

Abstract
Purpose
Mutations affecting the KRAS gene are an established negative predictor for anti-epidermal growth factor receptor (anti-EGFR) therapies in metastatic colorectal cancer (CRC). However, the role of KRAS mutation as a biomarker for anti-vascular endothelial growth factor (VEGF) remains controversial.
Materials and Methods
We analyzed retrospective data from 32 CRC patients who were available for KRAS mutation status and received cytotoxic chemotherapy plus bevacizumab as a first-line therapy. Six of 32 patients received anti-EGFR therapies. We used KRAS mutation status as a predictive or prognostic factor in CRC patients receiving bevacizumab.
Results
We observed mutations in KRAS in 59.4% of patients. Bevacizumab was used in combination with oxaliplatin based regimens. There was no significant difference for progression free survival (PFS) and overall survival (OS) in patients with oxaliplatin based cytotoxic chemotherapy plus bevacizumab according to the status of KRAS mutation. After first-line therapy, 28 patients (87.5%) received second-line therapy. In univariate analysis, KRAS mutations did not have a major prognostic value for PFS (hazard ratio, 1.007; 95% confidence interval [CI], 0.469 to 2.162; p>0.05) or OS (hazard ratio, 0.548; 95% CI, 0.226 to 1.328; p>0.05). In addition, anti-EGFR therapies did not affect the impact on OS.
Conclusion
KRAS mutation is neither a predictive for bevacizumab nor a prognostic for OS in CRC patients receiving anti-VEGF therapy.
Keywords: KRAS, Vascular endothelial growth factor, Colonic neoplasms
Introduction
Colorectal cancer (CRC) is the fourth most common cancer in men and third most common cancer in women worldwide [1]. Although patients diagnosed with early stage disease have a high cure rate, many present later when five year survival is 5-8%. Palliative chemotherapy is the only option for the majority of these patients. With advances in systemic chemotherapy for metastatic CRC, survival has increased from 12 months with fluorouracil (5-FU) monotherapy to about 2 years with the addition of oxaliplatin, irinotecan, and targeted or biologic agents [2-4].
The epithelial growth factor receptor (EGFR) and the vascular endothelial growth factor (VEGF) are related to the growth, survival, proliferation and metastasis of tumor cells. Thus, targeted agents that are capable of inhibiting signal transduction through EGFR and VEGF have been developed [5-8]. This led to the advent of many EGFR- and VEGF-inhibitors, of which cetuximab and bevacizumab are among the most common. Cetuximab efficacy for front line chemotherapy improves when administered with anti-EGFR agents, but this effect is restricted to patients with wild type Kirsten-ras (KRAS) gene [9,10]. Therefore, current guidelines recommend testing the status of KRAS mutation as a routine procedure to avoid unnecessary treatment potentially associated with significant toxicity. This patient selection engenders significant cost savings in health care systems [11]. Bevacizumab is humanized monoclonal antibody against VEGF, the major mediator of angiogenesis. Bevacizumab is the first drug developed as an inhibitor of angiogenesis to be approved by the Food and Drug Administration (FDA), based on the survival benefit seen in a landmark trial for first-line treatment of metastatic CRC when combined with conventional chemotherapy [12]. However, unlike cetuximab, despite intense research efforts, no biomarker that can identify the patients who would benefit from bevacizumab therapy has yet been found. The cost and toxicity of bevacizumab accentuate the need for predictive markers for both efficacy and toxicity.
VEGF is an important regulator of physiologic and pathologic angiogenesis and is overexpressed in many malignancies [13]. VEGF and EGFR pathways interact, increasing angiogenesis [14,15]. RAS pathway signaling increases expression of VEGF and represses negative regulators of angiogenesis, suggesting that aberrations in KRAS may influence the response to anti-angiogenic therapy [16-18]. However, the role of KRAS mutation as a biomarker for anti-VEGF remains controversial.
We evaluated the role for the status of KRAS mutation as predictive and prognostic marker in CRC with anti-VEGF therapy.
Materials and Methods
1. Patients
We reviewed the records of 32 CRC patients who were available for KRAS mutation status and treated with cytotoxic chemotherapy plus bevacizumab as a first-line therapy at the Korea University Anam Hospital, Seoul, Korea between April 2007 and January 2011. All patients had pathologically or cytologically proven metastatic or recurrent CRC. During treatment, all patients received bevacizumab and some patients with wild type KRAS were treated by anti-EGFR therapies. Clinical information collected from the medical records of each patient were physical examination, surgical and pathologic reports, and imaging. Medical information including chemotherapy regimens, response, the date of progression, date of last visit and deaths were collected.
2. Treatment
The decision whether chemotherapy was conducted or not depended, in all cases, on the discussion between physician and patient. The chemotherapy regimen was determined by the physician. All three chemotherapeutic agents (5-FU, oxaliplatin, and irinotecan) were used over the treatment course. Bevacizumab was always administered concomitantly with intravenous or oral 5-FU plus oxaliplatin (FOLFOX or XELOX) regimen for the first-line treatment. Some patients with wild type KRAS were treated by an anti-EGFR agent, but not combined with bevacizumab. All tumors were evaluated after every three or four cycles of chemotherapy, by computed tomography scan and other tests that were used initially to stage the tumor. Responses were classified according to the Response Evaluation Criteria in Solid Tumors (RECIST) ver. 1.0.
3. Mutation analysis
We extracted DNA from five paraffin sections of 10 µm thickness containing a representative portion of tumor tissue (Qiagen, Hilden, Germany). Fifty nanograms of DNA were amplified in a 20 µL reaction solution containing 10 µL of 2×concentrated Hot StarTaq Master Mix (Qiagen), including polymerase chain reaction buffer, 3 mM MgCl2, 400 µM each of dNTP, and 0.3 µM each of the primer pairs (codon 12, 13; F: 5'-CGTCTGCAGTCAACTGGAAT, R: 5'-GAGAATGGTCCTGCACCAGTAA). Amplifications were performed using a 15 minute initial denaturation at 95℃, followed by 35 cycles of 30 seconds at 94℃, 30 seconds at 59℃, 30 seconds at 72℃, and a 10 minute final extension at 72℃. The polymerase chain reaction (PCR) products were then 2% gel-purified using the QIAgen gel extraction kit (Qiagen).
DNA sequencing was conducted as follows. First, digested mutated DNA was used as a template for the second PCR, in which the primer Ras 3 antisense; (5'-GGATGGTCCTCCACCAGTAATATGGATATTA-3') was used instead of Ras 2 (3'). The PCR was run under the same conditions as the first PCR for 32 cycles. Because of the nested antisense primer (Ras 3), the second PCR generated a fragment of 152 bp. This mutated DNA was excised from 3% agarose gels. The amplicons were then purified using the High Pure PCR Product Purification kit (Boehringer-Mannheim, Mannheim, Germany). Five ng of the purified amplicons was used for sequencing, performed with the Big Dye RR Terminator reaction (ABI, Weiterstadt, Germany). The product was run on a 5% polyacrylamide gel in an ABI 373A Sequencer (ABI) and analyzed for point mutations of the respective a mplicons.
4. Statistical analysis
Treatment outcomes were estimated as response rate (RR), progression free survival (PFS), and overall survival (OS). Tumor response was determined according to RECIST ver. 1.0. PFS was defined as the time from the date of the diagnosis for recurrence or metastatic disease to the date of disease progression or death from any cause. OS was defined as the time between the date of the diagnosis for recurrence or metastatic disease and the date of death. The PFS for oxaliplatin-and irinotecan-based chemotherapy and the OS according to KRAS status were analyzed with the Kaplan-Meier method to estimate the probability of survival and survival difference with the log-rank test. The χ2-test or Fisher's exact test as appropriate compared categorical variables. Two-sided null hypotheses of no difference were rejected if p-values were less than 0.05, or, equivalently, if the 95% confidence interals (CIs) of risk point estimates excluded 1.
Cox proportional hazards regression model was employed in univariate and multivariate analyses to identify the significant independent prognostic factors of various clinical parameters for survival. Variables found significant at the 0.05 level in univariate analysis for OS were included in multivariate analysis.
Results
1. Patient characteristics
Thirty two patients who were diagnosed with metastatic or recurrent CRC between April 2007 and January 2011 were tested for KRAS mutation status and received oxaliplatin based chemotherapy plus bevacizumab as first-line therapy. Among 32 patients who experienced disease progression after starting first-line therapy, 28 (87.5%) were treated with irinotecan based chemotherapy. During treatment, six of 32 patients received anti-EGFR therapies, not combined with bevacizumab. KRAS mutations were detected in 59.3% of tested patients. Table 1 summarizes the patient characteristics by KRAS mutational status. The median age was 53.5 years (range, 20 to 70 years) at diagnosis, and the male/female ratio was 2.5/1.0. Patient characteristics were similar between the KRAS mutation and the KRAS wild type groups. Moderate/poor differentiated tumors were observed more frequently in those with KRAS wild type compared to KRAS mutant patients (p=0.05).
Table 1.
Patient demographic and clinical characteristics
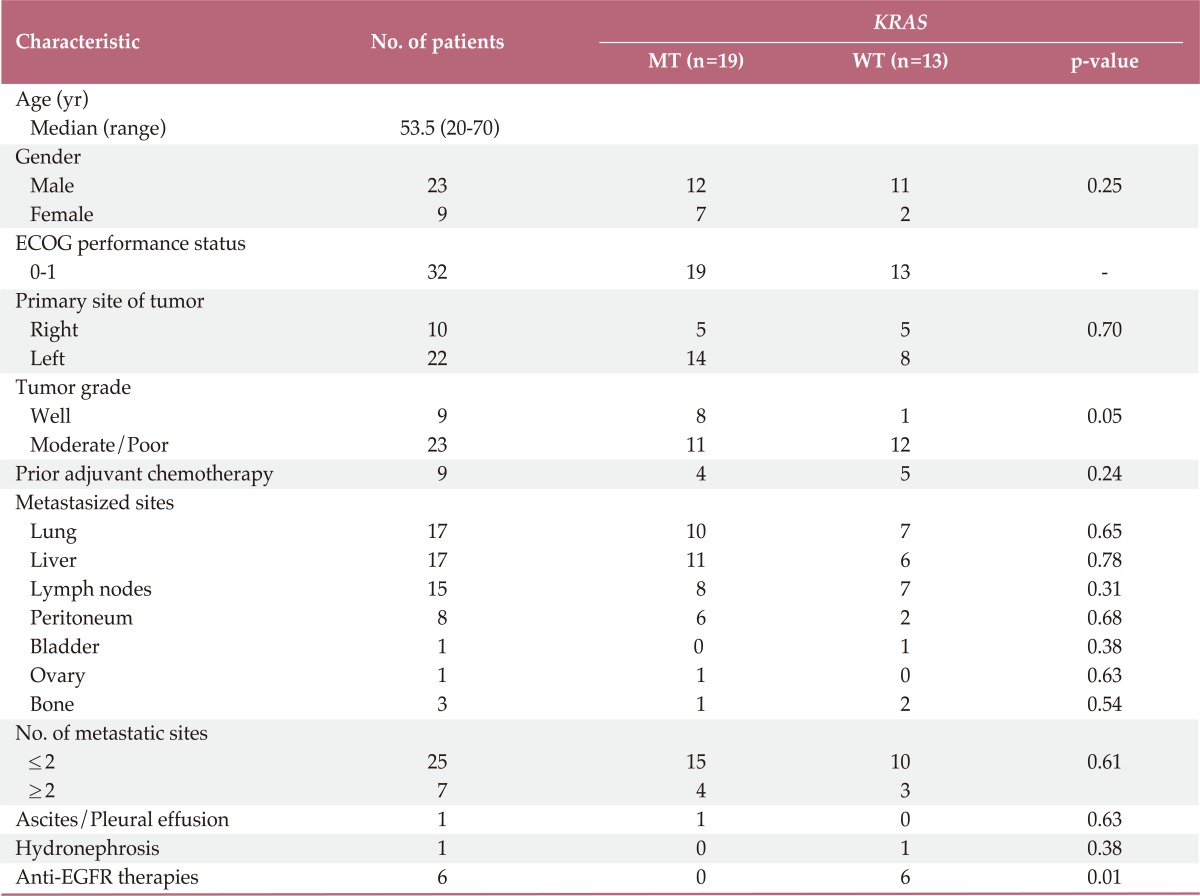
MT, mutant; WT, wild type; ECOG, Eastern Cooperative Oncology Group; EGFR, epidermal growth factor receptor.
2. Outcomes for chemotherapy
The overall RR of bevacizumab plus FOLFOX or XELOX was 50.0% and disease control rates 84.3% (Table 2). There was no significant difference for response (p=0.72) according to the status of KRAS mutation. Median PFS for first-line chemotherapy was 7.1 (95% CI, 5.8 to 8.4). There was no significant difference for PFS between KRAS mutation group and wild group (6.3 months; 95% CI, 3.2 to 9.4) vs. 8.5 months (95% CI, 4.5 to 12.4; p=0.98) (Fig. 1). In the second-line therapy, the overall RR was 25% and disease control rates 67.8%. According to the status of KRAS mutation, there was no significant difference of RR (Table 2). Moreover, patients with KRAS mutation showed similar OS as compared to patients without mutation (Fig. 2).
Table 2.
Response to chemotherapy according to KRAS mutation status
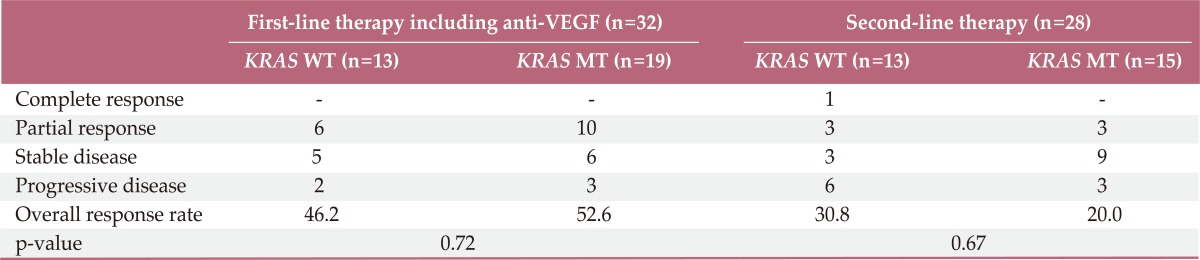
VEGF, vascular endothelial growth factor; WT, wild type; MT, mutant.
Fig. 1.

Progression free survival (PFS) to oxaliplatin chemotherapy plus anti-VEGF agents according to KRAS mutation status.
Fig. 2.

Overall survival (OS) according to KRAS mutation status.
3. Prognostic analysis
Results of prognostic analysis are provided in Table 3. When KRAS was assessed as a prognostic marker, there was no evidence as an independent factor for PFS to bevacizumab plus FOLFOX or XELOX in univariate analysis (hazard ratio, 1.007; 95% CI, 0.469 to 2.162; p=0.986). Similarly, there was no evidence that KRAS mutation was an independent prognostic factor for OS (hazard ratio, 0.548; 95% CI, 0.226 to 1.328; p=0.183). In univariate analysis of OS, the number of metastasizes was only statistically significant prognostic factor. In addition, anti-EGFR therapies did not influence OS.
Table 3.
Univariable survival analysis with proportional hazard regression in CRC patients with anti-VEGF therapies
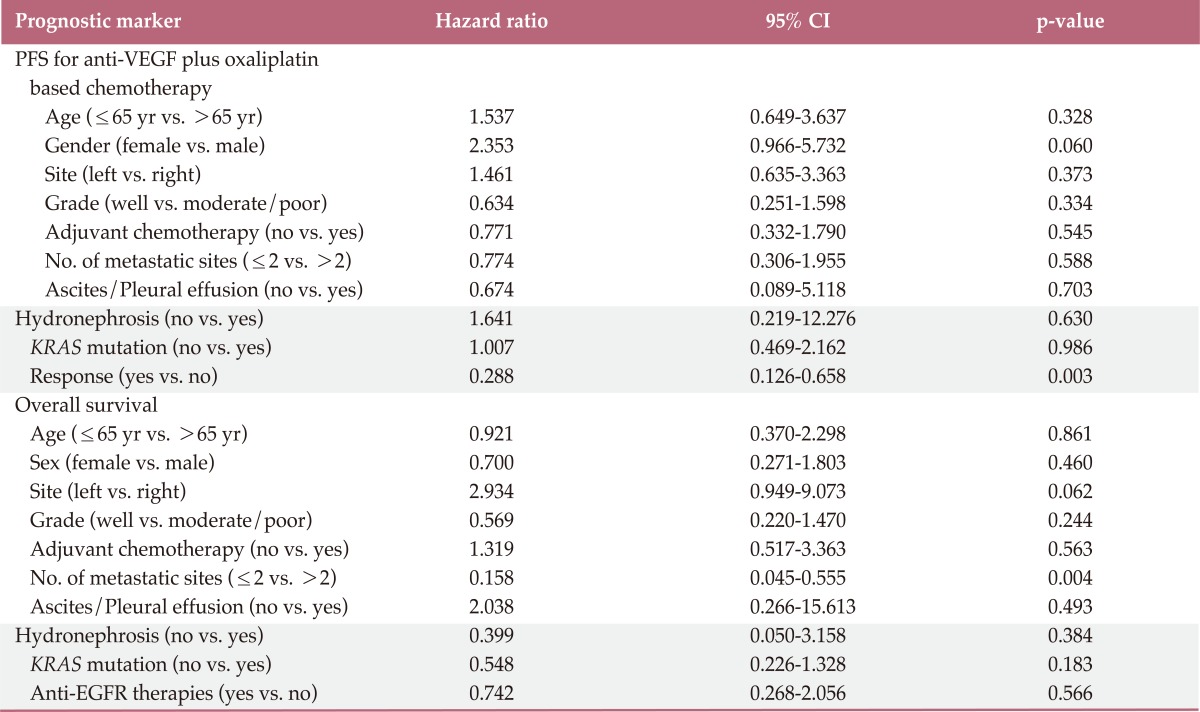
CRC, colorectal cancer; VEGF, vascular endothelial growth factor; CI, confidence interval; PFS, progression free survival; EGFR, epidermal growth factor receptor.
Discussion
For metastatic CRC, bevacizumab is clinically beneficial in combination with 5-FU alone or 5-FU combined with either oxaliplatin or irinotecan. However, no well-validated biomarkers have emerged to include or exclude patients from bevacizumab. It is unknown whether interactions in host-tumor microenvironment, biological features unique to the tumor, or features unique to the patients are most likely to yield predictors of therapy responsiveness [19].
Clinical and preclinical research indicate that alterations in the Ras/Raf/Mek/Erk pathway may have clinical relevance to the efficacy of anti-VEGF therapies [16-18,20]. In our study, there was no significant difference in response (p=0.72) or PFS (p=0.98) with the addition of bevacizumab to first-line chemotherapy by KRAS mutation status. This is consistent with previous retrospective studies. In the Hurwits study, there was no apparent interaction on the improvement PFS for KRAS status when bevacizumab was given in addition to irinotecan plus FU (IFL) chemotherapy [12]. However, there was a difference in RR with the addition of bevacizumab to IFL by KRAS mutation status. These findings were recently confirmed in the Australasian Gastrointestinal Trials Group Mitomycin Avastin Xeloda (AGITG MAX) study [21]. The MAX study reported that KRAS gene mutation status was neither prognostic for OS nor predictive of bevacizumab outcome in patients with advanced CRC. The relation between RR and KRAS mutation in MAX study was similar to that of the Hurwits study [12,21]. Although the Hurwitz study and MAX trial evaluated the role of KRAS mutation as predictive marker for the addition of bevacizumab to first-line chemotherapy, chemotherapy-backbones used in these studies were not suitable in metastatic CRC patients with relatively good performance status. There is no conclusive evidence that bevacizumab has superior activity when combined with specific chemotherapy, and rates of serious adverse events are similar for patients receiving FOLFOX, FOLFIRI, or XELOX. Thus all of these chemotherapy backbones may appropriate for combination with bevacizumab in the first-line treatment setting. In our study, all patients received bevacizumab plus FOLFOX as first-line therapy. Thus, our data set of patients receiving standard cytotoxic chemotherapy plus bevacizumab adds significant evidence to the lack of influence of KRAS status on the outcome of bevacizumab.
The prognostic values of KRAS mutation remain controversial in CRC. Our results indicate that KRAS mutation status has no major prognostic value for OS in patients with metastatic CRC with systemic therapy including bevacizumab. This finding was in accord with previous studies. The CO.17 study analyzed the prognostic impact by assessing the relation between KRAS status and survival in patients with best supportive care alone [10]. No significant difference was evident in median OS in patients with KRAS wild type or other mutations. Also, Reinacher-Schick et al. [22]. reported no evidence of KRAS gene status having prognostic value in metastatic CRC with bevacizumab containing therapy. In the MAX trial, KRAS gene mutation status was neither prognostic for PFS or OS in patients with advanced CRC [21]. However, conflicting findings were reported simultaneously in the two large collaborative Kristen Ras in Colorectal Cancer Collaborative Group (RASCAL) studies of 2,721 and 4,268 patients with CRC, respectively [23]. While the first RASCAL study showed an increased risk of recurrence and death linked to KRAS mutation, the second refined this observation to report significant prognostic value in failure free survival only with the G12V mutation in Duke's C patients. Thus, the clinical impact of our results is difficult to predict because many other factors might affect our finding. Furthermore, the subanalysis from the Hurwitz study showed that patients with KRAS gene mutation had a lower overall PFS and OS [12].
Our study was a retrospective analysis with small sample size. Also, the prognosis of CRC was affected by mutational status of many other genes as well as KRAS. The tumorigenesis and tumor progression of CRC result from multiple genetic and epigenetic abnormalities, including defective DNA mismatch repair and mutation of KRAS, NRAS, BRAF, PI3K, PIK3CA, and p5 [24,25]. These genetic and epigenetic changes may affect the survival of patients with CRC, although in this study we focused only the status of KRAS mutation to analyze the survival of CRC.
The use of bevacizumab in metastatic CRC has been a topic of much debate. Current research is not in agreement. Moreover, cost-effectiveness is unclear. These problems may be caused by the lack of a confirmative marker for the benefit of bevacizumab. Based on many studies including this one, the clinical benefit from bevacizumab appears to be independent of KRAS status. Nevertheless, researchers must make efforts to excavate predictive or prognostic biomarkers for biologic agents including bevacizumab. These efforts make us move closer to truly personalized medicine of cancer, ensuring maximum effectiveness with minimal toxicities and a minimum effect on resources.
Conclusion
VEGF is an important regulator of physiologic and pathologic angiogenesis and is overexpressed in many malignancies. VEGF and EGFR pathways interact, increasing angiogenesis. RAS pathway signaling increases expression of VEGF and represses negative regulators of angiogenesis, suggesting that aberrations in KRAS influence the response to anti-angiogenic therapy. However, the role of KRAS mutation as a biomarker for anti-VEGF remains controversial. Based on our analysis, KRAS mutation revealed neither a predictive for bevacizumab nor a prognostic for OS in CRC patients receiving anti-VEGF therapy.
Footnotes
Conflict of interest relevant to this article was not reported.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Comella P, Casaretti R, Sandomenico C, Avallone A, Franco L. Capecitabine, alone and in combination, in the management of patients with colorectal cancer: a review of the evidence. Drugs. 2008;68:949–961. doi: 10.2165/00003495-200868070-00005. [DOI] [PubMed] [Google Scholar]
- 3.de Gramont A, Louvet C, Andre T, Tournigand C, Krulik M. A review of GERCOD trials of bimonthly leucovorin plus 5-fluorouracil 48-h continuous infusion in advanced colorectal cancer: evolution of a regimen. Groupe d'Etude et de Recherche sur les Cancers de l'Ovaire et Digestifs (GERCOD) Eur J Cancer. 1998;34:619–626. doi: 10.1016/s0959-8049(97)00364-x. [DOI] [PubMed] [Google Scholar]
- 4.Capdevila J, Elez E, Peralta S, Macarulla T, Ramos FJ, Tabernero J. Oxaliplatin-based chemotherapy in the management of colorectal cancer. Expert Rev Anticancer Ther. 2008;8:1223–1236. doi: 10.1586/14737140.8.8.1223. [DOI] [PubMed] [Google Scholar]
- 5.Borgstrom P, Hillan KJ, Sriramarao P, Ferrara N. Complete inhibition of angiogenesis and growth of microtumors by anti-vascular endothelial growth factor neutralizing antibody: novel concepts of angiostatic therapy from intravital videomicroscopy. Cancer Res. 1996;56:4032–4039. [PubMed] [Google Scholar]
- 6.Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 7.Blick SK, Scott LJ. Cetuximab: a review of its use in squamous cell carcinoma of the head and neck and metastatic colorectal cancer. Drugs. 2007;67:2585–2607. doi: 10.2165/00003495-200767170-00008. [DOI] [PubMed] [Google Scholar]
- 8.McCormack PL, Keam SJ. Bevacizumab: a review of its use in metastatic colorectal cancer. Drugs. 2008;68:487–506. doi: 10.2165/00003495-200868040-00009. [DOI] [PubMed] [Google Scholar]
- 9.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 10.Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 11.Shankaran V, Luu TH, Nonzee N, Richey E, McKoy JM, Graff Zivin J, et al. Costs and cost effectiveness of a health care provider-directed intervention to promote colorectal cancer screening. J Clin Oncol. 2009;27:5370–5375. doi: 10.1200/JCO.2008.20.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 13.Jubb AM, Pham TQ, Hanby AM, Frantz GD, Peale FV, Wu TD, et al. Expression of vascular endothelial growth factor, hypoxia inducible factor 1alpha, and carbonic anhydrase IX in human tumours. J Clin Pathol. 2004;57:504–512. doi: 10.1136/jcp.2003.012963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim SJ, Uehara H, Karashima T, Shepherd DL, Killion JJ, Fidler IJ. Blockade of epidermal growth factor receptor signaling in tumor cells and tumor-associated endothelial cells for therapy of androgen-independent human prostate cancer growing in the bone of nude mice. Clin Cancer Res. 2003;9:1200–1210. [PubMed] [Google Scholar]
- 15.Ellis LM. Epidermal growth factor receptor in tumor angiogenesis. Hematol Oncol Clin North Am. 2004;18:1007–1021. doi: 10.1016/j.hoc.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 16.Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 17.Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, et al. Mutant ras oncogenes upregulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res. 1995;55:4575–4580. [PubMed] [Google Scholar]
- 18.Watnick RS, Cheng YN, Rangarajan A, Ince TA, Weinberg RA. Ras modulates Myc activity to repress thrombospondin-1 expression and increase tumor angiogenesis. Cancer Cell. 2003;3:219–231. doi: 10.1016/s1535-6108(03)00030-8. [DOI] [PubMed] [Google Scholar]
- 19.Jubb AM, Harris AL. Biomarkers to predict the clinical efficacy of bevacizumab in cancer. Lancet Oncol. 2010;11:1172–1183. doi: 10.1016/S1470-2045(10)70232-1. [DOI] [PubMed] [Google Scholar]
- 20.Rak J, Mitsuhashi Y, Sheehan C, Tamir A, Viloria-Petit A, Filmus J, et al. Oncogenes and tumor angiogenesis: differential modes of vascular endothelial growth factor up-regulation in ras-transformed epithelial cells and fibroblasts. Cancer Res. 2000;60:490–498. [PubMed] [Google Scholar]
- 21.Price TJ, Hardingham JE, Lee CK, Weickhardt A, Townsend AR, Wrin JW, et al. Impact of KRAS and BRAF gene mutation status on outcomes from the Phase III AGITG MAX Trial of capecitabine alone or in Combination with bevacizumab and mitomycin in advanced colorectal cancer. J Clin Oncol. 2011;29:2675–2682. doi: 10.1200/JCO.2010.34.5520. [DOI] [PubMed] [Google Scholar]
- 22.Reinacher-Schick A, Schulmann K, Modest DP, Bruns N, Graeven U, Jaworska M, et al. Effect of KRAS codon13 mutations in patients with advanced colorectal cancer (advanced CRC) under oxaliplatin containing chemotherapy: results from a translational study of the AIO colorectal study group. BMC Cancer. 2012;12:349. doi: 10.1186/1471-2407-12-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andreyev HJ, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, et al. Kirsten ras mutations in patients with colorectal cancer: the 'RASCAL II' study. Br J Cancer. 2001;85:692–696. doi: 10.1054/bjoc.2001.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leslie A, Pratt NR, Gillespie K, Sales M, Kernohan NM, Smith G, et al. Mutations of APC, K-ras, and p53 are associated with specific chromosomal aberrations in colorectal adenocarcinomas. Cancer Res. 2003;63:4656–4661. [PubMed] [Google Scholar]
- 25.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]