

Abstract
Chromatin modifications have been well-established to play a critical role in the regulation of genome function. Many of these modifications are introduced and removed by enzymes that utilize cofactors derived from primary metabolism. Recently, it has been shown that endogenous cofactors and metabolites can regulate the activity of chromatin-modifying enzymes, providing a direct link between the metabolic state of the cell and epigenetics. Here we review metabolic mechanisms of epigenetic regulation with an emphasis on their role in cancer. Focusing on three core mechanisms, we detail and draw parallels between metabolic and chemical strategies to modulate epigenetic signaling, and highlight opportunities for chemical biologists to help shape our knowledge of this emerging phenomenon. Continuing to integrate our understanding of metabolic and genomic regulatory mechanisms may help elucidate the role of nutrition in diseases such as cancer, while also providing a basis for new approaches to modulate epigenetic signaling for therapeutic benefit.
Keywords: epigenetics, metabolism, histones, posttranslational modification, IDH1, 2-hydroxyglutarate, genomics, chromatin
I. Introduction
“The one-dimensional script of the human genome, shared by essentially all cells in all tissues, contains sufficient information to provide for differentiation of hundreds of different cell types, and the ability to respond to a vast array of internal and external influences. Much of this plasticity results from the carefully orchestrated symphony of transcriptional regulation.”
-- Lander et al., Nature 2001
The above quote, from a report of the draft sequence of the human genome in 2001, remains a paean to the importance of transcriptional regulation in human biological processes.1 Transcriptional regulation underlies differentiation, the process responsible for cellular identity,2 while aberrant transcriptional regulation is a hallmark of many pathological states, including cancer.3, 4 One way in which transcription and other genomic processes are regulated is by controlling access to DNA through packaging in chromatin. Chromatin consists of DNA wrapped in 147-bp segments around histone octamers to form nucleosomes, and serves to compact the 3×109-bp human genome into a nucleus of a few hundred μm3. Chemical modifications of chromatin, including histone acetylation, histone methylation, and DNA methylation, can alter the dynamics of transcription by regulating the accessibility of the genome and facilitating the binding of trans-acting factors (Figure 1a).5–7 These chemical modifications of chromatin, the enzymes that implement them, and the phenotypic effects they elicit have all been termed “epigenetic”.
Figure 1.
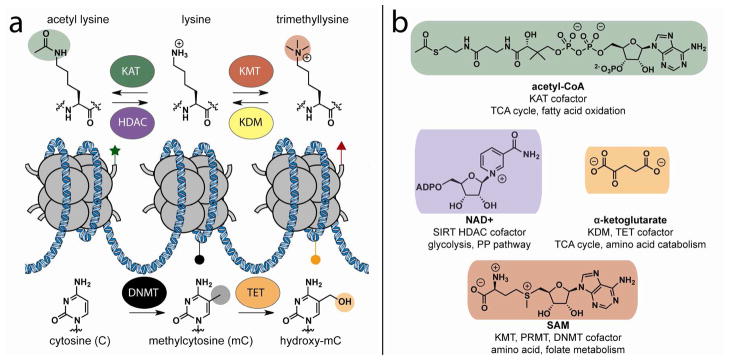
Regulation of genome function by cofactor-dependent enzymes. (a) Chromatin-modifying enzymes modulate the posttranslational modifications of histone amino acids and cytosine methylation state. These modifications can affect the physical accessibility of genomic loci, provide specific binding surfaces for effector proteins, or influence posttranslational modification of neighboring residues. (b) Enzyme cofactors, their associated chromatin modifiers, and examples of metabolic pathways that produce and consume each cofactor. Enzymes are abbreviated according to nomenclature established by Allis et al.117 KAT, lysine acetyltransferase; HDAC, sirtuin histone deacetylase; KMT, lysine methyltransferase; KDM, lysine demethylase; DNMT, DNA methyltransferase; TET, cytosine hydroxylase.
Although the precise definition of epigenetic has been the subject of much debate,8, 9 here we use the term simply to denote molecular events regulating transcription and other genomic functions that do not directly depend on the primary nucleotide sequence of DNA. Whether causal, heritable, or neither,10, 11 one indisputable fact is that chemical modifications of histones and DNA are biologically relevant. For example, Kouzarides and Dawson recently annotated 40 genes involved in the establishment or recognition of histone modifications that show recurrent mutation in cancer.12 These genetic errors can lead to the mistargeting or overactivity of chromatin modifiers due to translocation and gain-of-function mutations, and have been found to stimulate cellular transformation and proliferation in diverse cell lineages. 3, 12 Non-mutated chromatin modifiers also play a significant role in cancer, as oncogenic gene expression can demonstrate an absolute requirement for chromatin activities that are dispensable in phenotypically normal cells.13
“Big science” initiatives such as ENCODE,14 the Cancer Genome Atlas,15 and the Roadmap Epigenomics Project16 have provided an invaluable service discovering and cataloguing epigenetic drivers of disease. However, manipulating epigenetic processes requires moving from the macro-level of genome function to the micro-level of enzyme activity. At the molecular level, one characteristic that many KAT, HDAC, KMT, KDM, DNMT, and TET enzymes share in common is their use of cofactors derived from primary metabolism (Figure 1b). The binding sites for these cofactors have been used for the design of synthetic inhibitors and cofactor analogues targeting chromatin-modifying enzymes.17 More recent research has shown that endogenous cofactors and metabolites are also capable of directly influencing chromatin modifications. These studies were in part stimulated by the discovery that mutations in the metabolic enzymes SDH, FH, and IDH can cause cancer by producing metabolites that inhibit the activity of chromatin-modifying enzymes.18, 19 This has led to the hypothesis that the activity of chromatin-modifying enzymes may serve as central signal integrators, modulating genome function in response to rate-limiting levels of cofactors derived from metabolism. Several recent reviews have persuasively outlined the evidence supporting this hypothesis, and are highly recommended.20–23 To avoid redundancy, here we approach this topic from a different direction, focusing on the mechanistic basis for metabolic regulation of epigenetics. Limiting our scope to chromatin modifiers that utilize acetyl-CoA, NAD+, SAM, and α-KG as cofactors, we identify three mechanisms by which cellular metabolism can impact the activity of chromatin-modifying enzymes: i) competitive inhibition, ii) cofactor depletion, and iii) subcellular localization of cofactor biosynthesis. We detail case studies for each mechanism with an emphasis on their role in cancer, and then draw parallels between natural mechanisms and strategies devised by chemical biologists to modulate epigenetic states. Finally, we discuss future prospects for chemical biology to advance studies of genomic regulation, as well as to draw inspiration from metabolic strategies for new approaches to the targeted perturbation of genomic processes.
Note: much of this review assumes a working knowledge of chromatin modifications as well as their proposed role in transcription and associated genomic processes. For convenience, a glossary is provided at the end of the text summarizing some of this information. Chromatin-modifying enzyme activities are also defined in Figure 1. Further background and discussion can be found in recent reviews.24, 25
II. Regulation of Epigenetic Signaling by Cofactor Competitive Metabolites
The most straightforward mechanism by which metabolism can affect the activity of a chromatin modifier is through the production of a competitive inhibitor. Many chromatin-modifying enzymes are sensitive to feedback inhibition by the cofactor-derived product of their enzymatic reaction. For instance, the KAT enzyme Gcn5 binds acetyl-CoA and CoA with similar dissociation constants (Table 1). Cofactor competitive molecules can be produced as byproducts of enzymatic reactions that modify macromolecules, or directly as products of rewired metabolic pathways.26
Table 1.
Biochemical measurements of interactions between epigenetic modifiers and regulatory metabolites. Metabolic sensitivity indicates evidence for regulatory metabolites promoting or impeding chromatin modification in cells (y = yes, n = no, nd = not determined). References for data: Gcn5,148 p300,149, 150 pCAF,151 KDM4A/KDM4C,46 PHD2,31, 46, 48 EZH1/EZH2,152 CARM1,152, 153 SIRT1,154 CtBp.155 Gcn5 values from yeast homologue, Kd for CoA obtained with etheno-CoA; p300 Ac-CoA and CoA Kd and Ki obtained with acetonyl-CoA and desulfo-CoA respectively; SIRT1 Ki value from yeast homologue (Sir2). All values are given without standard deviations and have been rounded to the nearest decimal for simplicity.
| Epigenetic modifier | Regulatory metabolite | Km (μM) | Kd (μM) | IC50 or Ki (μM) | Chromatin mark | Metabolic sensitivity |
|---|---|---|---|---|---|---|
| Gcn5 | Ac-CoA | 2.5 | 8.5 | - | H3K14Ac | y |
| CoA | - | 5.1 | 6.7 | |||
| p300 | Ac-CoA | 1.2 | 0.7 | - | H3/H4Ac | nd |
| CoA | - | 7.3 | 8.1 | |||
| Pcaf | Ac-CoA | 1 | 0.6 | - | H3K9Ac | nd |
| CoA | - | - | 0.4 | |||
| KDM4A | a-KG | 6 | - | - | H3K9me3 (removal) | y |
| R-2-HG | - | - | 24 | |||
| KDM4C | a-KG | 4 | - | - | H3K9me3 (removal) | y |
| R-2-HG | - | - | 79 | |||
| PHD2 | a-KG | 1 | - | - | HIF-1 genomic occupancy reduced | y |
| R-2-HG | 210 | - | 7300 | |||
| succinate | - | - | 350 | |||
| fumarate | - | - | 80 | |||
| EZH1 | SAM | 2.5 | - | - | H3K27me | y |
| SAH | - | - | 8.3 | (me2/me3) | ||
| EZH2 | SAM | 1.2 | - | - | H3K27me | y |
| SAH | - | - | 7.5 | (me2/me3) | ||
| CARM1 | SAM | 0.2 | 0.8 | - | H3R17me2a | n |
| SAH | - | 6.9 | 0.1 | |||
| SIRT1 | NAD+ | 154 | - | - | H3K9Ac (removal) | y |
| NADH | - | - | 28000 | |||
| CtBP | NAD+ | - | 11 | - | co-repressor | y |
| NADH | - | 0.07 | - |
A spectacular example of the competitive inhibition mechanism is exhibited in cancers driven by mutations in the metabolic enzymes succinate dehydrogenase (SDH), fumarate hydratase (FH), and isocitrate dehydrogenase (IDH). Each of these mutations results in the cellular accumulation of a metabolite that antagonizes the activity of Fe(II)/α-KG-dependent dioxygenases. SDH and FH-driven cancers are associated with inactivating mutations.19 In both cases heterozygous germline mutations predispose an individual to cancer, but are not triggered until loss of the second allele occurs. This interrupts TCA cycle turnover and leads to accumulation of succinate and fumarate (Figure 2). SDH and FH-deficient tumor samples contain ~10–20 fold higher levels of succinate and fumarate than wild type tumors. 27 Notably, both metabolites contain a dicarboxylate moiety similar to that of the α-KG cofactor required for prolyl hydroxylase (PHD), KDM, and TET activity.
Figure 2.

Regulation of epigenetic signaling by cofactor competition. Diverse mechanisms of genomic regulation including transcription factor stability, and histone and cytosine methylation status are regulated by α-ketoglutarate-dependent dioxygenases (green arrow). Inactivating mutations in the TCA cycle enzymes FH and SDH or missense mutations in cytosolic IDH1 result in the production of high concentrations of fumarate, succinate, and (R)-2-hydroxyglutarate, which compete with α- ketoglutarate for enzyme active sites (red arrow). Metabolic enzymes are color-coded light blue, with those proteins mutated or overexpressed in cancer outlined in bold red. HIF, hypoxia-inducible factor; SDH, succinate dehydrogenase; FH, fumarate hydratase; IDH, isocitrate dehydrogenase.
Early studies centered on the ability of succinate and fumarate to activate the transcription factor HIF-1, a well-known oncogene in hereditary cancers driven by mutations to the Von-Hippel Lindau (VHL) tumor suppressor gene.28 Mechanistically this could proceed by succinate/fumarate inhibition of PHD2, a Fe(II)/α-KG-dependent prolyl hydroxylase whose activity promotes HIF-1 degradation. Indeed, cell-based models of SDH and FH inhibition demonstrated that accumulation of either TCA cycle intermediate could antagonize PHD2 activity, resulting in HIF-1 stabilization.29, 30 In vitro biochemical measurements indicate fumarate and succinate are mid- to high micromolar inhibitors of PHD2 activity, with fumarate possessing greater inhibitory activity than succinate (Table 1).31
Despite these compelling findings, more recent evidence suggests succinate and fumarate may exert their oncogenic effects through mechanisms unrelated to PHD2/HIF1. For example, when HIF1 is genetically inactivated in mice engineered to lack FH, tumorigenesis is actually accelerated.32 Similarly, paragangliomas associated with SDH and VHL mutants form two distinct gene expression subgroups, signifying they may function through different mechanisms.33 Furthermore, yeast deficient for SDH show increased levels H3K36me2, a histone mark normally removed by KDM activity.34 This same study found that succinate could inhibit human KDM4D in vitro, evoking a role for KDM enzymes in SDH-driven cancers. A recent survey of clinical tumor samples found that SDH-mutated tumors possess a distinct hypermethylated DNA profile that is conserved across developmentally distinct tumors harboring SDH mutations.35 This makes sense conceptually, as increased histone methylation – a consequence of KDM inhibition – can provide binding surfaces for plant homeodomain-containing proteins that can ultimately stimulate DNMT activity and lead to aberrant DNA methylation. Alternatively, increased DNA methylation could result from antagonizing active DNA demethylation processes mediated by TET enzymes.36 Supporting this, molecular pathology of SDH-deficient gastrointestinal stromal tumors has found these cancer exhibit decreased levels of 5-hydroxymethylcytosine.37 Succinate and fumarate accumulation may also impact the cell through mechanisms completely orthogonal to chromatin, such as chemical modification of proteins32 or interaction with membrane receptors.38 Interestingly, although individuals with inborn mutations to SDH and FH harbor these errors in every cell of their body, only a subset of tissues such as brain and kidney are predisposed to cancer. This suggests epigenetic reprogramming mediated by SDH and FH inactivation collaborates with tissue-specific genomic factors to generate the tumorigenic phenotype. However, the identity of these epigenetic collaborators, as well as exactly which Fe(II)/α-KG-dependent enzymes mediate the transformed phenotype in cancers driven by SDH and FH mutations, remain to be determined.
As with SDH and FH, mutations in IDH enzymes can also result in the production of a cofactor competitive metabolite. However, IDH mutants differ significantly in terms of occurrence, clinical impact, and mechanism. Cancers associated with mutations of cytosolic IDH1 and mitochondrial IDH2 span a wide variety of cell types and are relatively common compared to SDH/FH-driven cancers, occurring with particularly high incidence in cases of glioblastoma and acute myeloid leukemia (AML).39–42 For IDH1, highly specific somatic mutations affect a single amino acid in the active site, R132, and result in various amino acid substitutions, with R132H being the most common.43 Rather than inactivate IDH1, this mutation results in the mutant IDH1 gaining the abililty to convert α-KG to the R-enantiomer of 2-hydroxyglutarate (R-2HG; Figure 2).44 R-2HG shares the connectivity and dicarboxylate chemotype of α-KG. Similar to SDH and FH, early reports focused on the ability of R-2HG to inhibit PHD2 and stabilize HIF-1.45, 46 However, biochemical and cellular studies have now shown conclusively that R-2HG activates PHD2, while serving as an inhibitor of KDM and TET dioxygenases.47, 48 Although the inhibition constants of R-2HG for KDM and TET enzymes are weak (Table 1), R-2HG levels in tumor cells are increased ~1000-fold over wild type tumors (Table 2) making inhibition of dioxygenase activity biochemically feasible.41, 44 Genetic studies of AML patient blood samples found that IDH1 and TET2 mutations occur in a mutually exclusive manner, supporting the hypothesis that R-2HG production by mutant IDH phenocopies TET2 inactivation.49 Moreover, this study found that IDH1/2 and TET2 mutated tumors share similar genome-wide DNA hypermethylation profiles. This mirrors observations initially made in IDH1-mutated glioma, which also exhibits a hypermethylator phenotype.50, 51 In cell-based models, R-2HG itself is sufficient to block differentiation and promote growth factor independence, two hallmarks of leukemic transformation.52 These effects are recapitulated by TET2 knockdown, but are also dependent on the ability of R-2HG to activate PHD2 and induce HIF-1 degradation. Together these data implicate TET2 as a physiologically relevant epigenetic target that is inhibited by cofactor competition in IDH-mutated AML. Therapeutics that preferentially inhibit mutant IDH1 over the wild type enzyme induce changes in histone and DNA methylation, and have also shown promise in pre-clinical studies of IDH1-mutated AML.53, 54
Table 2.
Effect of metabolic perturbations on ratios and overall levels of cofactors used by epigenetic modifiers. Sources for perturbation data: Ac-CoA/CoA, liver levels in fed/fasted rats;60 α-KG/R-2HG, tumor levels in IDH1 R132 wild type/mutant tumors;44 SAM/SAH, cell lines in control/NNMT overexpressing cell lines.63 α-KG/R-2HG levels were calculated as the average of wild type/mutant samples from reference. All data are provided as unit-less ratios and fold-change to account for differences in units of measurement from each study, and have been rounded to the nearest decimal for simplicity.
| Epigenetic modifier | Regulatory metabolite | Perturbation | Native ratio | Perturbed ratio | Fold change - metabolite |
|---|---|---|---|---|---|
| KAT | Ac-CoA | fasting | 1.2 | 0.7 | 1.1 |
| CoA | 1.9 | ||||
| KDM | a-KG | IDH1 mutation | 0.2 | 0.002 | 0.9 |
| R-2-HG | 64.9 | ||||
| PMT | SAM | NNMT overexpression | 1.2 | 0.2 | 1.9 |
| SAH | 0.4 |
While seemingly a well-plowed field, recent data indicates the epigenetic consequences of IDH inhibition may still not be completely understood. For instance, at low doses inhibitors targeting mutant IDH1 do not affect histone or DNA methylation, yet still have anti-tumor activity in a mouse model of glioma. This suggests mutant IDH1 and/or IDH1 inhibitors are capable of acting independently of chromatin methylation.53 Studies of IDH1 mutations in other cell types have also implicated non-TET2 dioxygenases as physiologically relevant targets of R-2HG. Thompson and coworkers found that stable transfection of the IDH1-R132H mutant in immortalized astrocytes resulted in an accumulation of histone methylation on H3K9 and H3K27.55 R-2HG blocked differentiation in a cell-based model, an effect that was phenocopied by knockdown of the H3K9 demethylase KDM4C. This indicates R-2HG can utilize cell lineage-specific mechanisms to promote disease-relevant epigenetic change. Interestingly, although altered DNA methylation was observed in an IDH1 R132H conditional knock-in mouse, large changes in H3K9me3 levels were not observed.56
Cancers driven by mutants such as IDH1 R132H provide a fascinating example of how competitive metabolites can drive phenotypic change through epigenetic mechanisms. Further study is necessary to assess whether more transient changes in metabolism are also capable of exerting epigenetic effects. However, several lines of evidence suggest this may be the case. For example, in both yeast and human cells, changes in glucose availability can regulate transcription of growth-related genes through direct effects on the acetylation state of histones.57, 58 These histone acetylation events are mediated by Gcn5, a KAT enzyme. As mentioned above, Gcn5 is distinct from many KAT enzymes in that it possesses similar binding constants for Acetyl-CoA and CoA. Mutant studies of yeast Gcn5 suggest acetyl-CoA levels are sufficient to saturate Gcn5 KAT activity in vivo under normal growth conditions.59 However, animal models have shown that in many tissues Acetyl-CoA/CoA ratios fluctuate from >1 in fed to <1 in fasting states, even while overall CoA pools stay relatively constant.60 Together, these data support the premise that feedback inhibition by intracellular CoA may help regulate the ability of Gcn5 and other CoA-sensitive KATs to activate transcription of genes required for cell growth.61 Conclusive testing of this hypothesis will require integrated measurements of acetyl-CoA/CoA ratio, chromatin modifications, and gene expression under carefully defined cell growth conditions.57 Notably, several chromatin-modifying enzymes have Ki values for competitive metabolites that are estimated to be near their intracellular levels, suggesting competitive mechanisms may be operable in vivo (Table 1). Alternatively, chromatin modifications may be sensitive to transient changes in the combined level of competitive metabolites sharing a common chemotype (such as the dicarboxylates succinate/fumarate/2-HG) rather than any single competitive inhibitor alone.18 Finally, different chromatin modifications are introduced and removed with very different kinetics.62 Histone acetylations have a half-life on the order of minutes, while methylation persists on a time scale of hours. Thus, it is tempting to speculate that a wide-spectrum of metabolic sensitivity exists, in which some chromatin modifiers (such as KMT/KDM/TETs) may require only a short metabolic stimulus to elicit a sustained epigenetic response, while others (such as KAT/HDACs) may react more dynamically to changes in metabolism or nutritional status.
III. Regulation of Epigenetic Signaling by Cofactor Depletion
A second metabolic mechanism of epigenetic regulation is cofactor depletion. Cofactor depletion inhibits enzyme-catalyzed chromatin modifications by decreasing the concentration of enzyme cofactors to rate-limiting levels. Cofactor depletion can function independently of or collaborate with metabolic competition mechanisms, and disproportionately impacts chromatin-modifying enzymes with a high Km for their obligate cofactor. Cellular cofactors may be depleted by i) nutrient limitation of essential vitamin precursors, ii) inhibition of cofactor biosynthesis, or iii) overconsumption of the cofactor by a competing enzyme. An illustrative example of the latter is the inhibition of KMTs by nicotinamide-N-methyltransferase (NNMT).63
NNMT is a metabolic methyltransferase that is overexpressed in a variety of cancers and is phenotypically associated with increased tumor cell migration and invasion.64, 65 To understand how NNMT impacts cancer pathogenesis, Ulanovskaya et al. utilized an integrated profiling approach to study the effects of NNMT overexpression on metabolism, chromatin modification, and gene expression. Unbiased metabolomic profiling of NNMT overexpressing cells identified only two molecules as consistently increased: 1-methylnicotinamide (1MNA) and the cofactor byproduct SAH (Figure 3). Treatment of cells with 1MNA itself did not enhance tumor cell invasion or produce new metabolites, suggesting the primary effects of NNMT may derive from its ability to consume SAM and produce SAH. By coordinating NNMT overexpression with depletion of the SAM precursor methionine (Figure 3), the authors were able to distinguish between biological effects sensitive to SAM cofactor depletion (observed only with low methionine media) and cofactor competition caused by reduced SAH:SAM ratios (observed in low and high methionine media) (Table 2). Notably, increased SAH levels by themselves were not sufficient to reduce histone methylation, indicating that in this model KMTs are more sensitive to overall cellular concentrations of the SAM cofactor than the SAM/SAH ratio. The reduced levels of histone methylation correlated with changes in gene expression and increased migratory ability of tumor cells. At a molecular level NNMT overexpression leads to a ~40–50% decrease in H3K4me3, H3K9me2, and H3K27me3 levels, while having less effect on arginine and DNA methylation. This is consistent with biochemical data showing that some KMTs possess higher Km values for SAM than protein arginine methyltransferases (PRMTs), and thus might be expected to be disproportionately sensitive to cofactor depletion (Table 1). The finding that bulk DNA methylation was unchanged by NNMT overexpression is actually surprising based on the published Km and IC50 values of human DNMT1 for SAM and SAH;66 this may reflect the vagaries of cell culture conditions, or suggest changes in the DNA methylation machinery during NNMT overexpression.
Figure 3.
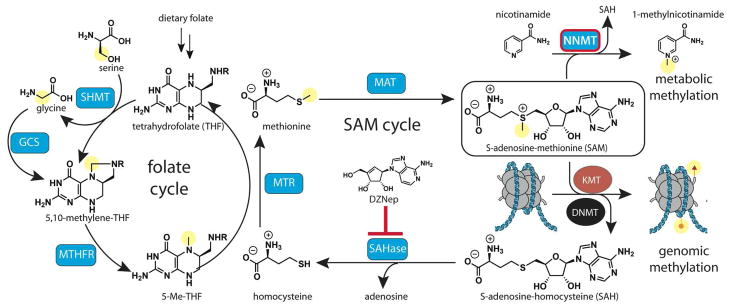
Regulation of epigenetic signaling by cofactor depletion. SAM functions as a universal methyl donor for methylation of macromolecules and metabolites. Overactivity of metabolic methyltransferases such as NNMT can result in depletion of SAM and decreased SAM/SAH ratios, thereby reducing KMT/DNMT activity and genomic methylation. Replenishment of SAM requires the activity of the folate and SAM cycles, which utilize methyl groups derived from folate or choline (not shown) to replenish intracellular methionine for SAM biosynthesis. Disruption of the SAM cycle by inhibitors has been shown to decrease the activity of KMT enzymes in cancer. Cofactors and byproducts of most enzymatic reactions have been omitted for simplicity. NNMT, nicotinamide N-methyltransferase; SAHase, S-adenosylhomocysteine hydrolase; MTR, 5-methyltetrahydrofolate-homocysteine methyltransferase; MTHFR, methylenetetrahydrofolate reductase; SHMT, serine hydroxymethyltransferase; MAT, methionine adenosyltransferase; DZNep, 3-deazaneplanocin A.
The integrated measurements of metabolism, chromatin modifications, and gene expression found in the NNMT study provide a textbook approach to dissecting metabolic mechanisms of epigenetic regulation. Although such efforts are still rare in the literature, evidence suggests other metabolic methyltransferases may also be capable of regulating chromatin methylation through cofactor depletion. Glycine-N-methyltransferase (GNMT) consumes SAM to methylate glycine, yielding sarcosine.67 As with NNMT, tumor cells often overexpress GNMT, creating an environment that inhibits histone and DNA methylation.67, 68 Hormone-induced expression of GNMT results in a global loss of DNA methylation.69 Catechol-O-methyltransferase (COMT) can reduce DNMT activity in vitro through similar mechanisms.70 Methylthioadenosine phosphorylase (MTAP) deletions are found in many tumors, and can inhibit protein arginine methylation by a combination of cofactor depletion (reducing methionine salvage) and competitive inhibition mechanisms (increased S-methylthioadenosine).71, 72
Contrasting with these cases, in some cancers cellular transformation and proliferation is stimulated by methylation-associated gene silencing.73 In these cancers, reducing methylation potential by cofactor depletion may provide a new avenue for therapy. For example, the SAH hydrolase inhibitor 3-deazaneplanocin A (DZNep) decreases protein and DNA methylation through disruption of SAM recycling (Figure 3).74 Inhibition of SAH hydrolase by DZNep globally inhibits protein and DNA methylation, indicating a severe disruption of cellular SAM and SAH levels.75 DZNep has shown promising activity as a single agent and combination therapy against hematological malignancies and solid tumors in preclinical models.76, 77
Cofactor depletion can also occur by limiting essential building blocks required for SAM biosynthesis. This manifests at the phenotypic level in yellow agouti mice, who acquire a brown coat when exposed to diets rich in folate, a SAM precursor, during development (Figure 3).78 This brown coat is the direct result of increased DNA methylation of the agouti gene, whose silencing allows mouse skin cells to produce a black pigment.79 The resulting brown coat phenotype is maintained for the entire life of the mouse and can even be inherited by the next generation. In addition to folate, changes in amino acid metabolism can also affect cofactor levels. For instance, glycine and serine provide methyl groups to SAM via the activities of serine hydroxymethyltransferase (SHMT) and the glycine cleavage system (GCS; Figure 3). Inhibition of threonine dehydrogenase (TDH) reduces glycine production in mouse embryonic stem cells, and has the downstream effect of depleting cells of the SAM cofactor.80 Cofactor depletion selectively reduced histone methylation at H3K4 (but not H3K9 or H3K27) and caused cells to differentiate due to an inability to induce the expression of genes necessary for self-renewal.80 While humans do not utilize TDH for glycine production, glycine and serine metabolism have been found to play critical roles in a number of cancers.81, 82 An important goal of future research will be to determine how the uptake and biosynthesis of amino acids, including non-essential amino acids such as proline,83 impact epigenetic states in disease.
Finally, while we have focused on SAM and chromatin methylation, cofactor depletion may also regulate other chromatin modifiers. Early studies of IDH1 mutation showed ectopic overexpression of IDH1 R132H could decrease cellular α-KG levels in cell lines, although the in vivo relevance of this effect is unclear.45 NAD+ levels are depleted by PARP enzymes in response to DNA damage, which may affect the activity of NAD+ utilizing HDACs such as SIRT1 (Table 1).84 NADH levels can also affect the activity of carboxyl-terminal binding protein (CtBP) a transcriptional corepressor active in breast cancer.85 In yeast and humans, histone acetylation is sensitive to reduced acetyl-CoA biosynthesis.58, 86, 87 Histone acetylation is also sensitive to inhibition of pantothenate kinase, the rate-limiting enzyme in CoA biosynthesis.88 In most of these cases, both how cofactor depletion collaborates with other metabolic mechanisms, and whether metabolic perturbations manifest global or targeted epigenetic effects, remain to be established.
IV. Regulation of Epigenetic Signaling by Localization of Cofactor Biosynthesis
All of the preceding examples involve regulation of epigenetic signaling by global changes in metabolite concentrations. Recent discoveries indicate chromatin-modifying enzymes may also be capable of responding to local changes in cofactor concentration. This emerging mechanism has been most well-studied in the context of MafK, a protein belonging to the Maf family of transcription factors whose overexpression is associated with poor prognosis in multiple myeloma.89 Spurred by proteomic studies that identified the SAM synthetase MATIIα as a MafK interaction partner in mouse cells, Katoh et al. analyzed how the genomic localization of MATIIα affected transcriptional repression by MafK.90 Chromatin immunoprecipitation showed that induction of the stress-inducible gene HO-1 led to displacement of MATIIα but not MafK from the HO-1 locus, suggesting MATIIα genomic occupancy is required for transcriptional repression. Transcriptional repression of HO-1 by MATIIα was dependent on an active catalytic domain, supporting a direct role for cofactor biosynthesis. Knockdown of MATIIα reduced levels of H3K4me2 and H3K9me2 at the HO-1 locus, but did not affect H3K36 or DNA methylation. Together, these observations support the hypothesis that KMTs may be capable of using increased SAM production at specific genomic loci as a signal to mediate discrete transcriptional outcomes (Figure 4). Genomic analyses of MATIIα occupancy and correlation with histone methylation levels will be required to understand whether this mechanism operates genome-wide or exclusively in the context of MafK transcription. However, several candidate KMTs were identified as interaction partners of MafK that may mediate this effect, including G9a, EHMT, and MLL1.90
Figure 4.

Regulation of epigenetic signaling by subcellular localization of cofactor biosynthesis. The SAM biosynthetic enzyme MAT associates with transcription factors at specific genomic loci, producing SAM that is used by KMT enzymes to methylate histones and establish a repressive heterochromatin state. Notably, addition of exogenous SAM does not stimulate the same effect, suggesting genomic-localized biosynthesis of SAM is required for transcriptional repression.
Genomic localization of cofactor biosynthesis is a relatively recent discovery and therefore has not been extensively studied. However, several observations support a potentially broad role for this mechanism. The KAT cofactor acetyl-CoA is produced by two main biosynthetic enzymes, ATP citrate-lyase (ACL) and acetyl-CoA synthetase (ACS). Both of these enzymes can be found in the nucleus in human cell lines.58 Furthermore, electron microscopy has found >95% of yeast ACS localizes inside the nucleus when cultured in rich media.86 The NAD+ synthase nicotinamide mononucleotide adenylyltransferase-1 (NMNAT-1) has been shown to co-localize in the nucleus with genes occupied by the HDAC SIRT1 and increase deacetylase activity.91 In addition to these examples, it has also been shown that primary metabolic enzymes directly regulate genome function without a cofactor intermediary. Pyruvate kinase M2 (PKM2), which catalyzes the final step in glycolysis, can directly phosphorylate histones at H3T11 using phosphoenolpyruvate as the phosphate group donor.92 This histone modification stimulates transcriptional activation at the MYC locus, and correlates with increased malignancy and progression in glioblastoma clinical isolates.
V. Mimicking Metabolic Mechanisms of Epigenetic Regulation with Synthetic Cofactors and Small Molecules
Understanding the metabolic basis of epigenetic regulation is not only an intellectual pursuit, but also one ultimately undertaken to provide a basis for therapeutic intervention in disease. Regulatory insights resulting from mechanistic study can be used to validate new targets, as in the case of mutant IDH1, or provide fodder for the development of new approaches to alter epigenetic signaling. Thus, it is useful to briefly compare metabolic strategies with those devised in the laboratory to study and perturb genome function.
The design and application of cofactor competitive inhibitors predates our knowledge that competitive metabolites may also regulate chromatin-modifying enzymes in cells, and is a topic worthy of its own discussion, provided by several superb review articles.17, 93, 94 However, consideration of a few examples is valuable. Synthetic cofactor analogues have been extremely useful for inhibition of KAT and KMT enzymes. Lys-CoA was one of the first designed KAT inhibitors, and consists of a high affinity bisubstrate formed by covalent linkage of the CoA cofactor to the ε-amine of lysine through a non-hydrolyzable acetyl bridge (Figure 5a).95 Lys-CoA inhibits the p300/CBP KAT reaction with a Ki of 19 nM in vitro, while related histone peptide-CoA conjugates show enhanced affinity and specificity for KATs in the pCAF and MYST families.95, 96 Rational design has also led to the development of potent cofactor-based inhibitors of the KMT DOT1L.97 These SAM analogues, exemplified by EPZ-5646 (Figure 5), take advantage of a unique open conformation DOT1L adopts upon SAM analogue binding to inhibit H3K79 methylation with high affinity (Ki = 0.08 nM) and selectivity.98 These SAM analogues reverse epigenetic patterning in cell and animal models, and show selective toxicity towards cancers harboring MLL gene fusions that are dependent on DOT1L activity.13, 97 A number of different molecular scaffolds containing the Fe(II)-chelating and dicarboxylate moieties of α-KG have been explored for inhibition of KDM and PHD enzymes, and show varying degrees of affinity and selectivity.99, 100 In contrast, aside from structural studies,101 inhibition of sirtuin-type HDACs by synthetic NAD+ analogues has been relatively unpursued.
Figure 5.

Mimicking metabolic mechanisms of epigenetic regulation with small molecules. (a) Synthetic cofactors applied for inhibition of chromatin-modifying enzymes. Cofactor-derived portions of each molecule are highlighted in gold. (b) NAD+ salvage pathway and NAMPT inhibitors. Blockade of NAD+ salvage has been shown to deplete cellular NAD+ levels and inhibit SIRT HDAC activity. (c) Chemical inducer of dimerization approach used to localize genomic function of HP1, which stimulates local KMT and DNMT activity. NAMPT, nicotinamide phosphoribosyltransferase; NMNAT, nicotinamide mononucleotide adenyltransferase; HP-1, heterochromatin binding protein.
In general, synthetic cofactor analogues show much higher affinity for KATs, KMTs, and KDMs than endogenous metabolites. However, due to their hydrophilic nature, one limitation synthetic cofactor analogues face is their low cell permeability. For example, Lys-CoA and related bisubstrate-CoA analogues must be delivered to cells using cell penetrating peptides or transfection agents.102 Cellular biosynthesis of Lys-CoA from a metabolic precursor has been explored, but appears to be incompatible with the substrate requirements of the human CoA biosynthetic pathway.103 SAM-based DOT1L inhibitors require prolonged administration to manifest biological effects,97 and cellular data for a number of highly active SAM-based KMT inhibitors have not been reported,104 both of which may reflect their poor cell penetrance. KDM inhibitors based on α-KG also suffer from poor cell permeability of dicarboxylates, a limitation that can be circumvented through their administration as esterase-labile prodrugs.93, 99 These challenges highlight one advantage endogenous metabolites have over synthetic cofactors as competitive inhibitors, namely that because their production occurs in the cell, high gradients of inhibitor can accumulate. This may be mimicked experimentally by using prodrug or metabolic precursor strategies to produce active species intracellularly. Another attractive approach is the discovery of chemotypes that directly compete with cofactors but are not structurally cofactor-based, such as the pyrimidone class of KMT inhibitors.105, 106
Cofactor depletion also provides fertile ground for epigenetic reprogramming by small molecules. Cofactors can be depleted pharmacologically by inhibition of salvage and recycling pathways, as with the aforementioned inhibition of SAH hydrolase by DZNep, which reduces histone and DNA methylation (Figure 3).75 Similar results can be elicited by treatment of cells with adenosine dialdehyde (Adox), an amine-reactive inhibitor of SAH hydrolase.75 Sirtuin HDAC activity is sensitive to cofactor depletion caused by small molecule inhibition of nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme of the NAD+ salvage pathway (Figure 5b).107 Treatment of cells with the NAMPT inhibitor FK866 can mimic effects observed during circadian oscillation of NAD+ levels, and increases acetylation of the regulatory protein BMAL to similar levels as observed in SIRT1 knockout mice.108 ATP-citrate lyase (ACL) inhibition reduces histone acetylation in cultured cells, although its effects on overall acetyl-CoA/CoA levels were not studied in detail.58 SAH, NAMPT, and ACL inhibitors all show potent antitumor activity.77, 109, 110 Further study will be required to understand what role inhibition of specific chromatin modifications plays in the effects of these pleiotropic agents.
In contrast to the previous two examples, no synthetic approach for controlling the genomic localization of cofactor biosynthesis has yet been developed. However, of relevance is a recent study in which approaches with a foundation in chemical biology have been applied to localize and study the effects of chromatin-modifying enzymes themselves.111 Hathway et al. recently developed an experimental system that applied chemical inducers of dimerization112 to control the dynamics of association of HP-1α and a transcriptional activator domain (TAD) at a designed Oct4 locus in mouse embryonic stem cells (Figure 5c).111 HP-1α is a KMT-recruiting protein involved in the establishment of transcriptionally silenced heterochromatin, while TAD domains stimulate transcription. Controlling the genomic localization of these two antagonistic protein domains with orthogonal small molecules (Figure 5c) allowed the experimental examination of how histone methylation propagates and is transmitted over multiple generations. Small molecule-induced recruitment of HP-1α silenced transcription from the Oct4 locus, and caused H3K9 trimethylation to spread ~10 kB from the site of targeting, similar to the span of natural heterochromatin territories. In mouse embryonic fibroblasts, induced histone methylation was inherited and maintained through multiple cell divisions even after removal of the chemical inducer of dimerization. These effects could be overcome by recruitment of the TAD domain, indicating that while HP-1α-induced histone methylation is self-propagating and persistent, they are also malleable. As methods for targeted genomic localization continue to develop,113 an interesting future question will be whether local epigenetic effects can also be stimulated by loci-specific recruitment of cofactor biosynthetic enzymes.
VI. Conclusion and Future Directions
It is becoming increasingly clear that metabolic control of epigenetic signaling is a real phenomenon that can contribute to phenotypic change and disease. While we have artificially limited our discussion here to a handful of well-studied chromatin-modifying enzymes that utilize acetyl-CoA, SAM, NAD+, and α-KG as cofactors, the constellation of protein and nucleic acid modifications is vast. Evidence supports a role for metabolism in regulating the activity of other cofactor-utilizing chromatin modifiers not discussed here, including non-sirtuin HDACs (flavin mononucleotide), PRMTs (SAM), PARPs (NAD+), histone threonine kinases (ATP), sirtuin lysine deacylases (succinyl-CoA, myristoyl-CoA), and O-GlcNAc-transferases (UDP-GlcNAc). 20–23, 114 Similarly, because acetylation and methylation of lysine residues block their modification by ubiquitin and ubiquitin-like modifiers, these modifications can also have a metabolic component.115 RNA methylation/hydroxymethylation is another cofactor-dependent (SAM/α-KG) epigenetic process that has been directly linked to obesity; however, the sensitivity of these enzymes to changes in cell metabolism has not been studied.116 Finally, chromatin modifiers underwent a nomenclature change in 2007 due to the realization that many of these enzymes modify and regulate the function of non-histone proteins.117 Understanding how metabolism co-regulates the posttranslational modification of histones,25 transcription factors,118 multiprotein complexes,119 and metabolic enzymes120 will be an important step to defining the feedback and feed-forward loops that connect metabolism and gene expression in vivo.
Looking forward, we see many valuable ways in which chemical biology can help explain how the metabolic state of the cell impacts epigenetic mechanisms. First and foremost, chemical biologists can accelerate our understanding of metabolic regulation of epigenetics by developing new tools to study biochemistry in living systems. The development and widespread application of genetically-encoded fluorescent metabolite sensors for imaging metabolite concentrations and gradients in living cells would provide a real-time view into how metabolism is rewired in response to nutrient uptake and/or metabolic perturbation.121, 122 Sensor-based approaches would complement traditional methods for measuring absolute metabolite concentration123 by differentiating free vs. bound cofactors and allowing metabolic compartmentalization to be directly visualized. A second crucial barrier is to correlate observed changes in metabolite concentration with enzyme activity. Activity-based protein profiling (ABPP) has proven a powerful approach to determine the activity of entire enzyme classes in living cells and cell lysates.124
Studies of kinases have demonstrated the feasibility of using active-site probes to profile the cofactor sensitivity of native cellular enzymes.125, 126 Notably, several active site-directed affinity probes for chromatin modifiers have been reported.127–130 As with metabolite profiling, the continued development of geneticallyencoded131, 132 and peptide-based133 reporters of chromatin-modifying enzyme activities will be an important complementary approach, providing insight into the dynamics and subcellular localization of specific posttranslational modification events. Real-time approaches may be especially valuable in determining how the kinetics and equilibrium of different chromatin marks relate to the metabolic sensitivity of their chromatin modifiers. Data from each of these methods will also provide valuable inputs for computational approaches to model the pleiotropic effects of metabolic perturbation.
Together with these approaches, understanding the functional consequences of metabolic flux and chromatin-modifying enzyme activity will also require new methods to profile the targets of cofactors and enzymes in living systems. Engineered “bump-hole” cofactor-enzyme pairs have been applied artfully to identify potential protein134, 135 and genomic136 targets of specific KMTs and PRMTs. Although this method does not currently report on endogenous KMT activities, this limitation is likely to be circumvented in the future through the use of genome editing tools to incorporate mutant KMTs into endogenous loci.113 From the perspective of metabolic regulation, it will be critical to find out whether KMT mutants differ from endogenous enzymes in their sensitivity to metabolites such as SAH and methylthioadenosine. Recent studies suggest chemical genetic approaches may also be useful for the identification of KAT substrates.137 This method faces the additional challenge of correcting for background due to the promiscuity of the KAT p300 for engineered cofactors138 as well as the high chemical reactivity of acyl-CoA species, which are known to non-enzymatically modify proteins.138, 139 Linked to this latter observation, recent studies that have shown that non-enzymatic modification of proteins by chemically reactive metabolites can directly modulate metabolic and transcriptional programs.32, 140 Therefore, another vital objective will be to understand how metabolism mediates protein modifications that result solely from chemical reactivity, and what, if any, downstream effects these modifications have on genome function.
A final way in which chemical biology can contribute to the studies of both metabolism and epigenetics is by continuing to develop new agents for the targeted perturbation of these processes. Organizations such as the Structural Genomics Consortium have made great strides in the development and dissemination of specific, high affinity probes for a variety of chromatin-modifying and chromatin-interacting proteins,141–143 and the identification of mutant IDH1 as an oncogenic driver has renewed interest in the development of specific small molecule inhibitors of metabolism.144 Cell permeable inhibitors and light-activated probes remain the gold standard for studying temporally defined effects of enzyme activation or inhibition.145 Combining carefully designed chemical agents with ensemble146 and single-cell methods147 for monitoring gene expression will aid the systematic identification of transcriptional programs sensitive to metabolic flux. Thus, as these techniques continue to develop and mature, it is our view that chemical biology may demonstrate a fine ear for “new melodies” in the carefully orchestrated symphony of transcriptional regulation.
Acknowledgments
We apologize to colleagues whose work could not be cited here due to space limitations. We thank members of the Meier laboratory (D. Montgomery, A. Sorum) and colleagues in the Chemical Biology Laboratory for careful reading of this manuscript and many helpful comments. JLM is supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Glossary
- Genome function
The functions of the genome are to serve as a template for replication and transcription, as well as to maintain fidelity of the template itself (repair) - this term refers to those three processes
- Epigenetic regulation
The regulation of transcription and other genomic functions by molecular events that are not directly dependent on the primary nucleotide sequence of DNA. Most commonly associated with histone/DNA modifications. Additional epigenetic modifiers beyond the scope of this review include ATP-dependent chromatin remodeling enzymes, non-coding RNAs and histone variants
- Chromatin (Heterochromatin, Euchromatin)
147-bp segments of DNA wrapped around histone octamers containing two copies each of the core histones H2A, H2B, H3, and H4. Open chromatin is referred to as euchromatin and is associated with transcriptionally active genes. Compacted chromatin is referred to as heterochromatin and is associated with transcriptionally inactive genes
- Histone acetylation
Posttranslational modification of histone lysine residues. Added by KATs, removed by HDACs. Commonly referred to by the name of the histone, residue modified, and modification. Example: H3K27Ac, monoacetylation of histone H3 on lysine 27. Lysine acetylation is associated with transcriptionally active euchromatin. Acetyl-lysine residues are bound by bromodomain-containing proteins
- Histone methylation
Posttranslational modification of histone lysine and arginine residues. Added by KMTs and PRMTs, removed by KDMs. Commonly referred to by the name of the histone, residue modified, number of modifications, and symmetry of modification (arginine only). Example: H3K9me3, trimethylation of histone H3 on lysine 4. H3R17me2a, dimethylation (assymetric) of histone H3 on lysine 4. Lysine methylation can promote DNA methylation through recruitment of repressive complexes; however, lysine methylation may be associated with either euchromatin or heterochromatin depending on the context. H3K9me3 residues interact with a variety of protein domains, including plant homeodomains
- DNA methylation
Methylation of the cytosine base at the C5 position. Added by DNMTs, oxidized by TETs to promote removal. Often occurs at genomic regions rich in 5′-CG-3′ sequences known as CpG islands. Associated with transcriptionally silenced heterochromatin. DNMTs can be recruited by plant homeodomain-containing proteins, thus linking them to histone lysine methylation
- Chromatin marks associated with transcriptionally active chromatin
Histone acetylation, H3K4me2, H3K4me3, H3K27Ac, H3K36me3, H3K79me2
- Chromatin marks associated with transcriptionally repressed chromatin
DNA methylation, H3K9me2, H3K9me3, H3K27me3
- Chromatin marks associated with bivalent states, poised for rapid induction of either transcription or repression
H3K4me3, H3K27me3
References
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Young RA. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 6.Schreiber SL, Bernstein BE. Signaling network model of chromatin. Cell. 2002;111:771–778. doi: 10.1016/s0092-8674(02)01196-0. [DOI] [PubMed] [Google Scholar]
- 7.Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20:259–266. doi: 10.1038/nsmb.2470. [DOI] [PubMed] [Google Scholar]
- 8.Ptashne M. Epigenetics: core misconcept. Proc Natl Acad Sci U S A. 2013;110:7101–7103. doi: 10.1073/pnas.1305399110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henikoff S, Shilatifard A. Histone modification: cause or cog? Trends Genet. 2011;27:389–396. doi: 10.1016/j.tig.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Whitehouse I, Smith DJ. Chromatin dynamics at the replication fork: there’s more to life than histones. Curr Opin Genet Dev. 2013;23:140–146. doi: 10.1016/j.gde.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 13.Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, Pollock RM, Richon VM, Kung AL, Armstrong SA. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, Farnham PJ, Hirst M, Lander ES, Mikkelsen TS, Thomson JA. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cole PA. Chemical probes for histone-modifying enzymes. Nat Chem Biol. 2008;4:590–597. doi: 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oermann EK, Wu J, Guan KL, Xiong Y. Alterations of metabolic genes and metabolites in cancer. Semin Cell Dev Biol. 2012;23:370–380. doi: 10.1016/j.semcdb.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frezza C, Pollard PJ, Gottlieb E. Inborn and acquired metabolic defects in cancer. J Mol Med (Berl) 2011;89:213–220. doi: 10.1007/s00109-011-0728-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab. 2012;16:9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaelin WG, Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153:56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krejci A. Metabolic sensors and their interplay with cell signalling and transcription. Biochem Soc Trans. 2012;40:311–323. doi: 10.1042/BST20110767. [DOI] [PubMed] [Google Scholar]
- 23.Donohoe DR, Bultman SJ. Metaboloepigenetics: interrelationships between energy metabolism and epigenetic control of gene expression. J Cell Physiol. 2012;227:3169–3177. doi: 10.1002/jcp.24054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 25.Dhall A, Chatterjee C. Chemical approaches to understand the language of histone modifications. ACS Chem Biol. 2011;6:987–999. doi: 10.1021/cb200142c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108:19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, Tomlinson IP. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 28.Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 2010;7:277–285. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 30.Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, Ratcliffe PJ, Linehan WM, Neckers L. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–153. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 31.Koivunen P, Hirsila M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem. 2007;282:4524–4532. doi: 10.1074/jbc.M610415200. [DOI] [PubMed] [Google Scholar]
- 32.Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K, Stevens M, Fischer R, Carmeliet P, Maxwell PH, Pugh CW, Frizzell N, Soga T, Kessler BM, El-Bahrawy M, Ratcliffe PJ, Pollard PJ. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524–537. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, Buffet A, Marcaillou C, Bertherat J, Amar L, Rustin P, De Reynies A, Gimenez-Roqueplo AP, Favier J. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 34.Smith EH, Janknecht R, Maher LJ., 3rd Succinate inhibition of alphaketoglutarate- dependent enzymes in a yeast model of paraganglioma. Hum Mol Genet. 2007;16:3136–3148. doi: 10.1093/hmg/ddm275. [DOI] [PubMed] [Google Scholar]
- 35.Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, Quezado M, Smith WI, Jr, Jahromi MS, Xekouki P, Szarek E, Walker RL, Lasota J, Raffeld M, Klotzle B, Wang Z, Jones L, Zhu Y, Wang Y, Waterfall JJ, O’Sullivan MJ, Bibikova M, Pacak K, Stratakis C, Janeway KA, Schiffman JD, Fan JB, Helman L, Meltzer PS. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3:648–657. doi: 10.1158/2159-8290.CD-13-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25:2436–2452. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mason EF, Hornick JL. Succinate dehydrogenase deficiency is associated with decreased 5-hydroxymethylcytosine production in gastrointestinal stromal tumors: implications for mechanisms of tumorigenesis. Mod Pathol. 2013 doi: 10.1038/modpathol.2013.86. [DOI] [PubMed] [Google Scholar]
- 38.He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, Chen JL, Tian H, Ling L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature. 2004;429:188–193. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 39.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, Abbott RM, Vickery TL, Reed JS, Robinson JS, Wylie T, Smith SM, Carmichael L, Eldred JM, Harris CC, Walker J, Peck JB, Du F, Dukes AF, Sanderson GE, Brummett AM, Clark E, McMichael JF, Meyer RJ, Schindler JK, Pohl CS, Wallis JW, Shi X, Lin L, Schmidt H, Tang Y, Haipek C, Wiechert ME, Ivy JV, Kalicki J, Elliott G, Ries RE, Payton JE, Westervelt P, Tomasson MH, Watson MA, Baty J, Heath S, Shannon WD, Nagarajan R, Link DC, Walter MJ, Graubert TA, DiPersio JF, Wilson RK, Ley TJ. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang P, Dong Q, Zhang C, Kuan PF, Liu Y, Jeck WR, Andersen JB, Jiang W, Savich GL, Tan TX, Auman JT, Hoskins JM, Misher AD, Moser CD, Yourstone SM, Kim JW, Cibulskis K, Getz G, Hunt HV, Thorgeirsson SS, Roberts LR, Ye D, Guan KL, Xiong Y, Qin LX, Chiang DY. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32:3091–3100. doi: 10.1038/onc.2012.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, Xiong Y. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, McDonough MA, King ON, Clifton IJ, Klose RJ, Claridge TD, Ratcliffe PJ, Schofield CJ, Kawamura A. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Travins J, Weiss S, Looper R, Ligon KL, Verhaak RG, Yan H, Kaelin WG., Jr Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, Levine R, Heguy A, Viale A, Morris LG, Huse JT, Mellinghoff IK, Chan TA. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG., Jr (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, Kunii K, Pedraza A, Schalm S, Silverman L, Miller A, Wang F, Yang H, Chen Y, Kernytsky A, Rosenblum MK, Liu W, Biller SA, Su SM, Brennan CW, Chan TA, Graeber TG, Yen KE, Mellinghoff IK. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, Yang H, Gross S, Artin E, Saada V, Mylonas E, Quivoron C, Popovici-Muller J, Saunders JO, Salituro FG, Yan S, Murray S, Wei W, Gao Y, Dang L, Dorsch M, Agresta S, Schenkein DP, Biller SA, Su SM, de Botton S, Yen KE. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 55.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, Harris IS, Holmes R, Wakeham A, Haight J, You-Ten A, Li WY, Schalm S, Su SM, Virtanen C, Reifenberger G, Ohashi PS, Barber DL, Figueroa ME, Melnick A, Zuniga-Pflucker JC, Mak TW. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488:656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell. 2011;42:426–437. doi: 10.1016/j.molcel.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langer MR, Fry CJ, Peterson CL, Denu JM. Modulating acetyl-CoA binding in the GCN5 family of histone acetyltransferases. J Biol Chem. 2002;277:27337–27344. doi: 10.1074/jbc.M203251200. [DOI] [PubMed] [Google Scholar]
- 60.Gao L, Chiou W, Tang H, Cheng X, Camp HS, Burns DJ. Simultaneous quantification of malonyl-CoA and several other short-chain acyl-CoAs in animal tissues by ion-pairing reversed-phase HPLC/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;853:303–313. doi: 10.1016/j.jchromb.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 61.Albaugh BN, Arnold KM, Denu JM. KAT(ching) metabolism by the tail: insight into the links between lysine acetyltransferases and metabolism. Chembiochem. 2011;12:290–298. doi: 10.1002/cbic.201000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barth TK, Imhof A. Fast signals and slow marks: the dynamics of histone modifications. Trends Biochem Sci. 2010;35:618–626. doi: 10.1016/j.tibs.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 63.Ulanovskaya OA, Zuhl AM, Cravatt BF. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat Chem Biol. 2013;9:300–306. doi: 10.1038/nchembio.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roessler M, Rollinger W, Palme S, Hagmann ML, Berndt P, Engel AM, Schneidinger B, Pfeffer M, Andres H, Karl J, Bodenmuller H, Ruschoff J, Henkel T, Rohr G, Rossol S, Rosch W, Langen H, Zolg W, Tacke M. Identification of nicotinamide N-methyltransferase as a novel serum tumor marker for colorectal cancer. Clin Cancer Res. 2005;11:6550–6557. doi: 10.1158/1078-0432.CCR-05-0983. [DOI] [PubMed] [Google Scholar]
- 65.Tang SW, Yang TC, Lin WC, Chang WH, Wang CC, Lai MK, Lin JY. Nicotinamide N-methyltransferase induces cellular invasion through activating matrix metalloproteinase-2 expression in clear cell renal cell carcinoma cells. Carcinogenesis. 2011;32:138–145. doi: 10.1093/carcin/bgq225. [DOI] [PubMed] [Google Scholar]
- 66.Bacolla A, Pradhan S, Larson JE, Roberts RJ, Wells RD. Recombinant human DNA (cytosine-5) methyltransferase. III Allosteric control, reaction order, and influence of plasmid topology and triplet repeat length on methylation of the fragile X CGG.CCG sequence. J Biol Chem. 2001;276:18605–18613. doi: 10.1074/jbc.M100404200. [DOI] [PubMed] [Google Scholar]
- 67.Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, Laxman B, Mehra R, Lonigro RJ, Li Y, Nyati MK, Ahsan A, Kalyana-Sundaram S, Han B, Cao X, Byun J, Omenn GS, Ghosh D, Pennathur S, Alexander DC, Berger A, Shuster JR, Wei JT, Varambally S, Beecher C, Chinnaiyan AM. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457:910–914. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 68.Guerinot F, Bohuon C. Glycine-N-methyltransferase levels in human breast cancer tissue. Eur J Cancer. 1977;13:1257–1259. doi: 10.1016/0014-2964(77)90033-0. [DOI] [PubMed] [Google Scholar]
- 69.Rowling MJ, McMullen MH, Schalinske KL. Vitamin A and its derivatives induce hepatic glycine N-methyltransferase and hypomethylation of DNA in rats. J Nutr. 2002;132:365–369. doi: 10.1093/jn/132.3.365. [DOI] [PubMed] [Google Scholar]
- 70.Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, Welsh W, Yang CS. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003;63:7563–7570. [PubMed] [Google Scholar]
- 71.Christopher SA, Diegelman P, Porter CW, Kruger WD. Methylthioadenosine phosphorylase, a gene frequently codeleted with p16(cdkN2a/ARF), acts as a tumor suppressor in a breast cancer cell line. Cancer Res. 2002;62:6639–6644. [PubMed] [Google Scholar]
- 72.Mowen KA, Tang J, Zhu W, Schurter BT, Shuai K, Herschman HR, David M. Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell. 2001;104:731–741. doi: 10.1016/s0092-8674(01)00269-0. [DOI] [PubMed] [Google Scholar]
- 73.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 74.Glazer RI, Hartman KD, Knode MC, Richard MM, Chiang PK, Tseng CK, Marquez VE. 3-Deazaneplanocin: a new and potent inhibitor of S-adenosylhomocysteine hydrolase and its effects on human promyelocytic leukemia cell line HL-60. Biochem Biophys Res Commun. 1986;135:688–694. doi: 10.1016/0006-291x(86)90048-3. [DOI] [PubMed] [Google Scholar]
- 75.Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE, Jones PA. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8:1579–1588. doi: 10.1158/1535-7163.MCT-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kemp CD, Rao M, Xi S, Inchauste S, Mani H, Fetsch P, Filie A, Zhang M, Hong JA, Walker RL, Zhu YJ, Ripley RT, Mathur A, Liu F, Yang M, Meltzer PA, Marquez VE, De Rienzo A, Bueno R, Schrump DS. Polycomb repressor complex-2 is a novel target for mesothelioma therapy. Clin Cancer Res. 2012;18:77–90. doi: 10.1158/1078-0432.CCR-11-0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Momparler RL, Idaghdour Y, Marquez VE, Momparler LF. Synergistic antileukemic action of a combination of inhibitors of DNA methylation and histone methylation. Leuk Res. 2012;36:1049–1054. doi: 10.1016/j.leukres.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 78.Cooney CA, Dave AA, Wolff GL. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J Nutr. 2002;132:2393S–2400S. doi: 10.1093/jn/132.8.2393S. [DOI] [PubMed] [Google Scholar]
- 79.Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet. 1999;23:314–318. doi: 10.1038/15490. [DOI] [PubMed] [Google Scholar]
- 80.Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, Asara JM, Daley GQ, Cantley LC. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science. 2013;339:222–226. doi: 10.1126/science.1226603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, Chen WW, Barrett FG, Stransky N, Tsun ZY, Cowley GS, Barretina J, Kalaany NY, Hsu PP, Ottina K, Chan AM, Yuan B, Garraway LA, Root DE, Mino-Kenudson M, Brachtel EF, Driggers EM, Sabatini DM. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Phang JM, Liu W, Hancock C. Bridging epigenetics and metabolism: role of non-essential amino acids. Epigenetics. 2013;8:231–236. doi: 10.4161/epi.24042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem. 2005;280:43121–43130. doi: 10.1074/jbc.M506162200. [DOI] [PubMed] [Google Scholar]
- 85.Di LJ, Byun JS, Wong MM, Wakano C, Taylor T, Bilke S, Baek S, Hunter K, Yang H, Lee M, Zvosec C, Khramtsova G, Cheng F, Perou CM, Miller CR, Raab R, Olopade OI, Gardner K. Genome-wide profiles of CtBP link metabolism with genome stability and epithelial reprogramming in breast cancer. Nat Commun. 2013;4:1449. doi: 10.1038/ncomms2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell. 2006;23:207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 87.Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012;48:612–626. doi: 10.1016/j.molcel.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Siudeja K, Srinivasan B, Xu L, Rana A, de Jong J, Nollen EA, Jackowski S, Sanford L, Hayflick S, Sibon OC. Impaired Coenzyme A metabolism affects histone and tubulin acetylation in Drosophila and human cell models of pantothenate kinase associated neurodegeneration. EMBO Mol Med. 2011;3:755–766. doi: 10.1002/emmm.201100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eychene A, Rocques N, Pouponnot C. A new MAFia in cancer. Nat Rev Cancer. 2008;8:683–693. doi: 10.1038/nrc2460. [DOI] [PubMed] [Google Scholar]
- 90.Katoh Y, Ikura T, Hoshikawa Y, Tashiro S, Ito T, Ohta M, Kera Y, Noda T, Igarashi K. Methionine adenosyltransferase II serves as a transcriptional corepressor of Maf oncoprotein. Mol Cell. 2011;41:554–566. doi: 10.1016/j.molcel.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 91.Zhang T, Berrocal JG, Frizzell KM, Gamble MJ, DuMond ME, Krishnakumar R, Yang T, Sauve AA, Kraus WL. Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J Biol Chem. 2009;284:20408–20417. doi: 10.1074/jbc.M109.016469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685–696. doi: 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rose NR, McDonough MA, King ON, Kawamura A, Schofield CJ. Inhibition of 2-oxoglutarate dependent oxygenases. Chem Soc Rev. 2011;40:4364–4397. doi: 10.1039/c0cs00203h. [DOI] [PubMed] [Google Scholar]
- 94.Copeland RA, Olhava EJ, Scott MP. Targeting epigenetic enzymes for drug discovery. Curr Opin Chem Biol. 2010;14:505–510. doi: 10.1016/j.cbpa.2010.06.174. [DOI] [PubMed] [Google Scholar]
- 95.Lau OD, Kundu TK, Soccio RE, Ait-Si-Ali S, Khalil EM, Vassilev A, Wolffe AP, Nakatani Y, Roeder RG, Cole PA. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol Cell. 2000;5:589–595. doi: 10.1016/s1097-2765(00)80452-9. [DOI] [PubMed] [Google Scholar]
- 96.Wu J, Xie N, Wu Z, Zhang Y, Zheng YG. Bisubstrate Inhibitors of the MYST HATs Esa1 and Tip60. Bioorg Med Chem. 2009;17:1381–1386. doi: 10.1016/j.bmc.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 97.Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, Jin L, Kuntz KW, Chesworth R, Moyer MP, Bernt KM, Tseng JC, Kung AL, Armstrong SA, Copeland RA, Richon VM, Pollock RM. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, Allain CJ, Klaus CR, Raimondi A, Scott MP, Waters NJ, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122:1017–1025. doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Luo X, Liu Y, Kubicek S, Myllyharju J, Tumber A, Ng S, Che KH, Podoll J, Heightman TD, Oppermann U, Schreiber SL, Wang X. A selective inhibitor and probe of the cellular functions of Jumonji C domain-containing histone demethylases. J Am Chem Soc. 2011;133:9451–9456. doi: 10.1021/ja201597b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.King ON, Li XS, Sakurai M, Kawamura A, Rose NR, Ng SS, Quinn AM, Rai G, Mott BT, Beswick P, Klose RJ, Oppermann U, Jadhav A, Heightman TD, Maloney DJ, Schofield CJ, Simeonov A. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS One. 2010;5:e15535. doi: 10.1371/journal.pone.0015535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Szczepankiewicz BG, Dai H, Koppetsch KJ, Qian D, Jiang F, Mao C, Perni RB. Synthesis of carba-NAD and the structures of its ternary complexes with SIRT3 and SIRT5. J Org Chem. 2012;77:7319–7329. doi: 10.1021/jo301067e. [DOI] [PubMed] [Google Scholar]
- 102.Zheng Y, Balasubramanyam K, Cebrat M, Buck D, Guidez F, Zelent A, Alani RM, Cole PA. Synthesis and evaluation of a potent and selective cell-permeable p300 histone acetyltransferase inhibitor. J Am Chem Soc. 2005;127:17182–17183. doi: 10.1021/ja0558544. [DOI] [PubMed] [Google Scholar]
- 103.Cebrat M, Kim CM, Thompson PR, Daugherty M, Cole PA. Synthesis and analysis of potential prodrugs of coenzyme A analogues for the inhibition of the histone acetyltransferase p300. Bioorg Med Chem. 2003;11:3307–3313. doi: 10.1016/s0968-0896(03)00265-7. [DOI] [PubMed] [Google Scholar]
- 104.Yao Y, Chen P, Diao J, Cheng G, Deng L, Anglin JL, Prasad BV, Song Y. Selective inhibitors of histone methyltransferase DOT1L: design, synthesis, and crystallographic studies. J Am Chem Soc. 2011;133:16746–16749. doi: 10.1021/ja206312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, Scott MP, Jin L, Smith JJ, Olhava EJ, Chesworth R, Moyer MP, Richon VM, Copeland RA, Keilhack H, Pollock RM, Kuntz KW. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 106.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, LaFrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK, Tummino PJ, Creasy CL. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 107.Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–7442. [PubMed] [Google Scholar]
- 108.Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–657. doi: 10.1126/science.1170803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311–321. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 110.Galli U, Travelli C, Massarotti A, Fakhfouri G, Rahimian R, Tron GC, Genazzani AA. Medicinal Chemistry of Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors. J Med Chem. 2013;56:6279–6296. doi: 10.1021/jm4001049. [DOI] [PubMed] [Google Scholar]
- 111.Hathaway NA, Bell O, Hodges C, Miller EL, Neel DS, Crabtree GR. Dynamics and memory of heterochromatin in living cells. Cell. 2012;149:1447–1460. doi: 10.1016/j.cell.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Corson TW, Aberle N, Crews CM. Design and Applications of Bifunctional Small Molecules: Why Two Heads Are Better Than One. ACS Chem Biol. 2008;3:677–692. doi: 10.1021/cb8001792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gaj T, Gersbach CA, Barbas CF., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lin H, Su X, He B. Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem Biol. 2012;7:947–960. doi: 10.1021/cb3001793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lin R, Tao R, Gao X, Li T, Zhou X, Guan KL, Xiong Y, Lei QY. Acetylation Stabilizes ATP-Citrate Lyase to Promote Lipid Biosynthesis and Tumor Growth. Mol Cell. 2013;51:506–518. doi: 10.1016/j.molcel.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zheng G, Dahl JA, Niu Y, Fu Y, Klungland A, Yang YG, He C. Sprouts of RNA epigenetics: The discovery of mammalian RNA demethylases. RNA Biol. 2013;10:915–918. doi: 10.4161/rna.24711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar R, Shilatifard A, Workman J, Zhang Y. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 118.Benayoun BA, Veitia RA. A post-translational modification code for transcription factors: sorting through a sea of signals. Trends Cell Biol. 2009;19:189–197. doi: 10.1016/j.tcb.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 119.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 120.Guan KL, Xiong Y. Regulation of intermediary metabolism by protein acetylation. Trends Biochem Sci. 2011;36:108–116. doi: 10.1016/j.tibs.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hung YP, Albeck JG, Tantama M, Yellen G. Imaging cytosolic NADH-NAD(+) redox state with a genetically encoded fluorescent biosensor. Cell Metab. 2011;14:545–554. doi: 10.1016/j.cmet.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Paige JS, Nguyen-Duc T, Song W, Jaffrey SR. Fluorescence imaging of cellular metabolites with RNA. Science. 2012;335:1194. doi: 10.1126/science.1218298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Evans MJ, Cravatt BF. Mechanism-based profiling of enzyme families. Chem Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- 125.Becher I, Savitski MM, Savitski MF, Hopf C, Bantscheff M, Drewes G. Affinity profiling of the cellular kinome for the nucleotide cofactors ATP, ADP, and GTP. ACS Chem Biol. 2013;8:599–607. doi: 10.1021/cb3005879. [DOI] [PubMed] [Google Scholar]
- 126.Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, Nordin B, Weissig H, Yang Q, Lee JD, Gray NS, Kozarich JW. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem Biol. 2011;18:699–710. doi: 10.1016/j.chembiol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hwang Y, Thompson PR, Wang L, Jiang L, Kelleher NL, Cole PA. A selective chemical probe for coenzyme A-requiring enzymes. Angew Chem Int Ed Engl. 2007;46:7621–7624. doi: 10.1002/anie.200702485. [DOI] [PubMed] [Google Scholar]
- 128.Cen Y, Falco JN, Xu P, Youn DY, Sauve AA. Mechanism-based affinity capture of sirtuins. Org Biomol Chem. 2011;9:987–993. doi: 10.1039/c0ob00774a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rotili D, Altun M, Kawamura A, Wolf A, Fischer R, Leung IK, Mackeen MM, Tian YM, Ratcliffe PJ, Mai A, Kessler BM, Schofield CJ. A photoreactive small-molecule probe for 2-oxoglutarate oxygenases. Chem Biol. 2011;18:642–654. doi: 10.1016/j.chembiol.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dalhoff C, Huben M, Lenz T, Poot P, Nordhoff E, Koster H, Weinhold E. Synthesis of S-adenosyl-L-homocysteine capture compounds for selective photoinduced isolation of methyltransferases. Chembiochem. 2010;11:256–265. doi: 10.1002/cbic.200900349. [DOI] [PubMed] [Google Scholar]
- 131.Lin CW, Jao CY, Ting AY. Genetically encoded fluorescent reporters of histone methylation in living cells. J Am Chem Soc. 2004;126:5982–5983. doi: 10.1021/ja038854h. [DOI] [PubMed] [Google Scholar]
- 132.Dancy BM, Crump NT, Peterson DJ, Mukherjee C, Bowers EM, Ahn YH, Yoshida M, Zhang J, Mahadevan LC, Meyers DJ, Boeke JD, Cole PA. Live-cell studies of p300/CBP histone acetyltransferase activity and inhibition. Chembiochem. 2012;13:2113–2121. doi: 10.1002/cbic.201200381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Baba R, Hori Y, Mizukami S, Kikuchi K. Development of a fluorogenic probe with a transesterification switch for detection of histone deacetylase activity. J Am Chem Soc. 2012;134:14310–14313. doi: 10.1021/ja306045j. [DOI] [PubMed] [Google Scholar]
- 134.Wang R, Zheng W, Yu H, Deng H, Luo M. Labeling substrates of protein arginine methyltransferase with engineered enzymes and matched S-adenosyl-L-methionine analogues. J Am Chem Soc. 2011;133:7648–7651. doi: 10.1021/ja2006719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Islam K, Bothwell I, Chen Y, Sengelaub C, Wang R, Deng H, Luo M. Bioorthogonal profiling of protein methylation using azido derivative of S-adenosyl-L-methionine. J Am Chem Soc. 2012;134:5909–5915. doi: 10.1021/ja2118333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang R, Islam K, Liu Y, Zheng W, Tang H, Lailler N, Blum G, Deng H, Luo M. Profiling genome-wide chromatin methylation with engineered posttranslation apparatus within living cells. J Am Chem Soc. 2013;135:1048–1056. doi: 10.1021/ja309412s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yang C, Mi J, Feng Y, Ngo L, Gao T, Yan L, Zheng YG. Labeling lysine acetyltransferase substrates with engineered enzymes and functionalized cofactor surrogates. J Am Chem Soc. 2013;135:7791–7794. doi: 10.1021/ja311636b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yang YY, Ascano JM, Hang HC. Bioorthogonal chemical reporters for monitoring protein acetylation. J Am Chem Soc. 2010;132:3640–3641. doi: 10.1021/ja908871t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tanner KG, Trievel RC, Kuo MH, Howard RM, Berger SL, Allis CD, Marmorstein R, Denu JM. Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J Biol Chem. 1999;274:18157–18160. doi: 10.1074/jbc.274.26.18157. [DOI] [PubMed] [Google Scholar]
- 140.Moellering RE, Cravatt BF. Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science. 2013;341:549–553. doi: 10.1126/science.1238327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, Liu F, Gao C, Huang XP, Kuznetsova E, Rougie M, Jiang A, Pattenden SG, Norris JL, James LI, Roth BL, Brown PJ, Frye SV, Arrowsmith CH, Hahn KM, Wang GG, Vedadi M, Jin J. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013 doi: 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.James LI, Barsyte-Lovejoy D, Zhong N, Krichevsky L, Korboukh VK, Herold JM, MacNevin CJ, Norris JL, Sagum CA, Tempel W, Marcon E, Guo H, Gao C, Huang XP, Duan S, Emili A, Greenblatt JF, Kireev DB, Jin J, Janzen WP, Brown PJ, Bedford MT, Arrowsmith CH, Frye SV. Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat Chem Biol. 2013;9:184–191. doi: 10.1038/nchembio.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, Dimaggio PA, Wasney GA, Siarheyeva A, Dong A, Tempel W, Wang SC, Chen X, Chau I, Mangano TJ, Huang XP, Simpson CD, Pattenden SG, Norris JL, Kireev DB, Tripathy A, Edwards A, Roth BL, Janzen WP, Garcia BA, Petronis A, Ellis J, Brown PJ, Frye SV, Arrowsmith CH, Jin J. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7:566–574. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10:671–684. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- 145.Lee HM, Larson DR, Lawrence DS. Illuminating the chemistry of life: design, synthesis, and applications of “caged” and related photoresponsive compounds. ACS Chem Biol. 2009;4:409–427. doi: 10.1021/cb900036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 147.Larson DR, Zenklusen D, Wu B, Chao JA, Singer RH. Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science. 2011;332:475–478. doi: 10.1126/science.1202142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tanner KG, Langer MR, Kim Y, Denu JM. Kinetic mechanism of the histone acetyltransferase GCN5 from yeast. J Biol Chem. 2000;275:22048–22055. doi: 10.1074/jbc.M002893200. [DOI] [PubMed] [Google Scholar]
- 149.Thompson PR, Kurooka H, Nakatani Y, Cole PA. Transcriptional coactivator protein p300. Kinetic characterization of its histone acetyltransferase activity. J Biol Chem. 2001;276:33721–33729. doi: 10.1074/jbc.M104736200. [DOI] [PubMed] [Google Scholar]
- 150.Liu X, Wang L, Zhao K, Thompson PR, Hwang Y, Marmorstein R, Cole PA. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008;451:846–850. doi: 10.1038/nature06546. [DOI] [PubMed] [Google Scholar]
- 151.Tanner KG, Langer MR, Denu JM. Kinetic mechanism of human histone acetyltransferase P/CAF. Biochemistry (Mosc) 2000;39:11961–11969. doi: 10.1021/bi001272h. [DOI] [PubMed] [Google Scholar]
- 152.Richon VM, Johnston D, Sneeringer CJ, Jin L, Majer CR, Elliston K, Jerva LF, Scott MP, Copeland RA. Chemogenetic analysis of human protein methyltransferases. Chem Biol Drug Des. 2011;78:199–210. doi: 10.1111/j.1747-0285.2011.01135.x. [DOI] [PubMed] [Google Scholar]
- 153.Sack JS, Thieffine S, Bandiera T, Fasolini M, Duke GJ, Jayaraman L, Kish KF, Klei HE, Purandare AV, Rosettani P, Troiani S, Xie D, Bertrand JA. Structural basis for CARM1 inhibition by indole and pyrazole inhibitors. Biochem J. 2011;436:331–339. doi: 10.1042/BJ20102161. [DOI] [PubMed] [Google Scholar]
- 154.Smith BC, Hallows WC, Denu JM. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal Biochem. 2009;394:101–109. doi: 10.1016/j.ab.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fjeld CC, Birdsong WT, Goodman RH. Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc Natl Acad Sci U S A. 2003;100:9202–9207. doi: 10.1073/pnas.1633591100. [DOI] [PMC free article] [PubMed] [Google Scholar]