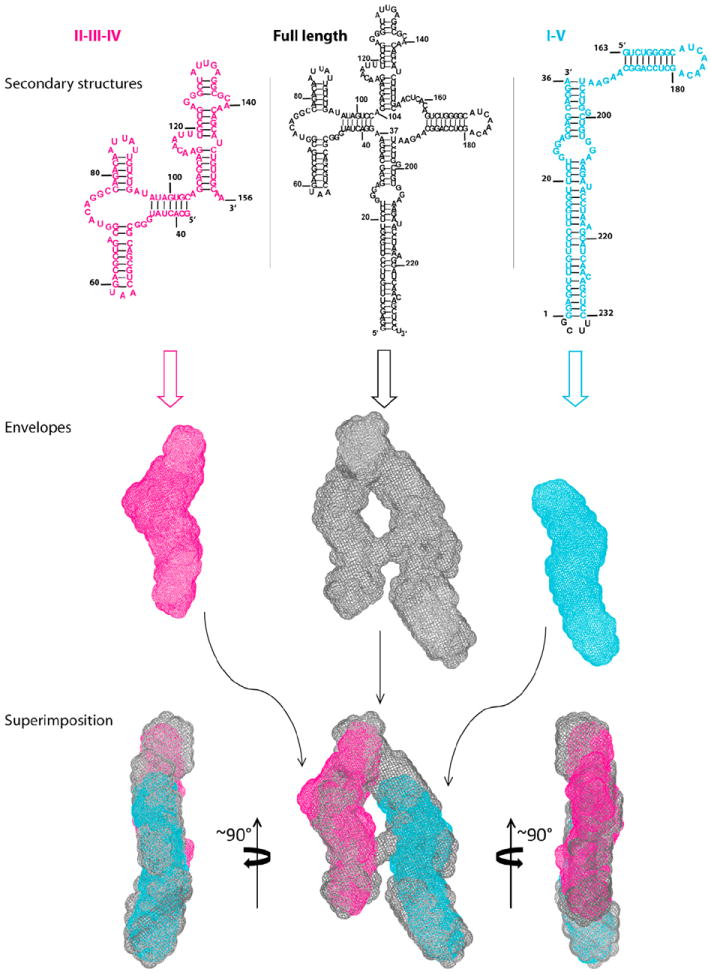
Top and Middle: the secondary structures and envelopes of the domain constructs and the RRE, respectively, where domains II, III and IV are highlighted in magenta, domains I and V are highlighted in cyan, and the full-length RRE is in gray. Bottom: three views of superimposition of the envelopes. See also Figure S2, Table S1, and Movies S2 and S3, which shows all views of the fits.
The construct II-III-IV-X with the 7 bp extension of stem-loop IIB (Figures S2A–S2D and Movie S3) was also used to determine whether stem-loop IIB is located at the top or the bottom of the shorter leg of the “A.” Comparison of the shape of this construct with that of the RRE envelope confirmed the location of IIB as the lower part of the short leg of the “A” (Figure S2A). Next, the location of domain IA was derived by the following rationale: excluding single bulges, the distance spanned by the 23 bp between the end of stem I and the beginning of the purine-rich IA internal asymmetrical loop (Figure 1A) is ~60 Å, assuming a standard A-form duplex (Figure S2E). This distance places site IA in the long leg, across from site IIB on the opposing leg of the “A” with a separation of ~55 Å (Figure 3A). A summary of the RRE domain locations is illustrated in Figure S2F. These combined SAXS envelopes, together with the approximate layout of the domains, make it possible to construct a three-dimensional structural model of the RRE using the software package G2G (Wang et al., 2009). An overlay of the three-dimensional model, drawn in ribbon diagrams, with the three-dimensional RRE SAXS envelope is shown in Figure 3A. The construction of the RRE atomic coordinates is shown in Movie S4.
Figure 3. The Putative Structural Model of the RRE, Fitness to the Experimental SAXS Data, Pairwise RMSD of the Top 20 Ensemble Structures, and Histograms of Characteristic Distance Distributions of the RRE RNA.

(A) Superimposition of the putative RRE structural model in red ribbon with the SAXS envelope in cyan mesh. The four-way junction, three-way junction, and the two known primary Rev-binding sites are marked. The distance between the two binding sites is about 55Å. The points A and B on the top of the envelope are also labeled. Domain II structure was generated based on its homology to an adenine riboswitch (Figure S4).
(B) Left: The back-calculated scattering curves with Ne = 3 (blue) superimposed on the experimental SAXS-WAXS (black with error bars) curves. The inset shows the χ2 between experimental and back-calculated SAXS-WAXS curves versus the ensemble size, Ne, used in the calculation; top right: an expanded high-q region of the superimposed scattering curves on the left; bottom right: residual differences in scattering curves between the top 20 structures in the ensemble and the average experimental data.
(D and E) Histograms of (D) the distance between the duet of the binding sites IIB and IA, Dduet and (E) between points A and B, DAB, at the top of the “A” of the ensembles (see Figure 3A). The distances between the centers of mass of phosphate atoms of residues (47G, 70G, 46G, 72A)IIB and (24G, 206A, 25G, 205G)IA are taken as the approximate distance between the IIB and IA duet of binding sites, while the locations of A and B are defined as the centers of the phosphates of residues 16G and 225U. The red lines are the best fits to the histograms assuming a Gaussian distribution. See also Figures S3 and S4 and Movies S3 and S4.
The Conformation Space of the RRE Characterized by SAXS
The RRE can be recognized by a chimeric Rev (Hoffmann et al., 2012), as well as by wild-type (WT) Rev. This suggests that the RRE structure retains some flexibility. We therefore investigated the possible conformation space of the RRE. SAXS has been used to characterize the conformation space of flexible or disordered proteins (Bernadó and Svergun, 2012; Rambo and Tainer, 2011) and DNA (Schwieters and Clore, 2007) using SAXS-restrained ensemble calculation approaches, where the scattering intensity is calculated as:
wi is the weight of ensemble member i, taken as 1/Ne here, and Ii(q) is the contribution to the scattering intensity from ensemble member i. If the surface-bound solvent contribution is omitted, the Ii(q) of an individual member i can be calculated using the Debye formula (Debye, 1915). A detailed description of the ensemble calculation is presented in Supplemental Information. Our calculation reveals that a minimum ensemble size of three is sufficient for a good fit with the experimental scattering data (Figure 3B), indicating conformational flexibility of the RRE RNA in solution. This flexibility may originate from pivot points, such as loops, internal loops, junctions and bulges (Bailor et al., 2011). Because of flexibility, the characteristic dimensional parameters of the RRE, such as radius of gyration Rg and maximum distance Dmax, are better represented by distance distributions than by single values (Figures 3D, 3F, and S3).
The pattern of the pairwise RMSD among the top 20 ensemble structures versus residue number coincides with that of flexibility probed by chemical reactivity (Figure 3C) (Legiewicz et al., 2008). The relative distance between the two known Rev-binding sites (Dduet in Figure 3D) distributes within a narrow range, 54 ± 4 Å, among all structures of the ensembles. The relatively restricted positioning of these two Rev-binding sites is likely due to the constraint imposed by the one-residue linker, A37, between domains I and II in the four-way junction (Figure 1A). Furthermore, histograms of distances between points A and B at the top of the “A” (Figure 3A) suggest a possible subset of conformations with an “H-like” topology, due to relative movement of the two legs of the “A” (Figure 3E). An animated possible conformation space of the RRE RNA is illustrated in Movie S5. The distance distribution between the two main structural segments varies between 45–60 Å, and these two structural segments may constitute two tracks of a scaffold for RRE-directed oligomerization of Rev (Daugherty et al., 2010a).
Structural Determinants for Formation of High-Order Rev-RRE Complexes
To test the significance of the RRE topology and its structural determinants, we characterized the binding and oligomerization of the Rev protein to several RRE mutants, as well as to WT RRE, by electrophoretic mobility shift assay (EMSA) and functional studies (see below). The RRE mutants included two truncation mutants (Figure 2) and three insertion mutants (Figure 4A). The insertion mutants were made by inserting 10 or 20 base pairs into the “crossbar” between the two legs of the “A.” SAXS analysis showed, as expected, that these mutant RREs retained their two-legged structure, but the insertions increased the distance between the two legs, presumably by forming a 10 or 20 bp double helix within the crossbar (Figures 4A and 4B). We also constructed a mutant RRE with a 15 bp insertion in the crossbar (Figures 4A and 4B); this insertion represents 1 1/2 turns of a double helix, so that one of the two legs is presumably inverted with respect to the other. This is confirmed by superimposition and comparison of the envelopes of domains II-III-IV and the 15 bp insertion mutant (Figures 4C and 4D). The two envelopes match in the domain II region only after rotating the envelope of domains II-III-IV by 180° around a horizontal axis (Figure 4C); without such a rotation, the envelopes do not match (Figure 4D). Figure 4B also shows that the distance between sites IA and IIB is increased in this mutant, as well as in the 10 and 20 bp insertion mutants. The agreement of these results with predictions is a strong validation of the overall SAXS-derived topology for the RRE.
Figure 4. The Secondary Structures and Molecular Envelopes of the Insertion Mutants.
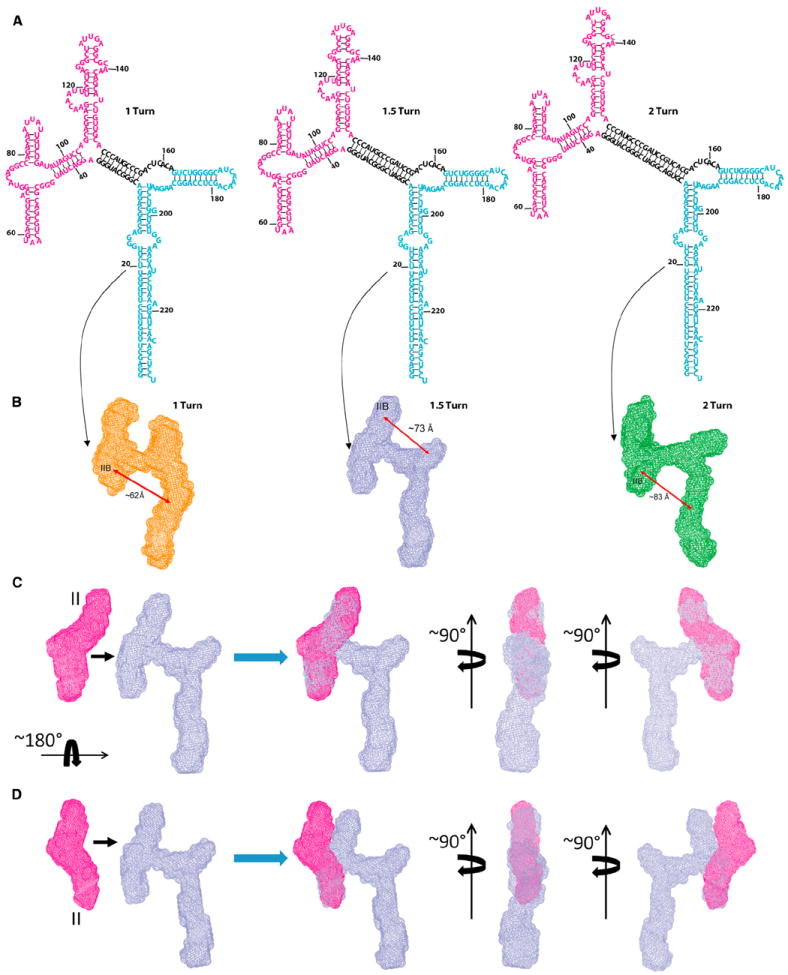
(A) Secondary structures of the three insertion mutants. Domains II-III-IV and I-V are colored in magenta and cyan, respectively and the inserted segments in black.
(B)The SAXS-derived envelopes of 1 turn (orange), 1.5 turns (light blue), and 2 turns (green) mutants. Double-headed arrows indicate approximate distances of the separations between the IIB site and the centers of the opposing envelopes of domains I-V. Note that domain II-III-IV in the 1.5 turn insertion mutant is rotated by 180° relative to domain I-V around the horizontal axis (see Figures 4C and 4D).
(C) Illustration of a 180° rotation of domains II-III-IV about the horizontal axis in the 1.5 turn insertion mutant. The envelope of domain II-III-IV in magenta (far left) can be fitted to that of the 1.5 turn insertion mutant only if the domains are rotated by 180° around the horizontal axis (three views on right).
(D) Without the rotation, the envelope of domain II-III-IV cannot be fitted to the short leg of the insertion mutant (three views on the right).
To assess the binding of Rev to these RREs, WT or mutant RRE RNAs were incubated with increasing amounts of the Rev protein. As shown in Figure 5A (top), some WT RRE RNA was shifted at Rev:RRE ratios as low as 2:1. The shifted RNA was resolved into 6–7 distinct bands. We have grouped these Rev-RRE complexes into low- (bands 1 and 2), mid- (bands 3-5), and high-order complexes (above band 5). At higher Rev:RRE ratios, the intensity of high-order bands is increased, and at ~30:1 stoichiometry, all or nearly all of the RRE has been shifted to high-order Rev-RRE complexes.
Figure 5. Rev-RRE Electrophoretic Mobility Shift Assays and Functional Studies.

(A) EMSA assays. Rev-binding reactions were loaded onto 6% or 10% nondenaturing TBE gels depending on the size of the RRE mutant (domain II-III-IV: 119 nt; domain I-V: 108 nt; 1 turn: 253 nt; 1.5 turn: 263 nt; 2 turn: 273 nt). The first eight lanes contain 0.5 pmol RRE, nonspecific competitor RNA (RiboA) at 20-fold mass excess, and titrating amounts of Rev at the molar ratio indicated above each lane. The last 2 lanes contain only competitor RNA, either in the absence of Rev or with Rev added to the same level as in the highest Rev:RRE stoichiometry. White lines indicate cropping of irrelevant lanes.
(B) Fractions of WT, truncated or mutant RRE RNAs engaged in high-order complexes are plotted as a function of Rev:RRE molar ratio. Data from three independent EMSA experiments are shown as mean ± SD.
(C) Plasmid pCMVgagpol-RRE (Srinivasakumar et al., 1997) was modified to simplify the RRE reporter assay. First, the pol gene was deleted; removal of the protease domain within pol prevents Gag cleavage, thus confining Gag to one band on a western blot. Second, the 233-nt “core” RRE was rendered identical in sequence to the NL4.3 RRE analyzed by SAXS. Third, the N-terminal glycine codon of gag was replaced by an alanine codon, preventing Gag myristylation, and thus, virion release.
(D) Western analysis of cell lysates using anti-p24CA and anti-β-actin antibodies. Top: pcDNA: cells transfected with neither pCMVRev nor reporter plasmid; Rev, cells transfected with pCMVRev alone; and remaining lanes contain reporter plasmids as indicated. WT: wild-type RRE; I-V: domains I-V (domains II-III-IV have been deleted); II-III-IV: domains II-III-IV (domains I-V have been deleted); 1 turn, 1.5 turns, and 2 turns: insertion mutants with one, one and a half and two turns of a duplex, respectively. Bottom: Quantification of the western analysis. Gag synthesis relative to WT (set at 100%): Gag levels were normalized to actin levels and to transfection efficiency, as measured by G. Luciferase assays. Gag synthesis is shown as the mean ± SD from three to five independent transfections.
A truncated RRE-containing domains II-III-IV, which harbors the primary high-affinity Rev-binding site on domain IIB, showed several faint shifted bands only at ratios of 16:1 to 32:1 (Figure 5A, second from top). However, the high-order complexes formed at high ratios with WT RRE were never seen with this mutant (Figure 5B, magenta plot). In dramatic contrast, with the I–V mutant RRE, which harbors the secondary binding site IA in domain I, a faint shifted band appeared at ~32:1, but no other bands were ever formed, even at higher stoichiometries (Figure 5A, third from top; Figure 5B, cyan plot). Thus, neither leg alone binds Rev with affinity or cooperativity comparable to that of the intact RRE: both legs are required for formation of high-order complexes. The difference between the two RRE fragments is also consistent with the well-established significance of site IIB for Rev binding.
We also performed EMSAs using the three RRE insertion mutants and noticed that they also began to shift at 2:1 stoichiometries (bottom three panels, Figure 5A). However, unlike the results with WT RRE, only low-order complexes appear at low Rev:RRE ratios, and intermediate complexes do not appear until the Rev:RRE ratio is ~30:1. At even higher Rev:RRE ratios, high-order complexes were observed, though often as a smear rather than discrete bands (orange, blue, and green plots, Figure 5B). Presumably, the impaired ability of these insertion mutants to form high-order complexes is due to the increased distance between the two legs. The residual binding affinity of Rev for the insertion mutants might be due to the presence of the major grooves in the inserted duplex, which are also positioned near the high-affinity IIB site. These major grooves might serve as structural analogs of the IA site, resulting in formation of a distorted initial Rev:RRE mutant complex. In summary, both legs of the “A” as well as the optimal separation of the two legs are important for formation of high-order Rev:RRE complexes.
Functional Implications of the RRE Structural Determinants
To further test the functional relevance of these topological arrangements, we determined the effects of these mutations on the nuclear export activity of the RRE in 293T human cells. As shown in Figures 5C and 5D, we devised an assay in which the Gag mRNA contained WT RRE, so that synthesis of Gag protein was completely dependent upon the coexpression of Rev. Gag protein production was measured by immunoblotting of cell lysates using antiserum against a fragment of Gag. Mutations in the reporter plasmid ensured that the Gag protein, which is the “readout” of the assay, is not cleaved in or exported from the cells, but remains as a single band in the immunoblot. Using this assay, we first measured the nuclear export activity of mutant RREs in which either domains II-III-IV or domains I-V had been deleted. Neither mutant exhibited detectable activity in our RRE activity assay (lanes under I-V and II-III-IV, Figure 5D). As these isolated domains both maintain the same overall structures as in the intact RRE (Figure 2) and since the II-III-IV RNA retains the IIB site and multiple major grooves, these results show that neither of the single legs alone is sufficient for RRE function in vivo; rather, both legs of the “A” are required.
We then tested the three insertion mutants in our functional RRE assay. Each insertion reduced the level of Gag synthesis by 6- to 9-fold (lanes under 1, 1.5, and 2 turns, Figure 5D). These results suggest that the 55 Å distance between the two legs of the “A” is optimal for RRE function in vivo. The residual activity might be attributed partly to the flexibility of the RRE structure and/or the flexibility of the Rev protein, as suggested by the differences in the angles and distances between the two ARMs observed in the two crystal structures of Rev dimer (Daugherty et al., 2010b; DiMattia et al., 2010). As noted above, the structural versatility of the RRE RNA is also evident in its ability to form functional complexes with chimeric proteins in vivo (Hoffmann et al., 2012). In summary, the in vivo functional assays data correlate very well with the in vitro results, suggesting that under our assay conditions, only the high-order complexes are capable of supporting RNA export; both the in vitro and in vivo studies illustrate that both legs of the “A” structure are required for the formation of high-order Rev:RRE complexes and optimal RRE function, with a preferred distance between the two legs of ~55 Å.
DISCUSSION
The “match” between the distance separating the primary Rev-binding sites in the RRE of this report and that between arginine-rich motifs in Rev dimers (Daugherty et al., 2010b; DiMattia et al., 2010) immediately suggests a solution to the conundrum of how Rev discriminates between RRE-containing RNAs and other RNAs, specifically promoting nuclear export of the former. We tested the role of the novel “A” shape in the specific binding of Rev to the RRE and in RRE function. We found that removal of either of the two legs of the “A” eliminated both specific binding (the second and third panels, Figure 5A) and RRE function (Figure 5D). Moreover, insertions in the “crossbar” of the RRE that increased the distance between the two binding sites impaired, but did not completely prevent, both specific binding (Figures 5A and 5B) and RRE function (Figure 5D). Taken together, these results provide strong support for the idea that the spatial opposition of the two sites is essential for Rev binding and function, and that the distance between them, corresponding to the “span” of the two ARMs in a Rev dimer, is optimal for binding and function.
Based on these observations and the structural features of the RRE presented here, we propose the following model for Rev-RRE interactions. The overall RRE architecture positions the IIB and IA Rev-binding sites at an optimal distance and orientation for initial binding of two Rev molecules. Subsequently, hydrophobic Rev-Rev interactions and general affinity of ARMs for the major grooves along the two legs facilitate the formation of a functional complex between an RRE RNA and multiple Rev molecules (Figure 6). A more realistic structural model is presented in Figure S5.
Figure 6. Models for Initial Rev Binding and Rev Oligomerization on the RRE RNA.
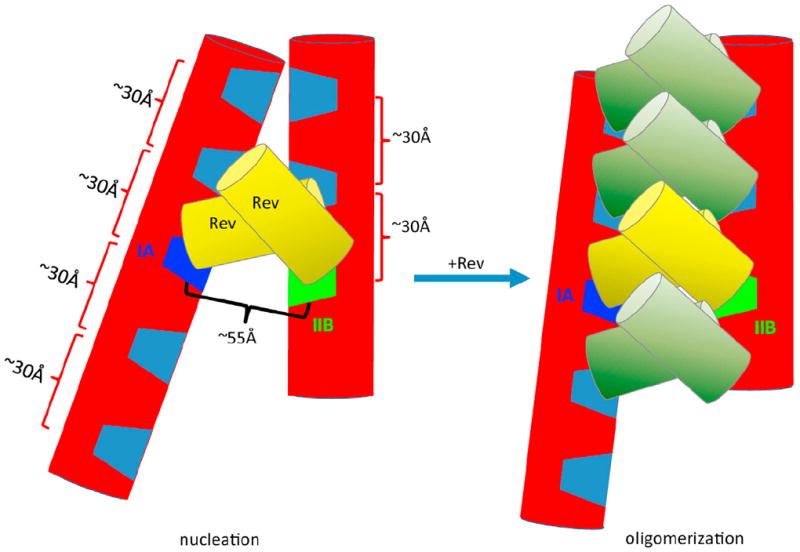
The RRE RNA is depicted in red cylinders with the IIB and IA Rev-binding sites highlighted in green and dark blue, respectively, and other major grooves shown in light blue. Initial binding of the first two Rev molecules (depicted in yellow cylinders) to the IIB and IA sites results in a nucleation site (left) for subsequent Rev oligomerization on the RRE RNA (right). This oligomerization is partially driven by hydrophobic interactions between the two Rev dimers and is constrained by the major groove spacing and the topological arrangement of the two major segments of the RRE. This oligomerization model illustrates the maximum number of Rev molecules that could potentially bind to this 233 nt RRE molecule based on spatial constraints. See also Figure S5 for a more realistic structural model.
Our results and this model are consistent with a number of properties of the Rev-RRE system. It is known that IIB alone is insufficient for initiation of nuclear export (Daugherty et al., 2008; Malim and Cullen, 1991; Mann et al., 1994) and that mutating a single residue in IA reduces Rev affinity for RRE ~5-fold (Daugherty et al., 2008), while affinity of Rev for the intact RRE is 500 times greater than its affinity for an isolated IIB motif (Daugherty et al., 2010a; Daugherty et al., 2008; Daugherty et al., 2010b). In fact, it has been speculated that a “composite binding site” may exist that contributes to specificity and cooperativity (Daugherty et al., 2010b; DiMattia et al., 2010). More specifically, two arginine-rich motifs in a Rev dimer have previously been proposed to bind to IIB on one side and an unknown stem loop, presumably IA, on the opposing side (DiMattia et al., 2010). Furthermore, two different sets of residues in Rev are involved in binding to the IIB and IA sites, suggesting a topological asymmetry of the two separate sites (Daugherty et al., 2008), consistent with the topologically asymmetrical setting of IIB and IA in our model. Our model also appears to be consistent with atomic force microscopy observations in which Rev oligomerization appears to take place on the entire RRE, including domain I (Pallesen et al., 2009). In conclusion, our results reveal the global structural constraints that appear to drive the specificity and cooperative binding of multiple Rev molecules required for RRE function.
As export of unspliced and partially spliced viral RNAs is essential for HIV-1 replication, it is a potential target for antiviral therapy. Indeed, there have been a few reports of compounds inhibiting Rev-RRE interaction (Chapman et al., 2002; Shuck-Lee et al., 2008). The structural information we have presented regarding the RRE should be useful in the search for other potential therapeutic compounds.
EXPERIMENTAL PROCEDURES
Plasmid Construction and RNA Sample Preparation
RRE-containing plasmids used for in vitro transcription or cell-based assays were modified as needed by site-directed mutagenesis or inverse PCR. Linearized plasmids were used as templates for in vitro transcription. RNA samples were gel-purified without refolding and washed extensively by filtration. Details are described in Extended Experimental Procedures.
SAXS Collection, Processing, Analysis and Ab Initio Three-Dimensional Shape Reconstructions
SAXS/WAXS data acquisition, analysis, including distance distribution and Rg calculations, are described in Supplemental Information. The procedures for data collection, processing, and analysis are similar to that previously described (Wang et al., 2009) and are in full conformity with the recently published recommended standards (Jacques et al., 2012). Data collection and processing used the in-house program package called NCI-SAXS, while analysis used the ATSAS program package by Svergun and coworkers (http://www.embl-hamburg.de/biosaxs/). Ab initio modeling was performed using the program DAMMIN (Svergun, 1999) and a two-step protocol (Movie S1). The two-step protocol is described as follows. The initial search volume was chosen as “sphere,” with a diameter 10–20 Ǻ greater than the Dmax of the corresponding construct, to avoid distortion caused by possible underestimation of Dmax. Thirty-two independent DAMMIN runs were performed in the “slow” mode. The parallelepiped search volume with defined length, width and height in “Expert” mode was further used. The average of the initial 32 bead models was calculated using the program package DAMAVER (Volkov and Svergun, 2003). In this program, the normalized spatial discrepancy (NSD) between each pair of models was computed, and the model with the lowest average NSD relative to the rest of the models was chosen as the reference model. The remaining models were superimposed onto the reference model using SUPCOMB (Kiozin and Svergun, 2001), except that possible outliers identified by NSD criteria were discarded. The calculation using the sphere as an initial search space indicated that the RRE has a dimension of ~30 × 90 × 180 Å, or is roughly a flat molecule with a thickness of ~30 Å on one of its sides. This thickness is a little more than the diameter of an A-form duplex and is also implied by the PDDF and its second derivative (Figure 1B). The flatness of the RRE molecule is not unexpected, as RNAs of similar sizes adopt a planar shape (Holbrook, 2008; Reiter et al., 2011). The thickness of an RNA shape is closely related to the number of duplexes packed on top of each other in a parallel or antiparallel configuration (Leontis et al., 2006), with a dimension of approximately n-fold of 30 Å. To reduce the degeneracy among the bead models and increase the resolution, we employed confined search space modeling based on the results of our initial calculation. Thus, knowing that the overall shape of the RRE envelope is flat, the second-round calculation was performed using a confined search space of a 60 × 120 × 220 Å rectangular shape, larger than the initial RRE model. The resulting structural models were subjected to averaging and the NSD score of the 32 models calculated using this confined search space was 1.00 ± 0.03.
SAXS/WAXS-Restrained Ensemble Calculations
The ensemble calculation was performed using an NCI-SAXS-WAXS module that is interfaced to the Xplor-NIH and a previously published protocol (Schwieters and Clore, 2007). The NCI-SAXS program allows a simultaneous calculation of fit between the experimental and back-calculated data in both SAXS and WAXS regions. The equally sparse SAXS/WAXS data, with q ranging from 0.006 to 2.3 Å−1 (a total of 82 data points), was used during the SAXS-restrained ensemble calculation. The difference in SAXS/WAXS curves between experimental and back-calculated data is expressed as χ2, as defined:
where c is a scaling factor, σ(qi) is the experimental error, and Iexp(qi) and Ical(qi) are the experimental and back-calculated scattering intensities of the ith data point of the total N data points. A weighted harmonic energy potential function was used:
where CSAXS is a scaling factor. During the calculation, the Rg restraint was not turned on so that the ensemble members were allowed to freely sample space. Additional restraints were also applied to maintain covalent geometry, prevent atomic overlap, and maintain canonical base pairing in duplexes. Knowledge-based restraints were applied to nucleic acid torsion angle conformations (Clore and Kuszewski, 2003) and to base-base packing (Kuszewski et al., 1997). The ensemble calculation was analyzed using an NCI-SAXS-WAXS data analysis module. All computation modules, scripts that contain all calculation parameters and conditions, and ensemble of the structures and SAXS data used for the calculation are provided upon request to the authors.
Gag Synthesis Measurements
Gag synthesis was detected in cell lysates by western analysis and quantified using secondary antibodies labeled with near-infrared dyes. Gag was normalized to actin levels (as a loading control) and transfection efficiency. Details are presented in Extended Experimental Procedures.
Electrophoretic Mobility Shift Assays
RRE RNAs were incubated in titrating amounts of full-length Rev protein and competitor RNA as described in Extended Experimental Procedures. Protein-RNA complexes were resolved in nondenaturing polyacrylamide gels and detected by SYBR Gold staining.
Construction of the RRE–Rev Dimer Complex Model
The Rev dimer structure was docked to the RRE as described in Extended Experimental Procedures.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Drs. Stephen Hughes, Alexander Wlodawer, and Prof. James Williamson for discussion. This work was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute (NCI) Center for Cancer Research; National Institute of Arthritis, Musculoskeletal and Skin Diseases; Center for Information Technology; and SAIC-Frederick under contract HHSN26120080001E. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Extended Experimental Procedures, five figures, five movies, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2013.10.008.
References
- Bailor MH, Mustoe AM, Brooks CL, 3rd, Al-Hashimi HM. Topological constraints: using RNA secondary structure to model 3D conformation, folding pathways, and dynamic adaptation. Curr Opin Struct Biol. 2011;21:296–305. doi: 10.1016/j.sbi.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battiste JL, Mao H, Rao NS, Tan R, Muhandiram DR, Kay LE, Frankel AD, Williamson JR. Alpha helix-RNA major groove recognition in an HIV-1 rev peptide-RRE RNA complex. Science. 1996;273:1547–1551. doi: 10.1126/science.273.5281.1547. [DOI] [PubMed] [Google Scholar]
- Bayer TS, Booth LN, Knudsen SM, Ellington AD. Arginine-rich motifs present multiple interfaces for specific binding by RNA. RNA. 2005;11:1848–1857. doi: 10.1261/rna.2167605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernadó P, Svergun DI. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol Biosyst. 2012;8:151–167. doi: 10.1039/c1mb05275f. [DOI] [PubMed] [Google Scholar]
- Chapman RL, Stanley TB, Hazen R, Garvey EP. Small molecule modulators of HIV Rev/Rev response element interaction identified by random screening. Antiviral Res. 2002;54:149–162. doi: 10.1016/s0166-3542(01)00222-4. [DOI] [PubMed] [Google Scholar]
- Clore GM, Kuszewski J. Improving the accuracy of NMR structures of RNA by means of conformational database potentials of mean force as assessed by complete dipolar coupling cross-validation. J Am Chem Soc. 2003;125:1518–1525. doi: 10.1021/ja028383j. [DOI] [PubMed] [Google Scholar]
- Cook KS, Fisk GJ, Hauber J, Usman N, Daly TJ, Rusche JR. Characterization of HIV-1 REV protein: binding stoichiometry and minimal RNA substrate. Nucleic Acids Res. 1991;19:1577–1583. doi: 10.1093/nar/19.7.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly TJ, Cook KS, Gray GS, Maione TE, Rusche JR. Specific binding of HIV-1 recombinant Rev protein to the Rev-responsive element in vitro. Nature. 1989;342:816–819. doi: 10.1038/342816a0. [DOI] [PubMed] [Google Scholar]
- Daly TJ, Doten RC, Rennert P, Auer M, Jaksche H, Donner A, Fisk G, Rusche JR. Biochemical characterization of binding of multiple HIV-1 Rev monomeric proteins to the Rev responsive element. Biochemistry. 1993;32:10497–10505. doi: 10.1021/bi00090a028. [DOI] [PubMed] [Google Scholar]
- Daugherty MD, D’Orso I, Frankel AD. A solution to limited genomic capacity: using adaptable binding surfaces to assemble the functional HIV Rev oligomer on RNA. Mol Cell. 2008;31:824–834. doi: 10.1016/j.molcel.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Booth DS, Jayaraman B, Cheng Y, Frankel AD. HIV Rev response element (RRE) directs assembly of the Rev homooligomer into discrete asymmetric complexes. Proc Natl Acad Sci USA. 2010a;107:12481–12486. doi: 10.1073/pnas.1007022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Liu B, Frankel AD. Structural basis for cooperative RNA binding and export complex assembly by HIV Rev. Nat Struct Mol Biol. 2010b;17:1337–1342. doi: 10.1038/nsmb.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debye P. Zerstreuung von Roentgenstrahlen. Annalen der Physik. 1915;46:809–823. [Google Scholar]
- DiMattia MA, Watts NR, Stahl SJ, Rader C, Wingfield PT, Stuart DI, Steven AC, Grimes JM. Implications of the HIV-1 Rev dimer structure at 3.2 A resolution for multimeric binding to the Rev response element. Proc Natl Acad Sci USA. 2010;107:5810–5814. doi: 10.1073/pnas.0914946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funari SS, Rapp G, Perbandt M, Dierks K, Vallazza M, Betzel C, Erdmann VA, Svergun DI. Structure of free Thermus flavus 5 S rRNA at 1.3 nm resolution from synchrotron X-ray solution scattering. J Biol Chem. 2000;275:31283–31288. doi: 10.1074/jbc.M004974200. [DOI] [PubMed] [Google Scholar]
- Hajdin CE, Ding F, Dokholyan NV, Weeks KM. On the significance of an RNA tertiary structure prediction. RNA. 2010;16:1340–1349. doi: 10.1261/rna.1837410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarskjold MH, Rekosh D. A long-awaited structure is revealed. Viruses. 2011;3:484–492. doi: 10.3390/v3050484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaphy S, Dingwall C, Ernberg I, Gait MJ, Green SM, Karn J, Lowe AD, Singh M, Skinner MA. HIV-1 regulator of virion expression (Rev) protein binds to an RNA stem-loop structure located within the Rev response element region. Cell. 1990;60:685–693. doi: 10.1016/0092-8674(90)90671-z. [DOI] [PubMed] [Google Scholar]
- Heaphy S, Finch JT, Gait MJ, Karn J, Singh M. Human immunodeficiency virus type 1 regulator of virion expression, rev, forms nucleoprotein filaments after binding to a purine-rich “bubble” located within the rev-responsive region of viral mRNAs. Proc Natl Acad Sci USA. 1991;88:7366–7370. doi: 10.1073/pnas.88.16.7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann D, Schwarck D, Banning C, Brenner M, Mariyanna L, Krepstakies M, Schindler M, Millar DP, Hauber J. Formation of transactivation competent HIV-1 Rev:RRE complexes requires the recruitment of multiple protein activation domains. PLoS ONE. 2012;7:e38305. doi: 10.1371/journal.pone.0038305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbrook SR. Structural principles from large RNAs. Annu Rev Biophys. 2008;37:445–464. doi: 10.1146/annurev.biophys.36.040306.132755. [DOI] [PubMed] [Google Scholar]
- Jacques DA, Guss JM, Trewhella J. Reliable structural interpretation of small-angle scattering data from bio-molecules in solution—the importance of quality control and a standard reporting framework. BMC Struct Biol. 2012;12:9. doi: 10.1186/1472-6807-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacrot B. The study of biological structures by neutron scattering from solution. Rep Prog Phys. 1976;39:43. [Google Scholar]
- Kiozin M, Svergun DI. Automated matching of high- and low-resolution structural models. J Appl Cryst. 2001;34:33–41. [Google Scholar]
- Kjems J, Brown M, Chang DD, Sharp PA. Structural analysis of the interaction between the human immunodeficiency virus Rev protein and the Rev response element. Proc Natl Acad Sci USA. 1991;88:683–687. doi: 10.1073/pnas.88.3.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MH, Vachette P, Svergun DI. Small-angle scattering: a view on the properties, structures and structural changes of biological macromolecules in solution. Q Rev Biophys. 2003;36:147–227. doi: 10.1017/s0033583503003871. [DOI] [PubMed] [Google Scholar]
- Kuszewski J, Gronenborn AM, Clore GM. Improvements and extensions in the conformational database potential for the refinement of NMR and X-ray structures of proteins and nucleic acids. J Magn Reson. 1997;125:171–177. doi: 10.1006/jmre.1997.1116. [DOI] [PubMed] [Google Scholar]
- Laing C, Schlick T. Analysis of four-way junctions in RNA structures. J Mol Biol. 2009;390:547–559. doi: 10.1016/j.jmb.2009.04.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Ramirez A, Daugherty MD, Frankel AD. A simple motif for protein recognition in DNA secondary structures. J Mol Biol. 2005;351:982–994. doi: 10.1016/j.jmb.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Legiewicz M, Badorrek CS, Turner KB, Fabris D, Hamm TE, Rekosh D, Hammarskjöld ML, Le Grice SF. Resistance to RevM10 inhibition reflects a conformational switch in the HIV-1 Rev response element. Proc Natl Acad Sci USA. 2008;105:14365–14370. doi: 10.1073/pnas.0804461105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontis NB, Lescoute A, Westhof E. The building blocks and motifs of RNA architecture. Curr Opin Struct Biol. 2006;16:279–287. doi: 10.1016/j.sbi.2006.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J, Ouellet J, Norman DG, Doniach S, Lilley DM. The complete VS ribozyme in solution studied by small-angle X-ray scattering. Structure. 2008;16:1357–1367. doi: 10.1016/j.str.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusvarghi S, Sztuba-Solinska J, Purzycka KJ, Pauly GT, Rausch JW, Le Grice SF. The HIV-2 Rev-response element: determining secondary structure and defining folding intermediates. Nucleic Acids Res. 2013;41:6637–6649. doi: 10.1093/nar/gkt353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH, Cullen BR. HIV-1 structural gene expression requires the binding of multiple Rev monomers to the viral RRE: implications for HIV-1 latency. Cell. 1991;65:241–248. doi: 10.1016/0092-8674(91)90158-u. [DOI] [PubMed] [Google Scholar]
- Malim MH, Hauber J, Le SY, Maizel JV, Cullen BR. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature. 1989;338:254–257. doi: 10.1038/338254a0. [DOI] [PubMed] [Google Scholar]
- Malim MH, Tiley LS, McCarn DF, Rusche JR, Hauber J, Cullen BR. HIV-1 structural gene expression requires binding of the Rev trans-activator to its RNA target sequence. Cell. 1990;60:675–683. doi: 10.1016/0092-8674(90)90670-a. [DOI] [PubMed] [Google Scholar]
- Mann DA, Mikaélian I, Zemmel RW, Green SM, Lowe AD, Kimura T, Singh M, Butler PJ, Gait MJ, Karn J. A molecular rheostat. Co-operative rev binding to stem I of the rev-response element modulates human immunodeficiency virus type-1 late gene expression. J Mol Biol. 1994;241:193–207. doi: 10.1006/jmbi.1994.1488. [DOI] [PubMed] [Google Scholar]
- McColl DJ, Honchell CD, Frankel AD. Structure-based design of an RNA-binding zinc finger. Proc Natl Acad Sci USA. 1999;96:9521–9526. doi: 10.1073/pnas.96.17.9521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra SH, Spring AM, Germann MW. Thermodynamic profiling of HIV RREIIB RNA-zinc finger interactions. J Mol Biol. 2009;393:369–382. doi: 10.1016/j.jmb.2009.07.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallesen J, Dong M, Besenbacher F, Kjems J. Structure of the HIV-1 Rev response element alone and in complex with regulator of virion (Rev) studied by atomic force microscopy. FEBS J. 2009;276:4223–4232. doi: 10.1111/j.1742-4658.2009.07130.x. [DOI] [PubMed] [Google Scholar]
- Pond SJ, Ridgeway WK, Robertson R, Wang J, Millar DP. HIV-1 Rev protein assembles on viral RNA one molecule at a time. Proc Natl Acad Sci USA. 2009;106:1404–1408. doi: 10.1073/pnas.0807388106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers. 2011;95:559–571. doi: 10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013;496:477–481. doi: 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter NJ, Chan CW, Mondragón A. Emerging structural themes in large RNA molecules. Curr Opin Struct Biol. 2011;21:319–326. doi: 10.1016/j.sbi.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters CD, Clore GM. A physical picture of atomic motions within the Dickerson DNA dodecamer in solution derived from joint ensemble refinement against NMR and large-angle X-ray scattering data. Biochemistry. 2007;46:1152–1166. doi: 10.1021/bi061943x. [DOI] [PubMed] [Google Scholar]
- Serganov A, Yuan YR, Pikovskaya O, Polonskaia A, Malinina L, Phan AT, Hobartner C, Micura R, Breaker RR, Patel DJ. Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem Biol. 2004;11:1729–1741. doi: 10.1016/j.chembiol.2004.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuck-Lee D, Chen FF, Willard R, Raman S, Ptak R, Hammarskjold ML, Rekosh D. Heterocyclic compounds that inhibit Rev-RRE function and human immunodeficiency virus type 1 replication. Antimicrob Agents Chemother. 2008;52:3169–3179. doi: 10.1128/AAC.00274-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasakumar N, Chazal N, Helga-Maria C, Prasad S, Hammarskjöld ML, Rekosh D. The effect of viral regulatory protein expression on gene delivery by human immunodeficiency virus type 1 vectors produced in stable packaging cell lines. J Virol. 1997;71:5841–5848. doi: 10.1128/jvi.71.8.5841-5848.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiley LS, Malim MH, Tewary HK, Stockley PG, Cullen BR. Identification of a high-affinity RNA-binding site for the human immunodeficiency virus type 1 Rev protein. Proc Natl Acad Sci USA. 1992;89:758–762. doi: 10.1073/pnas.89.2.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Cryst. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zuo X, Yu P, Xu H, Starich MR, Tiede DM, Shapiro BA, Schwieters CD, Wang YX. A method for helical RNA global structure determination in solution using small-angle x-ray scattering and NMR measurements. J Mol Biol. 2009;393:717–734. doi: 10.1016/j.jmb.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ma X, Yeh YS, Zhu Y, Daugherty MD, Frankel AD, Musier-Forsyth K, Barbara PF. Comparative analysis of RNA/protein dynamics for the arginine-rich-binding motif and zinc-finger-binding motif proteins encoded by HIV-1. Biophys J. 2010;99:3454–3462. doi: 10.1016/j.bpj.2010.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, Swanstrom R, Burch CL, Weeks KM. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460:711–716. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol. 2008;6:e96. doi: 10.1371/journal.pbio.0060096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Ellington AD. Anti-peptide aptamers recognize amino acid sequence and bind a protein epitope. Proc Natl Acad Sci USA. 1996;93:7475–7480. doi: 10.1073/pnas.93.15.7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapp ML, Hope TJ, Parslow TG, Green MR. Oligomerization and RNA binding domains of the type 1 human immunodeficiency virus Rev protein: a dual function for an arginine-rich binding motif. Proc Natl Acad Sci USA. 1991;88:7734–7738. doi: 10.1073/pnas.88.17.7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo X, Cui G, Merz KM, Jr, Zhang L, Lewis FD, Tiede DM. X-ray diffraction “fingerprinting” of DNA structure in solution for quantitative evaluation of molecular dynamics simulation. Proc Natl Acad Sci USA. 2006;103:3534–3539. doi: 10.1073/pnas.0600022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.