

Abstract
Predicting the 3D structures of small molecules is a common problem in chemoinformatics. Even the best methods are inaccurate for complex molecules, and there is a large gap in accuracy between proprietary and free algorithms. Previous work presented COSMOS, a novel, data-driven algorithm that uses knowledge of known structures from the Cambridge Structural Database, and demonstrated performance that was competitive with proprietary algorithms. However, dependence on the Cambridge Structural Database prevented its widespread use. Here we present an updated version of the COSMOS structure predictor, complete with a free structure library derived from open data sources. We demonstrate that COSMOS performs better than other freely-available methods, with a mean RMSD of 1.16 Å and 1.68 Å for organic and metal-organic structures, and a mean prediction time of 60 ms per molecule. This is a 17% and 20% reduction in RMSD compared to the free predictor provided by Open Babel, and ten times faster. The ChemDB webportal provides a COSMOS prediction webserver, as well as downloadable copies of the COSMOS executable and the library of molecular substructures.
Introduction
Accurate 3D structures are needed as a starting point for chemoinformatics calculations on small molecules, such as conformer generation1 and quantum mechanics models. A variety of proprietary and free software tools have been developed for this task,2–4 but there is no perfect solution that meets users’ demands for coverage, speed, and accuracy.
COordinates of Small MOleculeS (COSMOS) is a fast, data-driven algorithm that predicts 3D molecule geometries by matching substructures against a library of known structures, thus becoming more accurate with a larger data library. The algorithm attempts both exact matching and ’fuzzy’ matching of substructures, where a match is based on topology and general atom properties rather than specific elements or atom types. A complete structure is then created by joining rigid substructures according to known torsion angles, and attempts are made to resolve stereochemistry errors (if specified) through a series of transformations on the structure as a whole. In previous work,5 we demonstrated that the COSMOS algorithm had similar accuracy to the proprietary algorithm CORINA.6 Unfortunately, this version of COSMOS relied on crystal structure data from the Cambridge Structural Database (CSD),7 restricting how COSMOS could be used without a CSD license.8 In this paper, we introduce a new version of COSMOS that includes a free library of substructures derived by mining crystallography data from open databases.
Open Babel is an open-source chemoinformatics toolkit that includes one of the best free 3D structure predictors to our knowledge.9 The Open Babel predictor also uses a data-driven approach, but unlike the COSMOS structure library, which contains hundreds of thousands of unique substructures indexed by isomeric SMILES, the Open Babel predictor uses a small library containing only two thousand of the most common ring systems found in the NCI and ZINC databases.10 Furthermore, these are indexed by SMARTS, which only allow for fuzzy matching.
A free substructure data library
We present a library of 3D molecule substructures derived from two large sources of free X-ray crystallography data. The first is the Crystallography Open Database (COD),11 an open repository for crystal-structure files. The second is CrystalEye,12 a database created by mechanically crawling the websites of scientific journals and downloading structure files that accompany publications. The data obtained from these sources overlaps with the CSD because they are all based on published crystallography data. In order to curate this free data, a pipeline was developed to identify errors such as non-standard representations, partially-specified structures, and missing atoms. Some molecules with missing bonds or hydrogens were corrected, while other molecules were removed from the dataset. From these curated molecule structures, rigid substructures were extracted by removing rotatable bonds, and ring systems extracted by removing non-ring atoms. A single instance of each unique substructure was included in the substructure library with its 3D coordinates.
Table 1 compares the number of unique substructures in the free library to the number in the CSD. The free library covers only a fraction of the unique substructures found in the CSD, but the intersection substructures tend to be more common and therefore more useful for the prediction algorithm. Table 1 also includes the number of unique substructures used in the Open Babel predictor.
Table 1.
Unique substructures in each library, and percent coverage of CSD library.
| Total | Organic | Metal-Organic | |
|---|---|---|---|
|
| |||
| CSD | 697813 (100%) | 182075 (100%) | 515738 (100%) |
| Free | 446121 (9%) | 122909 (22%) | 323212 (4%) |
| Open Babel | 2236 | 2236 | 0 |
Experiments
We compared the coverage, accuracy, and speed of COSMOS to three other freely-available algorithms for structure prediction: Open Babel, RDKit, and Balloon. Predicted 3D structures were compared to ground-truth 3D structures from the CSD, both for a test set of organic molecules and a test set of metal-organic molecules. The test sets were constructed by randomly sampling 10,000 organic and 10,000 metal-organic molecules from the CSD, then removing molecules with any substructures contained in either of the data-derived libraries. This is necessary because both the test sets and the libraries contain structures from the same published crystallography data. Substructures were considered equivalent if they had the following three properties: 1) at least 7 heavy atoms, 2) the same isomeric SMILES code, and 3) an RMSD less than 0.001 Angstroms. After removing these, our clean test sets consisted of 9,184 organic molecules and 9,595 metal-organic molecules.
COSMOS relies on the OEChem software library,13,14 so the test molecules were represented as OEChem isomeric SMILES for COSMOS, RDKit, and Balloon. However, the Open Babel toolkit has its own functions for computing isomeric SMILES, so the Open Babel predictions were based on Open Babel isomeric SMILES computed directly from the 3D test structures.
Open Babel predictions were made using the OBBuild method and optimized using the UFF molecular force field15 for 500 steps. Because the Open Babel predictor uses a substructure library, we tested whether the predictor accuracy would increase with additional substructures from the crystallography data sources. As in the original Open Babel substructure library, the augmented libraries were sorted in decreasing size so that large substructures were matched first. Canonical SMILES were used to index the new substructures rather than the more general SMARTS because the SMARTS-matching routine was prohibitively slow with a large library. All SMILES were computed using Open Babel’s native routines.
RDKit16 is an open source chemoinformatics package that includes tools for generating 3D conformations of molecules from SMILES. RDKit relies on the combination of distance geometry methods with molecular mechanics force fields. It does not use a data-driven approach and has no substructure library. RDKit predictions were made by generating a single conformation, adding explicit hydrogens, then optimizing the structure with the UFF force field.15
Balloon4 is a freely available 3D conformer generation algorithm. Structures were predicted by generating a single conformer and performing the MMFF94 force field minimization provided with the software.17
Results
Coverage
Sometimes a prediction algorithm is unable to predict a good structure for a molecule and fails. In fact, none of the predictors were capable of predicting every molecule in either test set. Table 2 shows that RDKit and Balloon had significant difficulty predicting complex metal-organic structures, while COSMOS had the best coverage for both the organic and metal-organic test sets.
Table 2.
Fraction of test structures that were successfully predicted.
| Organic | Metal-Organic | |
|---|---|---|
|
| ||
| COSMOS (Free library) | 99% | 90% |
| Open Babel (OB library) | 98% | 85% |
| RDKit | 88% | 23% |
| Balloon | 89% | 58% |
Accuracy
Accuracy was measured as the RMSD of the heavy atoms. We account for differences in coverage by comparing the RMSD of the intersection set – the set of structures for which every method succeeded in predicting a structure. For the organic test set, this intersection set consists of 7,154 of 9,184 structures (Table 3); the metal-organic intersection consists of 5,053 of 9,595 structures (Table 4). RDKit and Balloon had very low coverage for the metal-organic molecules, so they were left out of the metal-organic comparison.
Table 3.
Mean RMSD (and standard deviation) in Å of predicted organic molecule structures, for different structure prediction algorithms (rows) and substructure libraries (columns).
| CSD | Free | OB | None | |
|---|---|---|---|---|
|
| ||||
| COSMOS | 1.04 (0.88) | 1.16 (0.88) | - | 1.24 (0.92) |
| Open Babel | 1.39 (0.96) | 1.39 (0.97) | 1.39 (0.97) | 1.65 (0.98) |
| RDKit | - | - | - | 1.36 (0.91) |
| Balloon | - | - | - | 1.47 (1.05) |
Table 4.
Mean RMSD (and standard deviation) in Å of predicted metal-organic molecule structures, for different structure prediction algorithms (rows) and substructure libraries (columns).
| CSD | Free | OB | None | |
|---|---|---|---|---|
|
| ||||
| COSMOS | 1.25 (1.07) | 1.68 (1.09) | - | 1.97 (1.07) |
| Open Babel | 2.10 (1.18) | 2.11 (1.18) | 2.11 (1.17) | 2.28 (1.22) |
Surprisingly, adding substructures to the Open Babel library had little effect on accuracy. Presumably this is because the substructures in the Open Babel library already cover a large portion of the substructures found in the test set, and the additional substructures have little impact on the final structure after force field minimization. A particular case is metallocenes, where force field minimization will give an inaccurate structure even when the initial structure is good (Figure 2). Thus, expanding the library may provide better initial structures for these complex structures, but the force field minimization will still result in the same, poor prediction.
Figure 2.
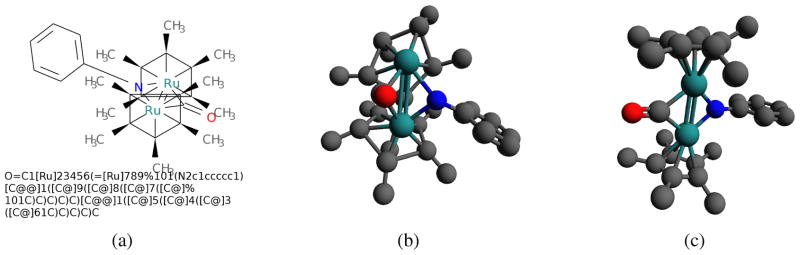
A typical metallocene complex: a) 2D structure and isomeric SMILES; b) 3D structure predicted by Open Babel; c) 3D structure predicted by COSMOS. The Open Babel and COSMOS RMSDs are 2.15 Å and 0.60 Å, respectively, and predictions took 2.15s and 1.21s.
Speed
COSMOS is significantly faster than the other prediction algorithms because it performs substructure matching with hash tables and uses a fast knowledge-based algorithm to optimize the final structure. Open Babel, although it uses a similar data-driven approach, is slower for two reasons: 1) it uses a SMARTS routine to match substructures with those in a library, and 2) it relies on a force field minimization on the final structure. RDKit and Balloon also use force field minimizations. Table 5 compares the average prediction time per molecule for the common set of successfully-predicted organic molecules. All experiments were carried out on an Intel Xeon 3.00 GHz processor with 48 Gb RAM.
Table 5.
Mean prediction time per molecule in seconds (with standard deviation).
| COSMOS with free library | Open Babel with OB library | RDKIT | Balloon |
|---|---|---|---|
| 0.06 (0.16) | 0.77 (0.74) | 0.19 (0.28) | 1.71 (3.85) |
Discussion
The COSMOS algorithm performs better than any other free prediction algorithm to our knowledge. The commonly-used CORINA algorithm may offer better performance on some types of molecules, but it is commercial. CORINA is also more limited in that it is unable to make predictions on metallocene complexes and any other molecule with a bond order greater than six.
COSMOS’s advantage over other structure predictors is that it makes heavy use of known molecular structures to predict new ones. It is therefore critical to have a large, varied data library to make accurate predictions over different areas of chemistry. We have shown here that a library of freely-available crystallography data, while a fraction of the size of the CSD, is nearly as good for predicting 3D molecular structures of both organic and metal-organic structures.
One approach we tried for expanding the substructure library even further was to predict library substructures using highly-accurate-but-computationally-expensive quantum mechanics methods. These methods are capable of modelling very complex molecular structures, both in gas phase or within a solvent,18 so substructures successfully predicted in this way are accurate enough to include in our data library. However, we found that geometry optimization with these methods was very sensitive to the initial structure, and was unreliable for the rough structures we could provide as input. Thus, high-throughput quantum mechanics modelling did not seem to be a fruitful approach to expanding the substructure library.
Conclusions
These results demonstrate that COSMOS is a versatile, fast, and accurate 3D structure predictor for drug-like molecules. Hashing substructures with isomeric SMILES keys has a significant speed advantage over Open Babel’s SMARTS-matching approach, and the knowledge-based assembly algorithm is more accurate than those that rely on molecular mechanics force fields. Furthermore, the accuracy of COSMOS will improve as more crystallography data becomes available and our substructure library grows. The latest substructure library, containing almost 500,000 unique molecule fragments, is available for download with the compiled COSMOS python code. COSMOS predictions can also be made through our online server, which is part of the ChemDB chemoinformatics web portal,19 http://cdb.ics.uci.edu.
Figure 1.
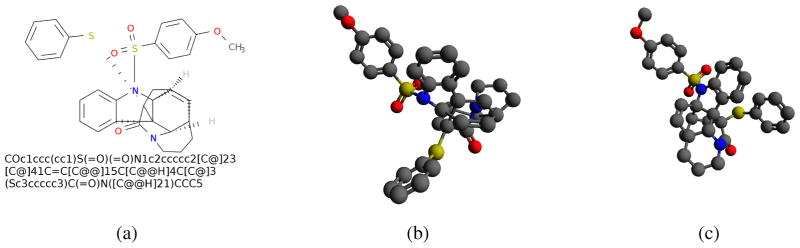
A complex organic molecule for which COSMOS performs better than Open Babel: a) 2D structure and isomeric SMILES; b) 3D structure predicted by Open Babel; c) 3D structure predicted by COSMOS. The Open Babel and COSMOS RMSDs are 3.23 Å and 2.44 Å, respectively, and predictions took 1.48s and 0.21s.
Figure 3.
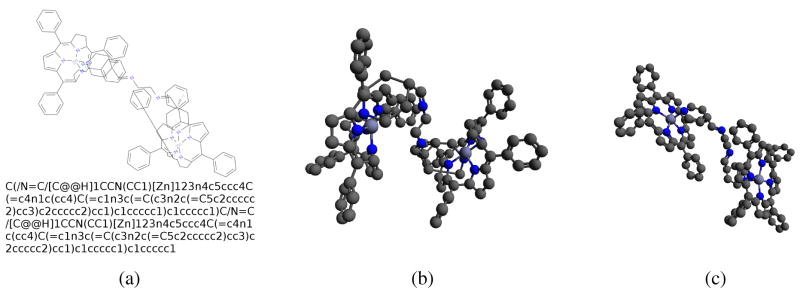
A typical large, metal-organic architecture: a) 2D structure and isomeric SMILES; b) 3D structure predicted by Open Babel; c) 3D structure predicted by COSMOS. The Open Babel and COSMOS RMSDs are 7.38 Å and 4.77 Å, respectively, and predictions took 6.66s and 0.37s.
Acknowledgments
This publication was made possible by grant number T15LM07443 from the National Library of Medicine at the National Institutes of Health. We wish to acknowledge OpenEye Scientific Software and ChemAxon for academic software licenses, and Jordan Hayes and Yuzo Kanomata for computing support.
References
- 1.O’Boyle NM, Vandermeersch T, Flynn CJ, Maguire AR, Hutchison GR. Confab -Systematic generation of diverse low-energy conformers. Journal of Cheminformatics. 2011;3:8. doi: 10.1186/1758-2946-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. Journal of Chemical Information and Modeling. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leite TB, Gomes D, Miteva M, Chomilier J, Villoutreix B, Tuffery P. Frog: a FRee Online druG 3D conformation generator. Nucleic Acids Research. 2007;35:W568–W572. doi: 10.1093/nar/gkm289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vainio MJ, Johnson MS. Generating Conformer Ensembles Using a Multiobjective Genetic Algorithm. Journal of Chemical Information and Modeling. 2007;47:2462–2474. doi: 10.1021/ci6005646. [DOI] [PubMed] [Google Scholar]
- 5.Andronico A, Randall A, Benz RW, Baldi P. Data-driven high-throughput prediction of the 3-D structure of small molecules: review and progress. Journal of chemical information and modeling. 2011;51:760–776. doi: 10.1021/ci100223t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadowski J, Gasteiger J, Klebe G. Comparison of Automatic Three-Dimensional Model Builders Using 639 X-ray Structures. Journal of Chemical Information and Computer Sciences. 1994;34:1000–1008. [Google Scholar]
- 7.Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta crystallographica Section B, Structural science. 2002;58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 8.Baldi P. Data-Driven High-Throughput Prediction of the 3-D Structure of Small Molecules: Review and Progress. A Response to the Letter by the Cambridge Crystallographic Data Centre. Journal of Chemical Information and Modeling. 2011;51:3029–3029. doi: 10.1021/ci200460z. [DOI] [PubMed] [Google Scholar]
- 9.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. Journal of Cheminformatics. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Irwin JJ, Shoichet BK. ZINC–a free database of commercially available compounds for virtual screening. Journal of chemical information and modeling. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grazulis S, Daskevic A, Merkys A, Chateigner D, Lutterotti L, Quiros M, Sere-bryanaya NR, Moeck P, Downs RT, Le Bail A. Crystallography Open Database (COD): an open-access collection of crystal structures and platform for world-wide collaboration. Nucleic Acids Research. 2011;40:D420–D427. doi: 10.1093/nar/gkr900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Day N, Downing J, Adams S, England NW, Murray-Rust P. CrystalEye : automated aggregation, semantification and dissemination of the world’s open crystallographic data. Journal of Applied Crystallography. 2012;45:316–323. [Google Scholar]
- 13.OEChem TK version 1.7.2.4. OpenEye Scientific Software; Santa Fe, NM: http://www.eyesopen.com/docs/toolkits/ http://www.eyesopen.com. [Google Scholar]
- 14.SZYBKI version 1.4.0. OpenEye Scientific Software; Santa Fe, NM: http://www.eyesopen.com. [Google Scholar]
- 15.Rappe AK, Casewit CJ, Colwell KS, Goddard WA, Skiff WM. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. Journal of the American Chemical Society. 1992;114:10024–10035. [Google Scholar]
- 16.Landrum G. RDKit: Open-source chemoinformatics. 2012 http://www.rdkit.org.
- 17.Halgren TA. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. Journal of Computational Chemistry. 1996;17:520–552. [Google Scholar]
- 18.Klamt A, Schüürmann G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. Journal of the Chemical Society, Perkin Transactions. 1993;2:799–805. [Google Scholar]
- 19.Chen J, Swamidass SJ, Dou Y, Baldi P. ChemDB: a public database of small molecules and related chemoinformatics resources. Bioinformatics. 2005;21:4133–4139. doi: 10.1093/bioinformatics/bti683. [DOI] [PubMed] [Google Scholar]