

Abstract
Receptor tyrosine kinases (RTKs) are cell-surface transmembrane receptors that contain regulated kinase activity within their cytoplasmic domain and play a critical role in signal transduction in both normal and malignant cells. Besides B-cell receptor (BCR) signaling in CLL, multiple RTKs have been reported to be constitutively active in CLL B-cells resulting in enhanced survival and resistance to apoptosis of the leukemic cells induced by chemotherapeutic agents. In addition to increased plasma levels of various types of cytokines/growth factors in CLL, we and others have detected that CLL B-cells spontaneously produce multiple cytokines in vitro which may constitute an autocrine loop of RTK activation on the leukemic B-cells. Moreover, aberrant expression and activation of non-RTKs, for example Src/Syk kinases, induce resistance of the leukemic B-cells to therapy. Based on current available knowledge, we detailed the impact of aberrant activities of various RTKs/non-RTKs on CLL B-cell survival and the potential of using these signaling components as future therapeutic targets in CLL therapy.
Keywords: CLL, Signal Transduction, RTK, Non-RTK, Apoptosis, Kinase Inhibitor, Therapy
INTRODUCTION
While treatment approaches in the past were based on disease control that largely employed single agents that for the most part achieved a chronic indolent disease, treatment goals nowadays are aimed at achieving long-term remissions for at least in low risk patients(1) with the use of chemoimmunotherapy (CIT). The treatment of B-cell chronic lymphocytic leukemia (CLL) is in the process of substantial changes as the novel therapies increasingly turn to oral drugs that attack signal pathways in the leukemic B-cell. The complex signaling pathways particularly those transmitted via various receptor tyrosine kinases (RTKs) are responsible for the enhanced survival and apoptotic resistance in CLL(2–6). Activation of B-cell receptor (BCR) signaling pathway either via antigen or “tonic signaling” plays an important pro-survival role in CLL B-cells even without any somatic mutation in the immunoglobulin heavy chain variable region gene (IGHV) which encodes part of the antigen-binding domain of the BCR. However, this review is primarily focused on non-BCR RTK signaling pathways in CLL irrespective of the IGHV mutational status of the leukemic clone.
RTKs constitute one of the largest classes of signaling molecules and have long served as a model for elucidating cellular signaling networks(7). However, oncogenic mutations and gene fusions generated by chromosomal translocations in RTKs are frequently observed in human cancers, leading to their constitutive activation(7). Moreover, oncogenic mutations or overexpression of RTKs can promote misfolding and aggregation of these proteins and impair trafficking to the cell surface(8). Although CLL B-cells from CLL patients of various risk for progression have been reported to express a number of non-BCR RTKs (2–6), their precise role in CLL B-cell biology and therapeutic applications directed at the RTKs have not been explored extensively. Here, we will focus on the expression and activation status of the RTKs and non-receptor kinases known to be expressed in CLL B-cells. We will, where possible, discuss current of future approaches to target these kinases in order to treat CLL patients. Our final section focuses on potential treatment approaches to CLL using the knowledge of these RTKs.
Membrane RTKs in CLL B-cells
This section discusses relatively more well-studied membrane RTKs that have known involvement in CLL B-cell survival, well described signal pathways and selected in vitro and or in vivo attempts to interfere with these pathways in CLL.
Insulin-like growth factor receptor and insulin receptor
Insulin-like growth factor-I (IGF-I) produced by bone-marrow stromal cells is involved, as a paracrine factor, in the differentiation of normal pro-B to pre-B lymphocytes, stimulating μ-heavy chain expression(9). IGF-I plays a role in maintaining hematopoietic cells by increasing the proliferation of progenitor cells(10) and by preventing the apoptosis of interleukin (IL)-3-deprived cells(11). IGF-I receptor (IGF-IR) is undetectable in CD34+ cells but is expressed in committed precursors(12) and in mature B-lymphocytes(13).
It is now known that IGF-I and IGF-IR are involved in the genesis of cancer. IGF-IR expression is a prerequisite for the development of several tumors because it facilitates transformation by viral and cellular oncogenes(14). The IGF-IR is a phylogenetically conserved RTK and belongs to the insulin receptor family, involving also the insulin receptor (IR) (see below), hybrid receptors and the IGF-2R/mannose 6-phosphate receptor. The function of the hybrid receptor is still not well understood(15). The IGF-2R/mannose 6-phosphate receptor is a monomeric receptor without TK activities(15). Both IGF-IR and IR are preformed dimeric TK receptors made up by two extracellular α-subunits and two β-subunits involving a small extracellular domain, an intramembraneous one and an intracellular domain(16). The latter includes the juxtamembraneous domain, the TK domain and the C-terminal domain. Interestingly, the IGF-IR is primarily involved in regulation of cell proliferation, apoptotic resistance, differentiation and cell motility, while IR is mostly involved in the control of glucose uptake and metabolism(15). In contrast to IR, IGF-IR is ubiquitously expressed in tissues in which it plays a role in tissue growth, mostly via growth hormone, which liberates IGF-I to activate IGF-IR. However current evidence suggests that IGF-IR is not an absolute requirement for normal growth (14).
The ligand-receptor interaction results in phosphorylation of tyrosine residues in the IGF-IR TK domain (spanning amino acid 973-1229) of the β-subunit. In the unstimulated receptor state, the activation loop (a-loop), containing the critical tyrosine (Y) residues 1131, 1135 and 1136, behaves as a pseudo substrate that blocks the active site. However, there are numerous intracellular adaptor proteins (e.g,, Shc, Grb2, CrkII, CrkL, etc) that link receptor signaling to downstream pathways(17–21). After ligand-binding, phosphorylation of Y1131 and Y1135 destabilizes the auto inhibitory conformation of the a-loop, whereas phosphorylation of Y1136 stabilizes the catalytically optimized conformation of the RTK(22). In turn, phosphorylation of the adapter proteins insulin receptor substrate 1 - 4 (IRS-1- 4) and Shc leads to activation of the phosphatidyl inositol-3 kinase (PI3K), the mitogen-activated protein kinase (MAPK) and the 14-3-3 pathways(23).
The first demonstration of IGF-IR expression in CLL B-cells from a subgroup of CLL patients was reported in 2005(6). IGF-IR protein and mRNA were shown to be present in CLL B-cells in 44% and 59% of CLL patients, respectively. Importantly, IGF-IR expression in CLL patients was positively correlated with the expression of the anti-apoptotic protein Bcl-2 and was involved in CLL cell survival in vitro(6). IGF-IR expression in CLL cells has been shown to be associated with CD38 expression, a marker associated with cells with poor response to treatment and shorter patient survival. Interestingly, serum IGF-I was elevated in CLL patients, but growth hormone (GH), an inducer of IGF-I expression, was normal(6). Therefore, local tissue site production of IGF-I by CLL B-cells may account for the increased levels of serum IGF-I, independent of GH, and may be related to paracrine/autocrine control of leukemic lymphocyte-survival by binding to and activating IGF-IR(6). This information highlights the importance of this growth factor receptor signaling as a possible therapeutic target in CLL. Indeed, blocking of IGF-IR with a neutralizing antibody induces apoptosis in CLL B-cells, but not in normal cells, in vitro(6). Indeed, IGF-IR inhibition using IGF-IR antibodies and tyrosine kinase inhibitors has been reported to enhance the tumor-cell killing effects of numerous conventional chemotherapeutic agents such as gemcitabine, irinotecan, etoposide, carboplatin, adriamycin, ifosfamide, navelbine, 5-fluorouracil and vincristine both in vitro and in vivo in various types of human malignancies(24).
More recently, detection of differential expression of the insulin receptor has been reported in CLL cases with higher levels in the majority of CLL with 11q chromosomal abnormalities (11q-del)(25). Indeed, a mean of about 10-fold higher IR mRNA expression level was documented in CLL with 11q-del cases as compared to CLL cases with other genomic categories(25). This study also found that exogenous addition of insulin stimulated canonical IR-signaling pathways including AKT/mTOR and Ras/Raf/Erk in CLL B-cells in vitro. Importantly, this study demonstrates a positive correlation of IR expression levels in CLL cells with shorter time to first therapy and shorter overall survival(25), suggesting a biologically meaningful link between IR expression levels in the leukemic B-cells and clinical course of the disease in a subset of CLL patients.
Vascular Endothelial Growth Factor Receptors
In humans, vascular endothelial growth factor (VEGF) ligand family consists of five members, VEGF A, B, C, D, and placenta growth factor (PLGF). These ligands bind in an overlapping pattern to three RTKs, VEGF receptor (VEGFR1), VEGFR2 and VEGFR3 as well as to their co-receptors. VEGFA, B and placental growth factor (PLGF) bind to VEGFR1, VEGFA binds to VEGFR2, and VEGFC and D bind to VEGFR3 however, proteolytic processing of the human VEGFC and D allows for binding to VEGFR2 albeit at much lower affinity than VEGFR3(26) (Fig. 1). The VEGFRs are members of the RTK superfamily and they belong to the same subclass as receptors for platelet-derived growth factor and fibroblast growth factors (FGFs). VEGFR1 is a positive regulator of monocyte and macrophage migration, and has been described as a positive and negative regulator of VEGFR2 signaling capacity. Negative regulation is exerted, at least in part, by an alternatively spliced soluble VEGFR1 variant that binds to VEGF and thereby prevents VEGF from binding to VEGFR2. VEGFR2 is implicated in many aspects of normal and pathological conditions, whereas VEGFR3 is important for lymphatic-endothelial-cell development and function(26).
Fig. 1. VEGF ligand and receptor-binding properties and signaling complexes.
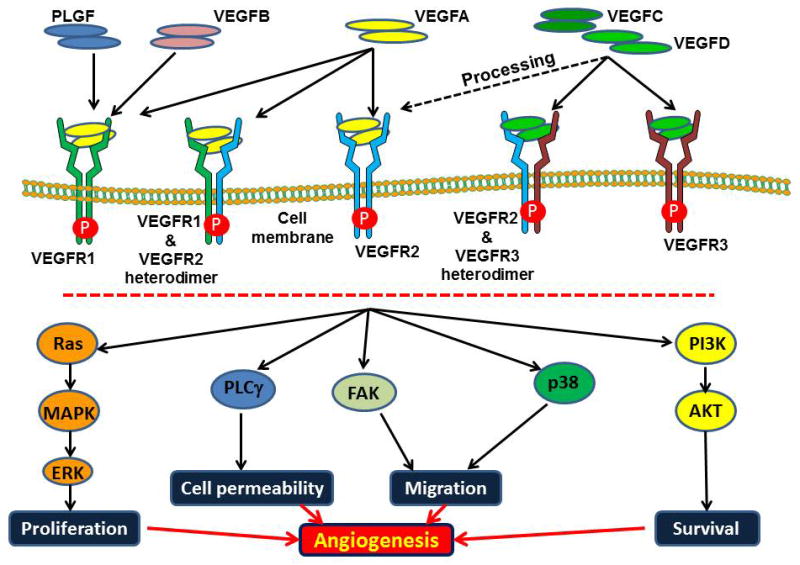
Mammalian VEGF ligands bind to the three VEGF receptor tyrosine kinases, leading to the formation of VEGFR homodimers or heterodimers. Proteolytic processing of VEGFC and VEGFD allows for binding to VEGFR2. Upon ligation with the ligand, VEGFRs transmit signals to transcribe the target cells via various intermediate components which also depend on the cellular context. Thus, activation of the specific VEGFR (via ligand binding or activating mutation) results in cell migration, permeability, proliferation and survival leading to angiogenesis.
The VEGFRs contain an approximately 750 amino-acid-residue extracellular domain, followed by a single transmembrane region, a juxta-membrane domain, a split tyrosine kinase domain that is interrupted by a 70-amino-acid kinase insert, and a C-terminal tail. Interestingly, alternative splicing or proteolytic processing of VEGFRs gives rise to secreted variants of VEGFR1(27) and VEGFR2(28), and in humans, to a C-terminal truncated VEGFR3(29). Guided by the binding properties of the ligands, the VEGFRs are able to form both homodimers and heterodimers(30). Dimerization of receptors is accompanied by activation of the receptor-kinase activity that leads to the autophosphorylation of the receptors. Phosphorylated receptors recruit interacting proteins and induce the activation of signaling pathways including Ras, Src, PI3K, focal adhesion kinase (FAK), phospholipase C (PLC)-γ, leading to proliferation, vascular permeability, cell migration and cell survival(26, 31).
In CLL, the pro-angiogenic factor VEGF (VEGFA) acts as an important survival factor for the leukemic B-cells, at least in part, by activating the STAT1/STAT3 signaling pathway and upregulating the critical anti-apoptotic protein, myeloid cell leukemia-1 (Mcl-1)(5). Indeed in a limited number of CLL patients (n=88), a strong correlation between Mcl-1 and VEGF mRNA expression levels was found(5). Angiogenesis and signaling via angiogenic cytokines have increasingly been recognized as an important process in the growth of both solid tumors(32) and hematologic malignancies(33), including CLL(34). This latter work has invoked the well-known “angiogenic switch” as a factor in CLL progression(35). Early work in CLL demonstrated that the CLL B-cell synthesizes and secretes pro-angiogenic molecules(36) (i.e. VEGF and bFGF) as well as anti-angiogenic molecules but the balance favors a pro-angiogenic environment. In addition, bone marrow microvessel density, a marker of angiogenesis, correlates with CLL disease stage(37, 38) and identifies patients with a shorter progression-free survival(39). Other reports also suggest that serum and urine levels of pro-angiogenic factors VEGF and bFGF are increased in CLL(40). Indeed, increased levels of serum VEGF or bFGF have been found to be associated with disease progression in patients with early-stage CLL(41).
CLL B-cells express VEGF receptors (R1 and R2)(42–44), and these receptors are constitutively phosphorylated(2). Culture of CLL B-cells with exogenous VEGF is associated with increased levels of the anti-apoptotic proteins MCL-1 and XIAP, as well as a reduction in both spontaneous and drug-induced apoptosis(2, 45). VEGF has also been implicated in CLL B-cell migration(46, 47), and can modulate the expression of B-cell receptor signaling through effects on protein kinase CβII(48). In addition, clinical studies found that patients with early-stage CLL who had higher serum VEGF levels had significantly shorter progression-free survival (40), Interestingly, VEGF levels in pretreatment plasma were associated with response to CIT treatment in patients with CLL(49). While these receptors were shown to be expressed on tumor cells and are likely to be involved in both autocrine survival and/or neovascularization in tumor models, there is increasing evidence that another VEGF receptor, neuropilin-1 (NRP-1), is critical in tumor angiogenesis and most likely involved in VEGF-mediated resistance to apoptosis(50). Aberrant NRP-1 expression has been shown in acute myeloid leukemia (AML) and associated with shortened overall survival of the AML patients(51). Importantly, it has also been reported that a subset of CLL B-cells, but not normal B-lymphocytes, express NRP-1(52). However, since VEGF supports an autocrine pathway that promotes CLL B-cell survival (2, 45, 53) and NRP-1 expression is limited to a subset of CLL patients, it will be critical to establish a relationship of NRP-1 expression with the known CLL prognostic factors. In addition, most recently our unpublished observations has detected the expression of VEGFR3 in CLL B-cells leading to the possibility that all three VEGF-receptors may be part of a network that results in the enhanced survival of the leukemic B-cells (unpublished observations: Kay and Ghosh). Consistent with this, we have also found that VEGF-C levels in early stage CLL (Rai stage 0) are comparable with that obtained from normal, healthy individuals but higher than in more advanced stages of CLL (Fig. 2A) suggesting that VEGF-C could be mediating disease progression in the early stage CLL patient. Interestingly, we see a reverse trend for VEGF-D with highest levels in the plasma of late stage CLL (Rai stages 3/4) when compared to that in normal plasma and lower stage CLL (Fig 2B) (unpublished observations: Kay and Ghosh). Importantly, we found that VEGF-A and -C are both produced by CLL B-cells via ELISA assays of their culture medium (data not shown: unpublished observations).
Fig. 2. CLL plasma contain both VEGF-C and VEGF-D.
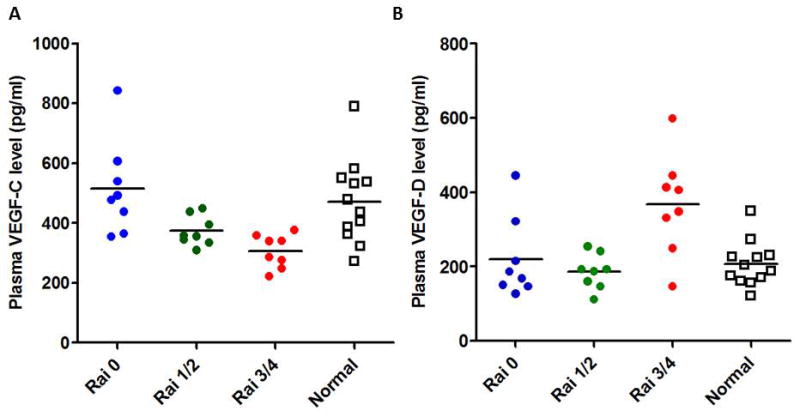
Plasma levels of VEGF-C and VEGF-D were measured in previously untreated CLL patients of various disease stages as indicated or age-matched healthy subjects using specific ELISA kits. Individual values are presented. Horizontal lines indicate the mean values. Although a trend of decrease VEGF-C levels were discernible with the disease progression, a sharp increase in VEGF-D levels were detected in advanced stages of CLL.
In total, these results suggest that signaling via the VEGF receptor signaling pathway may be an important process in the pathogenesis of CLL and could provide an important therapeutic target for patients with this disease.
Although various in vitro experiments on VEGF/VEGFR axis underscore a pro-survival role of this axis in CLL in addition to in vivo correlation of serum VEGF with early-stage CLL progression, it is important to note that a phase II clinical trial using anti-VEGF agents targeting VEGF or VEGFR (single agent) in relapsed/refractory CLL patients (n=46) has shown minimal clinical activity in this cohort of patients(54) (see below for detail). Information obtained from that clinical study also suggests that VEGF-VEGFR axis may not likely be the primary or predominant pro-survival axis in CLL.
Axl
It was originally detected in 1988 from patients with chronic myelogenous leukemia (CML) as an unidentified transforming gene and later was cloned from patients with CML and chronic myeloproliferative disorders(55). The name “Axl” was derived from the Greek word “”anexelekto” which meant “uncontrolled”. The human Axl gene is located on chromosome 19q13.2(55) and encodes a protein of molecular mass between 100 and 140 kD (depending on the extent of post-translational modifications) that contains an extracellular (N-terminal) domain and an intracellular (C-terminal) tyrosine kinase domain(56). Axl is a highly conserved gene across species (20 exons), but has two alternative variants due to a splicing site in exon 10 within the transmembrane domain(57–59). The promoter region of Axl is GC-rich and contains recognition sites for a variety of transcription factors, including Sp1 (specificity protein 1), AP2 (activating protein 2) and CREB (cAMP-response-element-binding protein)(60). Indeed, Axl is regulated by the Sp1/Sp3 transcription factors and methylation of CpG sites within specific Sp1 motifs(61). Given this, post-transcriptional regulations play a critical role in modifying and stabilizing the protein levels depending on cellular context. In addition, PKCα, PKCβ and constitutive activation of the Erk1/2 pathway have been reported to be critical for the overexpression of Axl in tyrosine kinase inhibitor-resistant cell lines(62).
Axl is a member of the TAM receptor tyrosine kinase family that also includes Tyro3 and Mer(63). Axl is composed of two immunoglobulin-like domains and dual fibronectin type III repeats in the extracellular region, a single transmembrane and a cytoplasmic domain with kinase activity(55). Axl is ubiquitously expressed in a wide variety of organs and cells, including hippocampus and cerebellum, monocytes, macrophages, platelets, endothelial cells, heart, skeletal muscle, liver, kidney and testis(58, 64, 65). However, Axl overexpression has been reported in several human cancers including colon, esophageal, thyroid, breast, lung, liver, and astrocytoma-glioblastoma(66–72).
Protein S and growth arrest specific gene 6 (Gas6) are the ligands for Axl, where the latter has very high-affinity to the Axl receptor(73, 74). Axl activation and signaling have been implicated in multiple cellular responses, including cell survival, proliferation, migration, adhesion and angiogenesis(75–79).
We identified Axl in CLL B-cells during our reported work on microvesicles in CLL plasma where we detected that CLL microvesicles carry the Axl RTK. CLL B-cells from the majority of CLL patients showed expression of constitutively phosphorylated and functionally active Axl RTK(3). Importantly, Axl RTK is physically associated with multiple non-receptor kinases and enzymes including Lyn (a member of the Src family kinases), Syk/ZAP70, PLC-γ2 and PI3K(3). In particular, the PI3K/AKT axis is a critical signaling pathway in many human malignancies including CLL and that over expression and increased activity of Lyn kinase has been reported in CLL. Interestingly, although CLL B-cells express c-Src, Axl showed very little affinity to bind to c-Src but did exhibit a very high affinity towards Lyn [Fig. 2B of ref(3)]. Our study suggests that Axl RTK is likely to be the primary RTK as inhibition of Axl induced massive cell death in CLL B-cells(3).
We have examined Axl expression on CLL B-cell surface from over 200 previously untreated CLL patients and detected variable levels of Axl expression (Kay and Ghosh: unpublished observations). However, we did not find any correlation of Axl expression with the known novel cell based prognostic factors in CLL (data not shown). In a related study most recently, we identified a miR-34a binding site on the Axl 3′-untranslated region (UTR). Interestingly, miR-34a is a direct target of the tumor suppressor p53 which has been reported to be inactive in many human cancers including CLL(80–82). Indeed, findings from a series of experiments suggest that miR-34a targets Axl 3′-UTR in response to p53 activation suggesting the existence of an inverse relationship between p53 functionality and regulation of Axl RTK expression in CLL(83).
Although Axl expression appears to be a predominant pro-survival signaling pathway in CLL, its relation or association with the CLL clinical course is yet to be established.
c-MET
The RTK c-MET, originally identified as a TRP-MET fusion gene from a human osteosarcoma cell line, encodes a prototypic member of the c-MET RTK subfamily(84). The tyrosine kinase c-MET is the high affinity receptor for hepatocyte growth factor (HGF)/scatter factor, a multifunctional cytokine with pleiotropic effects. The HGF/c-MET signaling pathway is one of the most frequently dysregulated pathways in human cancers. Aberrant HGF/c-MET signaling has been reported in a wide range of human malignancies, including bladder, breast, cervical, colorectal, endometrial, esophageal, gastric, head and neck, kidney, liver, lung, nasopharyngeal, ovarian, pancreatic, prostate and thyroid cancers, as well as cholangiocarcinoma, osteosarcoma, rhabdomyosarcoma, synovial sarcoma, Kaposi’s sarcoma, leiomyosarcomas and MFH/fibrosarcoma(85). In addition, abnormal HGF and/or c-MET expression has also been reported in hematological malignancies such as acute myelogenous leukemia, adult T-cell leukemia, chronic myeloid leukemia, lymphomas and multiple myeloma, as well as other tumors like melanoma, mesothelioma, Wilms’ tumor, glioblastoma, astrocytomas and CLL(85, 86).
The c-MET RTK subfamily is structurally distinct from most other RTK subfamilies. The mature form of the c-MET receptor is a disulfide-linked heterodimer containing an extracellular α-chain and a transmembrane β-chain, both of which result from the proteolytic cleavage of the same precursor protein(87). The β-chain consists of an extracellular domain, a transmembrane domain and a cytoplasmic portion containing juxtamembrane and kinase domains, and a C-terminal tail that is essential for substrate docking and downstream signaling(88–91). The binding of HGF ligand to functionally mature c-MET leads to receptor dimerization or multimerization, phosphorylation of multiple tyrosine residues in the intracellular region, catalytic activation, and downstream signaling through docking of a number of substrates(85) including RAS-MAPK, PI3K-AKT, STATs, PLCγ, and c-Src (88–90, 92). The c-Met signaling pathway has been shown to affect a wide range of biological activities, including cell motility, proliferation and protection from apoptosis. HGF/c-Met pathway is necessary for the normal growth and development of various cell types, including hematopoietic progenitors in embryonic life and adults(93, 94). Prior studies indicate that the signaling pathways of HGF/c-Met system and integrin family of adhesion molecules are linked and can cross-modulate their separate functions(95).
Recently, a group of investigators has reported that CLL B-cells express increased levels of c-METα and c-METβ while no expression was detected on normal CD19+ B-cells. Interestingly, this increase was found to be inversely correlated with decreased expression of adhesion molecules(86). In addition, serum level of HGF in CLL was reported to be increased(86). In vitro studies demonstrate that expressions of critical signaling molecules shared by adhesion molecules VLA-4 and HGF/c-MET systems including Bcl-xL, AKT, PI3K and phosphor-BAD136 following HGF stimulations of CLL B-cells have been found to be increased(86). These findings suggest that c-MET activation plays an important role in enhanced survival and apoptotic resistance of the leukemic B-cells. However, critical involvement of the HGF/c-MET signaling axis in CLL pathobiology or the prognostic relevance of HGF/c-MET expression in CLL B-cells remains to be investigated.
Novel Membrane RTKs in CLL
This section discusses more recently discovered or less well studied membrane RTKs that are likely involved in CLL B-cell survival.
Fibroblast Growth Factor Receptors
The FGF factor family and their four receptor tyrosine kinases, FGFR1/2/3/4, mediate multiple physiologic processes including cell migration, proliferation, survival and differentiation. All the four FGFRs are encoded by distinct genes and their structural variability is increased by alternative splicing(96). FGFRs are expressed on nearly every cell type of hematopoietic origin and deregulation of FGFR gene expression and/or gene mutation has been found in hematologic malignancies(97). Given the importance and critical roles of the FGF/FGFR signaling pathway, it is not surprising that aberrant FGFR signaling is detected in many human malignancies including multiple myeloma, gastric, endometrial, prostate, and breast(98, 99). For example, FGFR1 amplification in about 20% of squamous non-small cell lung carcinoma(100) and about 10% of breast cancers(101) has been reported. The FGFR2 gene is amplified in some cases of gastric cancer, resulting in a highly over expressed and constitutively active RTK(102, 103). On the other hand, t(4;14)(p16;q32) chromosomal translocation detected in 15% of multiple myeloma patients often results in overexpression of FGFR3(104–106). The overexpressed FGFR3 is usually wild type; sensitive to ligand-binding and the activated FGFR3 has a role in myelomagenesis(107). Amplification of FGFR4 has been detected in rhabdomyosarcoma and activating mutations characterized in 7% of cases(108). The affinity of bFGF with various FGFRs is different, and the downstream signaling pathways of different FGFRs are also varied(109), although the signaling domains of FGFRs are highly conserved. Several signaling pathways can be activated by FGFRs, such as the PLC-g, Src, Crk, and SNT-1/FRS2(110).
We and others have found that CLL B-cells constitutively produce the pro-angiogenic basic fibroblast growth factor (bFGF) in vitro(36, 111, 112). Increased levels of bFGF have also been reported in blood and urine of CLL patients(37, 111, 112). It is likely that the leukemic cells are the primary source of bFGF in vivo. Interestingly, higher plasma levels of VEGF and bFGF (FGF-2) have been reported to be predictors of longer survival in acute lymphoblastic leukemia (ALL)(113), while Bairey and co-investigators(114) showed that Bcl-2 expression correlates positively with serum bFGF and negatively with cellular VEGF in patients with CLL. Indeed an in vitro study using CLL-derived cell lines showed bFGF upregulates Bcl-2 expression resulting in delaying apoptosis(115). Interestingly, a recent study established a functional link between FGF- and VEGF-signaling pathways(116). This latter finding underscores that inhibition of both bFGF and VEGF signaling pathways may be necessary to sufficiently impair CLL B-cell survival.
A gene expression study using leukemic B-cells from CLL patients detected FGFR1 transcript with higher expression levels in CLL B-cells with unmutated IgVH status(117). However, this study did not demonstrate any expression of FGFR2, FGFR3 or FGFR4 in CLL B-cells. Most recently, our laboratory has indeed detected expression of FGFR1 and FGFR3, but not FGFR2 and FGFR4, in CLL B-cells from previously untreated CLL patients by both flow cytometric and Western blot analyses (Kay and Ghosh: unpublished observations). Constitutively phosphorylated FGFRs were also detected in CLL B-cells suggesting the existence of a paracrine/autocrine loop for activation of this FGF/FGFR-signaling pathway. However, at present whether this RTK-signaling pathway is critical for CLL B-cell survival and apoptotic resistance remains unknown.
ROR
Receptor tyrosine kinase-like orphan receptor (ROR) proteins are a conserved family of RTKs that function in developmental processes including skeletal and neuronal development, cell movement and cell polarity. Recent studies suggest that depending on cellular context, Ror proteins can either activate or repress transcription of Wnt target genes and can modulate Wnt signaling by sequestering Wnt ligands(118). It is not surprising that deregulated RTKs cause severe developmental defects and diseases like cancers. Thus, ROR proteins are no exception and disruption of human ROR proteins are associated with skeletal deformities and with increased incidence of leukemia(118).
Vertebrates express two ROR family members encoded by ROR1 and ROR2 genes(119). Ror proteins are type-I transmembrane RTKs and located predominantly in the plasma membrane(120). The extracellular region of Ror proteins contains an immunoglobulin (Ig) domain, a Cys-rich domain (CRD), also called Frizzled domain, a Kringle (Kr) domain, an intracellular tyrosine kinase domain and a proline-rich domain (PRD) straddled by two Ser/The-rich domains, Ser/Thr1 and Ser/Thr2(119). However, in humans, normal functions of the Ror protein are known to be related primarily for skeletal development(121–124).
Gene expression profiling studies showed a 43.8-fold increase of the ROR1 in CLL B-cells(125). Ror receptors participate in signal transduction, cell-cell interaction, regulation of cell proliferation, differentiation, cell metabolism and survival(119, 126). The ROR1 gene is located on human chromosome 1p31.3, a region where chromosomal aberrations are not frequently detected in hematological malignancies(127). The human ROR1 is expressed in heart, lung and kidney but less in placenta, pancreas and skeletal muscles(128). Truncated ROR1 (t-ROR1) has also been reported in fetal and adult human central nervous system, human leukemias, lymphoma cell lines and in a variety of human cancers derived from neuroectoderm(128). CLL cells have been reported to express ROR1 at the mRNA and protein levels uniformly, but not in normal B-lymphocytes(4, 127). Expression of ROR1 on CLL B-cells has been found to independent of disease stages, IGVH mutational status, and B-cell activation status(4, 127). Of note, expression of ROR2 was not detected on CLL B-cells(4). In total, unique expression pattern of Ror1 on CLL B-cells, not in normal B-lymphocytes, makes it an attractive target in CLL. However, whether ROR1 is critical for CLL progression or enhanced survival remains to be investigated.
Signaling in CLL B cells via Non-Receptor Tyrosine Kinases that are independent of BCR-Stimulation
This section discusses the relevant relationships of non-RTKs and their signal events to leukemic B-cell biology.
Lyn kinase
The members of Src-family kinases (SFKs) consist of Src, Fyn, Yes, Lck, Hck, Fgr, Lyn, Blk and Yrk. Each of these proteins are about 60 kD in molecular weight and have a common structure consisting of an N-terminal unique domain, followed by Src homology (SH) domain 3, SH2 and tyrosine kinase domains(129). SFKs can act as an upstream or downstream modulator of several receptors, as well as non-RTKs, which are responsible for robustness and persistence of RTK-signaling(130). SFKs participate in the activation of various downstream signaling pathways through molecular interactions with growth factor receptors such as the epidermal growth factor receptor (EGFR) family, MET, integrin cell adhesion receptors, steroid hormone receptors, G protein-coupled receptors, focal adhesion kinase (FAK) and cytoskeleton components(130, 131). SFKs can activate PI3K/AKT, growth factor receptor-bound protein 2 (Grb2)-Ras-Raf-mitogen activated protein kinase (MAPK), Jak-signal transducers and activation of transcription (STAT) and FAK-paxillin-p130-Crk-associated substrate (Cas) cascades that are most crucial for cell cycle progression, survival and proliferation(132–137).
Lyn, a member of the SFKs, is reported to be robustly overexpressed at the protein level in leukemic B-cells from CLL patients as compared to normal B-lymphocytes, with a substantial aliquot of the kinase anomalously present in the cytosol(138). While in normal B-lymphocytes Lyn activation is dependent on B-cell receptor stimulation, in resting malignant cells, the constitutive activity of the kinase accounts for high basal level protein tyrosine phosphorylation and low responsiveness to IgM ligation suggesting that it is independent of BCR-stimulation(138). Interestingly, the evidence that Lyn mRNA level was comparable in normal and neoplastic B-cells demonstrates the anomalous protein expression was not related to differences in gene transcription and/or mRNA stability. A possible explanation for this might be deregulated protein turnover in leukemic B-cells(138). However, treatment of CLL B-cells with the Lyn kinase inhibitors PP2 and SU6656 induces apoptosis, suggesting a direct correlation between high basal Lyn activity and defects in the induction of apoptosis in leukemic B-cells(138). In total, these findings support a critical role for Lyn in CLL pathogenesis and identify this non-RTK as a potential therapeutic target.
Syk Kinase
The protein tyrosine kinase spleen tyrosine kinase (Syk) represents a key mediator of proximal BCR signaling, providing proliferation and survival signals in a variety of hematopoietic cells(139). After BCR-stimulation, Syk is recruited to BCR and becomes activated by sequential phosphorylation at conserved tyrosine residues. Once activated, Syk propagates signals by associating with the critical signaling intermediates such as, VAV, PLCγ2, Bruton’s tyrosine kinase (Btk) and B-cell linker protein. The signaling cascade then proceeds with the activation of further downstream signaling molecules including extracellular signal regulated kinase ½ (Erk1/2) and p38(140). Translocations involving Syk have been identified in myelodysplastic syndromes and T-cell lymphoma, indicating that Syk may also function as a proto-oncogene(141, 142).
Gene expression profiling identified increased expression of Syk and downstream pathways in CLL compared with normal B-cells from healthy individuals. Western blot analysis showed increased expression and constitutive phosphorylation of Syk, and its downstream PLCγ2, signal transducers and activators of transcription 3 (STAT3), and Erk1/2 in CLL B-cells as compared to normal B-cells(143, 144). Indeed, Syk has been reported to be overexpressed in CLL B-cells at both mRNA and protein levels versus normal B-cells and pharmacological inhibition of Syk activity induced massive apoptotic leukemic B-cell death, regardless of clinical and biological status of the CLL patients(143, 144), emphasizing the potential clinical utility of Syk inhibition in hematological malignancies like CLL.
Potential of tyrosine kinase inhibitors in future CLL therapy
Multiple tyrosine kinases in the form of receptors and non-receptors have been detected in CLL as constitutively active and for the most part related to CLL B-cell survival. We believe that constitutively active RTKs in CLL B-cells constitute a network where one RTK acts as the predominant one, while others work as secondary RTKs, and that a functional interplay between multiple RTKs where a common converging signaling point is, for example, AKT. In this scenario then it is likely that inhibition of the primary RTK in leukemic B-cells may promote activation of a secondary RTK that maintains the survival signaling in the cells as most RTKs share the same downstream signal intermediates, like Src, PI3K/AKT (Fig. 3). Thus, effectively targeting multiple RTKs should have a better impact in CLL therapy. Nevertheless we wish to describe here prior clinical trials in CLL that have used a strategy of single RTK inhibition in the trial design. Because these were usually phase 2 trials all patients treated with RTK inhibition were relapsed/refractory CLL.
Fig. 3. Tyrosine kinase network in CLL.
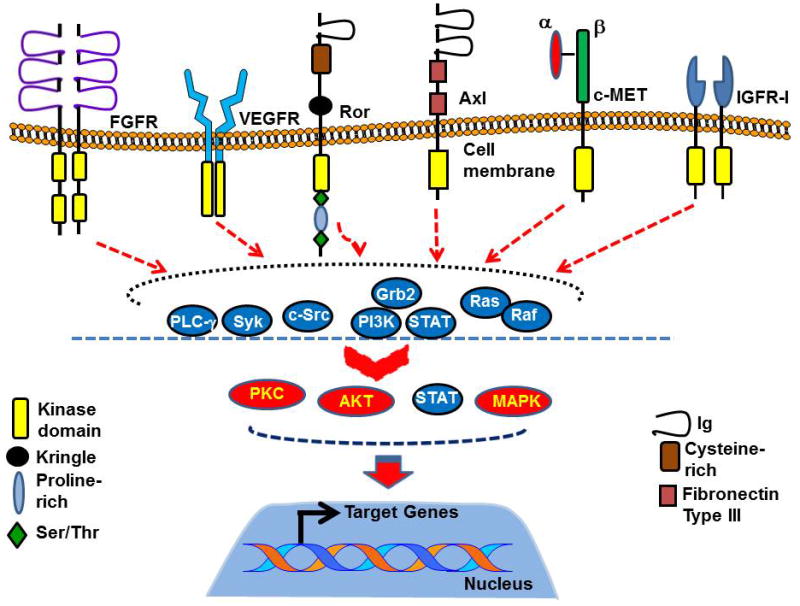
Leukemic B-cells from CLL patients express multiple RTKs which may directly or indirectly participate in the “Cell Survival” signaling network. As most of the RTK signaling pathways share common intermediate signaling components, for example, Src, PI3K/AKT, we believe that in this “RTK-Network”, one RTK plays the role of the “Predominant RTK” while others play a secondary role likely depending on the risk-factors of the cells. In CLL, upon binding specific ligands, these RTKs may activate multiple signaling intermediates including Src, Syk, Grb2/PI3K, Ras/Raf, PLC-γ leading to activation of the downstream effector signaling components, for example, AKT, MAPK, PKC or STATs, which ultimately activates various specific target genes resulting into cell survival, proliferation and apoptosis resistance. However, expression of constitutively active RTKs in CLL B-cells results into uncontrolled activation of the downstream signaling molecules leading to increased cell survival and apoptotic resistance to therapeutic agents. One such constitutively RTK in CLL we detected was Axl.
Targeting VEGF/VEGFR axis
To test the efficacy of anti-VEGF therapy in CLL, we initiated and completed separate phase II clinical testing of three different anti-VEGF therapies for patients with relapsed/refractory CLL: AZD2171 (a potent, oral, pan VEGF receptor inhibitor), bevacizumab (a recombinant humanized monoclonal antibody to VEGF), and sunitinib malate (a multi-targeted, small molecule inhibitor of RTKs involved in tumor proliferation and angiogenesis including VEGFR-1, VEGFR-2, VEGFR-3, and platelet-derived growth factor receptor [PDGFR])(54). Overall, 10 (71%) patients in the AZD2171 trial, 4 (33%) in the bevacizumab trial, and 16 (89%) in the sunitinib malate trial experienced a grade 3 or higher adverse event attributed to study medication. In the AZD2171 trial, the most frequent grade ≥3 adverse events were thrombocytopenia (5/14 patients), fatigue (5/14 patients), diarrhea (3/14 patients), muscle weakness (3/14 patients), and hypertension (3/14 patients). In the bevacizumab trial, the most frequent grade ≥3 adverse events were proteinuria (2/12 patients) and fatigue (2/12 patients). In the sunitinib malate trial, the most frequent grade ≥3 adverse events were thrombocytopenia (10/18 patients), fatigue (6/18 patients), neutropenia (5/18 patients), and anorexia (4/18 patients).
All three trials were closed early due to lack of efficacy. Although no complete or partial responses were obtained, 5/14 patients on AZD2171, 10/12 patients on bevacizumab, and 10/18 patients on sunitinib had stabilization of disease for a median duration of 2.7, 2.9, and 4.4 months, respectively. Thus, the absolute lymphocyte count (ALC) values declined by, at least, 10% during treatment for 5/14 patients on AZD2171, 3/12 patients on bevacizumab, and 6/18 patients on sunitinib malate.
Despite the lack of clinical activity observed in these trials, our and others work on the biology of VEGF and other related angiogenic events play a role in CLL(34). These include recent studies indicating that marrow vascular density is significantly higher in patients with CLL with high-risk FISH and CD38 positivity(145), a pro-angiogenic profile favors disease progression(146), circulating endothelial cells correlate with more advanced disease stage(147), proangiogenic molecules such as angiopoietin-2 and matrix metalloproteinase 9 are associated with progressive CLL(148, 149), and use of combination chemoimmunotherapy may work in part via antiangiogenic effects(150). Newer VEGF receptor RTK inhibitors have also recently demonstrated activity against CLL B-cells in vitro as well as in a xenograft model, and appear to increase the efficacy of purine nucleoside analogs against CLL on in vitro testing(151). These observations suggest that VEGF inhibition remains a potential therapeutic target in CLL and suggest that combining anti-VEGF therapy with more traditional therapeutic agents may be a useful strategy for patients with this disease. Indeed, we and others have already initiated clinical trials exploring the benefits of this approach as part of efforts to improve outcomes for patients with CLL.
Targeting Syk
The first clinical trial targeting Syk non-RTK used fostamatinib disodium (an oral Syk inhibitor) in a phase I/II studies in patients with relapsed/refractory non-hodgkin lymphoma (NHL) and CLL(152). Dose-limiting toxicity in the phase I portion was neutropenia, diarrhea, and thrombocytopenia, and 200 mg twice daily was chosen for the phase 2 study. In this phase of the trial the most common toxicities were reversible cytopenias, fatigue, diarrhea, and hypertension. Interestingly, 6 of 11 CLL patients (55%) achieved a partial response and the response rate in CLL was the highest amongst the patients with other NHL. However, to date no follow-up studies of fostamatinib in B-cell malignancies have been initiated in spite of a recently completed randomized phase III study in rheumatoid arthritis that showed significant activity and good tolerability of the drug(153).
Targeting Lyn-kinase
Dasatinib is an oral multikinase inhibitor targeting Src and Abl kinases which was approved for use in imatinib resistant chronic myelogenous leukemia (CML). It has been reported recently that dasatinib not only inhibits Lyn-kinase but also Btk at low nanomolar concentrations(154). However, in vitro data demonstrates that dasatinib induces variable degrees of apoptosis in leukemic B-cells with no correlation between response and inhibition of Lyn phosphorylation(155).
A phase II study of 140mg dasatinib once daily in a small cohort of relapsed/refractory CLL patients (n=15) reported an overall response rate of 20% with a progression-free survival of 7.5 months(156). However, 5 patients exhibited >50% reduction in lymphadenopathy. Myelosuppression was the primary toxicity with grade 4 neutropenia and thrombocytopenia occurring in 40% and 13% of the CLL patients, respectively(155).
Impact of Axl inhibitor in vitro
Axl RTK plays a critical role likely by regulating activity of multiple cellular kinases including non-RTKs like Lyn, Syk and lipid kinases like PI3K, PLCγ2 in CLL B-cells to modulate survival of the leukemic B-cells(3). We believe that Axl is acting as the predominant RTK in CLL B-cells (Fig. 3). This hypothesis is based on the fact that Axl inhibition induces robust apoptotic cell death in CLL B-cells from CLL patients with various disease stages, prognostic profiles and risk factors at very low LD50 doses (0.25 – 2.0 μM) of the high-affinity Axl inhibitors (ref and unpublished observations: Kay and Ghosh)(3). Indeed, a high-affinity, oral Axl-inhibitor BGB328 (BergenBio), formerly known as R428(157), reduced breast tumors in a mouse xenograft model with favorable toxicity profiles. A single administration of the agent in female BALB/c mice by oral gavage resulted in high plasma exposures (Cmax of approximately 2.6 and 6.8 μM/L with doses of 25 and 75 mg/kg, respectively) with linear dose proportionality up to 100 mg/kg body weight(157). Importantly, the Axl inhibitor exhibited a long plasma half-life (4 hours at 25 mg/kg; 13 hours at 75 mg/kg) and distributed effectively to tissues(157). Information from this pre-clinical study emphasized the potential use of the Axl-inhibitor in CLL patients in future phase I/II studies.
Acknowledgments
Part of the research information on CLL used in this review chapter was generated in our laboratories supported by the NIH research fund CA95241 (to NEK) and Eagles Cancer Research fund (to AKG). We acknowledge Ms. Tammy Hughes for her excellent secretarial help on this chapter.
References
- 1.Pleyer L, Egle A, Hartmann TN, Greil R. Molecular and cellular mechanisms of CLL: novel therapeutic approaches. Nature reviews. 2009 Jul;6(7):405–18. doi: 10.1038/nrclinonc.2009.72. [DOI] [PubMed] [Google Scholar]
- 2.Lee YK, Shanafelt TD, Bone ND, Strege AK, Jelinek DF, Kay NE. VEGF receptors on chronic lymphocytic leukemia (CLL) B cells interact with STAT 1 and 3: implication for apoptosis resistance. Leukemia. 2005 Apr;19(4):513–23. doi: 10.1038/sj.leu.2403667. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh AK, Secreto C, Boysen J, Sassoon T, Shanafelt TD, Mukhopadhyay D, et al. The novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of nonreceptor kinases: implications for therapy. Blood. 2011 Feb 10;117(6):1928–37. doi: 10.1182/blood-2010-09-305649. In Vitro Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baskar S, Kwong KY, Hofer T, Levy JM, Kennedy MG, Lee E, et al. Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008 Jan 15;14(2):396–404. doi: 10.1158/1078-0432.CCR-07-1823. Research Support, N.I.H., Intramural. [DOI] [PubMed] [Google Scholar]
- 5.Veronese L, Tournilhac O, Verrelle P, Davi F, Dighiero G, Chautard E, et al. Strong correlation between VEGF and MCL-1 mRNA expression levels in B-cell chronic lymphocytic leukemia. Leukemia Research. 2009 Dec;33(12):1623–6. doi: 10.1016/j.leukres.2009.05.003. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 6.Schillaci R, Galeano A, Becu-Villalobos D, Spinelli O, Sapia S, Bezares RF. Autocrine/paracrine involvement of insulin-like growth factor-I and its receptor in chronic lymphocytic leukaemia. Br J Haematol. 2005 Jul;130(1):58–66. doi: 10.1111/j.1365-2141.2005.05579.x. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 7.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001 May 17;411(6835):355–65. doi: 10.1038/35077225. Research Support, Non-U.S. Gov’t Review. [DOI] [PubMed] [Google Scholar]
- 8.Choudhary C, Olsen JV, Brandts C, Cox J, Reddy PN, Bohmer FD, et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell. 2009 Oct 23;36(2):326–39. doi: 10.1016/j.molcel.2009.09.019. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 9.Landreth KS, Narayanan R, Dorshkind K. Insulin-like growth factor-I regulates pro-B cell differentiation. Blood. 1992 Sep 1;80(5):1207–12. Research Support, U.S. Gov’t, P.H.S. [PubMed] [Google Scholar]
- 10.Wang LM, Myers MG, Jr, Sun XJ, Aaronson SA, White M, Pierce JH. IRS-1: essential for insulin- and IL-4-stimulated mitogenesis in hematopoietic cells. Science. 1993 Sep 17;261(5128):1591–4. doi: 10.1126/science.8372354. Research Support, U.S. Gov’t, P.H.S. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez-Tarduchy G, Collins MK, Garcia I, Lopez-Rivas A. Insulin-like growth factor-I inhibits apoptosis in IL-3-dependent hemopoietic cells. J Immunol. 1992 Jul 15;149(2):535–40. Research Support, Non-U.S. Gov’t. [PubMed] [Google Scholar]
- 12.Ratajczak MZ, Kuczynski WI, Onodera K, Moore J, Ratajczak J, Kregenow DA, et al. A reappraisal of the role of insulin-like growth factor I in the regulation of human hematopoiesis. The Journal of clinical investigation. 1994 Jul;94(1):320–7. doi: 10.1172/JCI117324. Research Support, U.S. Gov’t, P.H.S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kooijman R, Willems M, De Haas CJ, Rijkers GT, Schuurmans AL, Van Buul-Offers SC, et al. Expression of type I insulin-like growth factor receptors on human peripheral blood mononuclear cells. Endocrinology. 1992 Nov;131(5):2244–50. doi: 10.1210/endo.131.5.1425423. [DOI] [PubMed] [Google Scholar]
- 14.Baserga R. The IGF-I receptor in cancer research. Exp Cell Res. 1999 Nov 25;253(1):1–6. doi: 10.1006/excr.1999.4667. Research Support, U.S. Gov’t, P.H.S. Review. [DOI] [PubMed] [Google Scholar]
- 15.Larsson O, Girnita A, Girnita L. Role of insulin-like growth factor 1 receptor signalling in cancer. Br J Cancer. 2005 Jun 20;92(12):2097–101. doi: 10.1038/sj.bjc.6602627. Research Support, Non-U.S. Gov’t Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams TE, Epa VC, Garrett TP, Ward CW. Structure and function of the type 1 insulin-like growth factor receptor. Cellular and molecular life sciences : CMLS. 2000 Jul;57(7):1050–93. doi: 10.1007/PL00000744. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hernandez-Sanchez C, Blakesley V, Kalebic T, Helman L, LeRoith D. The role of the tyrosine kinase domain of the insulin-like growth factor-I receptor in intracellular signaling, cellular proliferation, and tumorigenesis. J Biol Chem. 1995 Dec 8;270(49):29176–81. doi: 10.1074/jbc.270.49.29176. [DOI] [PubMed] [Google Scholar]
- 18.Kim B, Cheng HL, Margolis B, Feldman EL. Insulin receptor substrate 2 and Shc play different roles in insulin-like growth factor I signaling. J Biol Chem. 1998 Dec 18;273(51):34543–50. doi: 10.1074/jbc.273.51.34543. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. [DOI] [PubMed] [Google Scholar]
- 19.Beitner-Johnson D, LeRoith D. Insulin-like growth factor-I stimulates tyrosine phosphorylation of endogenous c-Crk. J Biol Chem. 1995 Mar 10;270(10):5187–90. doi: 10.1074/jbc.270.10.5187. Research Support, U.S. Gov’t, P.H.S. [DOI] [PubMed] [Google Scholar]
- 20.D’Ambrosio C, Hongo A, Li S, Baserga R. The role of Grb2 in the growth and transformation of mouse embryo cells. Oncogene. 1996 Jan 18;12(2):371–8. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. [PubMed] [Google Scholar]
- 21.Butler AA, Blakesley VA, Koval A, deJong R, Groffen J, LeRoith D. In vivo regulation of CrkII and CrkL proto-oncogenes in the uterus by insulin-like growth factor-I. Differential effects on tyrosine phosphorylation and association with paxillin. J Biol Chem. 1997 Oct 31;272(44):27660–4. doi: 10.1074/jbc.272.44.27660. [DOI] [PubMed] [Google Scholar]
- 22.Favelyukis S, Till JH, Hubbard SR, Miller WT. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat Struct Biol. 2001 Dec;8(12):1058–63. doi: 10.1038/nsb721. Research Support, U.S. Gov’t, P.H.S. [DOI] [PubMed] [Google Scholar]
- 23.Baserga R. The contradictions of the insulin-like growth factor 1 receptor. Oncogene. 2000 Nov 20;19(49):5574–81. doi: 10.1038/sj.onc.1203854. Research Support, U.S. Gov’t, P.H.S. Review. [DOI] [PubMed] [Google Scholar]
- 24.Tao Y, Pinzi V, Bourhis J, Deutsch E. Mechanisms of disease: signaling of the insulin-like growth factor 1 receptor pathway--therapeutic perspectives in cancer. Nature clinical practice. Oncology. 2007 Oct;4(10):591–602. doi: 10.1038/ncponc0934. Review. [DOI] [PubMed] [Google Scholar]
- 25.Saiya-Cork K, Collins R, Parkin B, Ouillette P, Kuizon E, Kujawski L, et al. A pathobiological role of the insulin receptor in chronic lymphocytic leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011 May 1;17(9):2679–92. doi: 10.1158/1078-0432.CCR-10-2058. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nature reviews. 2006 May;7(5):359–71. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 27.Kendall RL, Thomas KA. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci U S A. 1993 Nov 15;90(22):10705–9. doi: 10.1073/pnas.90.22.10705. In Vitro. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ebos JM, Bocci G, Man S, Thorpe PE, Hicklin DJ, Zhou D, et al. A naturally occurring soluble form of vascular endothelial growth factor receptor 2 detected in mouse and human plasma. Molecular cancer research : MCR. 2004 Jun;2(6):315–26. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. [PubMed] [Google Scholar]
- 29.Hughes DC. Alternative splicing of the human VEGFGR-3/FLT4 gene as a consequence of an integrated human endogenous retrovirus. J Mol Evol. 2001 Aug;53(2):77–9. doi: 10.1007/s002390010195. [DOI] [PubMed] [Google Scholar]
- 30.Dixelius J, Makinen T, Wirzenius M, Karkkainen MJ, Wernstedt C, Alitalo K, et al. Ligand-induced vascular endothelial growth factor receptor-3 (VEGFR-3) heterodimerization with VEGFR-2 in primary lymphatic endothelial cells regulates tyrosine phosphorylation sites. J Biol Chem. 2003 Oct 17;278(42):40973–9. doi: 10.1074/jbc.M304499200. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 31.Ivy SP, Wick JY, Kaufman BM. An overview of small-molecule inhibitors of VEGFR signaling. Nature reviews Clinical oncology. 2009 Oct;6(10):569–79. doi: 10.1038/nrclinonc.2009.130. Review. [DOI] [PubMed] [Google Scholar]
- 32.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 Jan 7;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 33.Vacca A, Ribatti D, Ruco L, Giacchetta F, Nico B, Quondamatteo F, et al. Angiogenesis extent and macrophage density increase simultaneously with pathological progression in B-cell non-Hodgkin’s lymphomas. Br J Cancer. 1999 Feb;79(5–6):965–70. doi: 10.1038/sj.bjc.6690154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shanafelt TD, Kay NE. The clinical and biologic importance of neovascularization and angiogenic signaling pathways in chronic lymphocytic leukemia. Semin Oncol. 2006 Apr;33(2):174–85. doi: 10.1053/j.seminoncol.2006.01.008. Review. [DOI] [PubMed] [Google Scholar]
- 35.Kay NE, Shanafelt TD, Strege AK, Lee YK, Bone ND, Raza A. Bone biopsy derived marrow stromal elements rescue chronic lymphocytic leukemia B-cells from spontaneous and drug induced cell death and facilitates an “angiogenic switch”. Leuk Res. 2007 Jul;31(7):899–906. doi: 10.1016/j.leukres.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kay NE, Bone ND, Tschumper RC, Howell KH, Geyer SM, Dewald GW, et al. B-CLL cells are capable of synthesis and secretion of both pro- and anti-angiogenic molecules. Leukemia. 2002 May;16(5):911–9. doi: 10.1038/sj.leu.2402467. [DOI] [PubMed] [Google Scholar]
- 37.Kini AR, Kay NE, Peterson LC. Increased bone marrow angiogenesis in B cell chronic lymphocytic leukemia. Leukemia. 2000 Aug;14(8):1414–8. doi: 10.1038/sj.leu.2401825. [DOI] [PubMed] [Google Scholar]
- 38.Szmigielska-Kaplon A, Lech-Maranda E, Jesionek-Kupnicka D, Gora-Tybor J, Blonski JZ, Kasznicki M, et al. Prognostic value of the bone marrow microvessel density in progressive B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 2010 Jul;51(7):1351–3. doi: 10.3109/10428194.2010.486092. [DOI] [PubMed] [Google Scholar]
- 39.Molica S, Vacca A, Ribatti D, Cuneo A, Cavazzini F, Levato D, et al. Prognostic value of enhanced bone marrow angiogenesis in early B-cell chronic lymphocytic leukemia. Blood. 2002 Nov 1;100(9):3344–51. doi: 10.1182/blood-2002-01-0084. [DOI] [PubMed] [Google Scholar]
- 40.Molica S, Vitelli G, Levato D, Gandolfo GM, Liso V. Increased serum levels of vascular endothelial growth factor predict risk of progression in early B-cell chronic lymphocytic leukaemia. Br J Haematol. 1999 Dec;107(3):605–10. doi: 10.1046/j.1365-2141.1999.01752.x. [DOI] [PubMed] [Google Scholar]
- 41.Molica S, Vitelli G, Levato D, Ricciotti A, Digiesi G. Clinicoprognostic implications of increased serum levels of vascular endothelial growth factor and basic fibroblastic growth factor in early B-cell chronic lymphocytic leukaemia. Br J Cancer. 2002 Jan 7;86(1):31–5. doi: 10.1038/sj.bjc.6600022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aguayo A, Manshouri T, O’Brien S, Keating M, Beran M, Koller C, et al. Clinical relevance of Flt1 and Tie1 angiogenesis receptors expression in B-cell chronic lymphocytic leukemia (CLL) Leuk Res. 2001 Apr;25(4):279–85. doi: 10.1016/s0145-2126(00)00139-9. [DOI] [PubMed] [Google Scholar]
- 43.Bairey O, Boycov O, Kaganovsky E, Zimra Y, Shaklai M, Rabizadeh E. All three receptors for vascular endothelial growth factor (VEGF) are expressed on B-chronic lymphocytic leukemia (CLL) cells. Leuk Res. 2004 Mar;28(3):243–8. doi: 10.1016/s0145-2126(03)00256-x. [DOI] [PubMed] [Google Scholar]
- 44.Ferrajoli A, Manshouri T, Estrov Z, Keating MJ, O’Brien S, Lerner S, et al. High levels of vascular endothelial growth factor receptor-2 correlate with shortened survival in chronic lymphocytic leukemia. Clin Cancer Res. 2001 Apr;7(4):795–9. [PubMed] [Google Scholar]
- 45.Lee YK, Bone ND, Strege AK, Shanafelt TD, Jelinek DF, Kay NE. VEGF receptor phosphorylation status and apoptosis is modulated by a green tea component, epigallocatechin-3-gallate (EGCG), in B-cell chronic lymphocytic leukemia. Blood. 2004 Aug 1;104(3):788–94. doi: 10.1182/blood-2003-08-2763. [DOI] [PubMed] [Google Scholar]
- 46.Till KJ, Spiller DG, Harris RJ, Chen H, Zuzel M, Cawley JC. CLL, but not normal, B cells are dependent on autocrine VEGF and alpha4beta1 integrin for chemokine-induced motility on and through endothelium. Blood. 2005 Jun 15;105(12):4813–9. doi: 10.1182/blood-2004-10-4054. [DOI] [PubMed] [Google Scholar]
- 47.Ugarte-Berzal E, Redondo-Munoz J, Eroles P, Del Cerro MH, Garcia-Marco JA, Terol MJ, et al. VEGF/VEGFR2 interaction down-regulates matrix metalloproteinase-9 via STAT1 activation and inhibits B chronic lymphocytic leukemia cell migration. Blood. 2010 Jan 28;115(4):846–9. doi: 10.1182/blood-2009-08-239426. [DOI] [PubMed] [Google Scholar]
- 48.Abrams ST, Brown BR, Zuzel M, Slupsky JR. Vascular endothelial growth factor stimulates protein kinase CbetaII expression in chronic lymphocytic leukemia cells. Blood. 2010 Jun 3;115(22):4447–54. doi: 10.1182/blood-2009-06-229872. [DOI] [PubMed] [Google Scholar]
- 49.Shanafelt TD, Byrd JC, La PB, Zent CS, Call T, Secreto C, et al. Pretreatment angiogenic cytokines predict response to chemoimmunotherapy in patients with chronic lymphocytic leukaemia. Br J Haematol. 2009 Sep;146(6):660–4. doi: 10.1111/j.1365-2141.2009.07811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ellis LM. The role of neuropilins in cancer. Molecular cancer therapeutics. 2006 May;5(5):1099–107. doi: 10.1158/1535-7163.MCT-05-0538. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review. [DOI] [PubMed] [Google Scholar]
- 51.Kreuter M, Woelke K, Bieker R, Schliemann C, Steins M, Buechner T, et al. Correlation of neuropilin-1 overexpression to survival in acute myeloid leukemia. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2006 Nov;20(11):1950–4. doi: 10.1038/sj.leu.2404384. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 52.Nowakowski GS, Mukhopadhyay D, Wu X, Kay NE. Neuropilin-1 is expressed by chronic lymphocytic leukemia B cells. Leuk Res. 2008 Oct;32(10):1634–6. doi: 10.1016/j.leukres.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghosh AK, Shanafelt TD, Cimmino A, Taccioli C, Volinia S, Liu CG, et al. Aberrant regulation of pVHL levels by microRNA promotes the HIF/VEGF axis in CLL B cells. Blood. 2009 May 28;113(22):5568–74. doi: 10.1182/blood-2008-10-185686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shanafelt T, Zent C, Byrd J, Erlichman C, Laplant B, Ghosh A, et al. Phase II trials of single-agent anti-VEGF therapy for patients with chronic lymphocytic leukemia. Leuk Lymphoma. 2010 Dec;51(12):2222–9. doi: 10.3109/10428194.2010.524327. Clinical Trial, Phase II Multicenter Study Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O’Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B, Prokop C, et al. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. 1991 Oct;11(10):5016–31. doi: 10.1128/mcb.11.10.5016. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Korshunov VA. Axl-dependent signalling: a clinical update. Clin Sci. 2012 Apr;122(8):361–8. doi: 10.1042/CS20110411. Research Support, N.I.H., Extramural Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Faust M, Ebensperger C, Schulz AS, Schleithoff L, Hameister H, Bartram CR, et al. The murine ufo receptor: molecular cloning, chromosomal localization and in situ expression analysis. Oncogene. 1992 Jul;7(7):1287–93. Research Support, Non-U.S. Gov’t. [PubMed] [Google Scholar]
- 58.Neubauer A, Fiebeler A, Graham DK, O’Bryan JP, Schmidt CA, Barckow P, et al. Expression of axl, a transforming receptor tyrosine kinase, in normal and malignant hematopoiesis. Blood. 1994 Sep 15;84(6):1931–41. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. [PubMed] [Google Scholar]
- 59.Lu Q, Gore M, Zhang Q, Camenisch T, Boast S, Casagranda F, et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature. 1999 Apr 22;398(6729):723–8. doi: 10.1038/19554. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. [DOI] [PubMed] [Google Scholar]
- 60.Schulz AS, Schleithoff L, Faust M, Bartram CR, Janssen JW. The genomic structure of the human UFO receptor. Oncogene. 1993 Feb;8(2):509–13. [PubMed] [Google Scholar]
- 61.Mudduluru G, Allgayer H. The human receptor tyrosine kinase Axl gene--promoter characterization and regulation of constitutive expression by Sp1, Sp3 and CpG methylation. Biosci Rep. 2008 Jun;28(3):161–76. doi: 10.1042/BSR20080046. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 62.Dufies M, Jacquel A, Belhacene N, Robert G, Cluzeau T, Luciano F, et al. Mechanisms of AXL overexpression and function in Imatinib-resistant chronic myeloid leukemia cells. Oncotarget. 2011 Nov;2(11):874–85. doi: 10.18632/oncotarget.360. Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lai C, Lemke G. An extended family of protein-tyrosine kinase genes differentially expressed in the vertebrate nervous system. Neuron. 1991 May;6(5):691–704. doi: 10.1016/0896-6273(91)90167-x. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. [DOI] [PubMed] [Google Scholar]
- 64.Angelillo-Scherrer A, de Frutos P, Aparicio C, Melis E, Savi P, Lupu F, et al. Deficiency or inhibition of Gas6 causes platelet dysfunction and protects mice against thrombosis. Nat Med. 2001 Feb;7(2):215–21. doi: 10.1038/84667. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 65.Graham DK, Bowman GW, Dawson TL, Stanford WL, Earp HS, Snodgrass HR. Cloning and developmental expression analysis of the murine c-mer tyrosine kinase. Oncogene. 1995 Jun 15;10(12):2349–59. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. [PubMed] [Google Scholar]
- 66.Craven RJ, Xu LH, Weiner TM, Fridell YW, Dent GA, Srivastava S, et al. Receptor tyrosine kinases expressed in metastatic colon cancer. International journal of cancer Journal international du cancer. 1995 Mar 16;60(6):791–7. doi: 10.1002/ijc.2910600611. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. [DOI] [PubMed] [Google Scholar]
- 67.Nemoto T, Ohashi K, Akashi T, Johnson JD, Hirokawa K. Overexpression of protein tyrosine kinases in human esophageal cancer. Pathobiology. 1997;65(4):195–203. doi: 10.1159/000164123. [DOI] [PubMed] [Google Scholar]
- 68.Ito T, Ito M, Naito S, Ohtsuru A, Nagayama Y, Kanematsu T, et al. Expression of the Axl receptor tyrosine kinase in human thyroid carcinoma. Thyroid. 1999 Jun;9(6):563–7. doi: 10.1089/thy.1999.9.563. [DOI] [PubMed] [Google Scholar]
- 69.Meric F, Lee WP, Sahin A, Zhang H, Kung HJ, Hung MC. Expression profile of tyrosine kinases in breast cancer. Clin Cancer Res. 2002 Feb;8(2):361–7. [PubMed] [Google Scholar]
- 70.Shieh YS, Lai CY, Kao YR, Shiah SG, Chu YW, Lee HS, et al. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia. 2005 Dec;7(12):1058–64. doi: 10.1593/neo.05640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsou AP, Wu KM, Tsen TY, Chi CW, Chiu JH, Lui WY, et al. Parallel hybridization analysis of multiple protein kinase genes: identification of gene expression patterns characteristic of human hepatocellular carcinoma. Genomics. 1998 Jun 15;50(3):331–40. doi: 10.1006/geno.1998.5338. Comparative Study Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 72.Vajkoczy P, Knyazev P, Kunkel A, Capelle HH, Behrndt S, von Tengg-Kobligk H, et al. Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc Natl Acad Sci U S A. 2006 Apr 11;103(15):5799–804. doi: 10.1073/pnas.0510923103. Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stitt TN, Conn G, Gore M, Lai C, Bruno J, Radziejewski C, et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell. 1995 Feb 24;80(4):661–70. doi: 10.1016/0092-8674(95)90520-0. [DOI] [PubMed] [Google Scholar]
- 74.Varnum BC, Young C, Elliott G, Garcia A, Bartley TD, Fridell YW, et al. Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature. 1995;373(6515):623–6. doi: 10.1038/373623a0. [DOI] [PubMed] [Google Scholar]
- 75.Hafizi S, Dahlback B. Signalling and functional diversity within the Axl subfamily of receptor tyrosine kinases. Cytokine & growth factor reviews. 2006 Aug;17(4):295–304. doi: 10.1016/j.cytogfr.2006.04.004. Review. [DOI] [PubMed] [Google Scholar]
- 76.Korshunov VA, Mohan AM, Georger MA, Berk BC. Axl, a receptor tyrosine kinase, mediates flow-induced vascular remodeling. Circ Res. 2006 Jun 9;98(11):1446–52. doi: 10.1161/01.RES.0000223322.16149.9a. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 77.Collett GD, Sage AP, Kirton JP, Alexander MY, Gilmore AP, Canfield AE. Axl/phosphatidylinositol 3-kinase signaling inhibits mineral deposition by vascular smooth muscle cells. Circ Res. 2007 Mar 2;100(4):502–9. doi: 10.1161/01.RES.0000258854.03388.02. Comparative Study Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 78.Melaragno MG, Fridell YW, Berk BC. The Gas6/Axl system: a novel regulator of vascular cell function. Trends Cardiovasc Med. 1999 Nov;9(8):250–3. doi: 10.1016/s1050-1738(00)00027-x. Review. [DOI] [PubMed] [Google Scholar]
- 79.Ghosh AK, Secreto CR, Knox TR, Ding W, Mukhopadhyay D, Kay NE. Circulating microvesicles in B-cell chronic lymphocytic leukemia can stimulate marrow stromal cells: implications for disease progression. Blood. 2010 Mar 4;115(9):1755–64. doi: 10.1182/blood-2009-09-242719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004 Mar 2;101(9):2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network--another piece in the tumour-suppression puzzle. Nature reviews Cancer. 2007 Nov;7(11):819–22. doi: 10.1038/nrc2232. Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, Non-P.H.S. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zenz T, Mohr J, Eldering E, Kater AP, Buhler A, Kienle D, et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood. 2009 Apr 16;113(16):3801–8. doi: 10.1182/blood-2008-08-172254. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 83.Ghosh AK, Boysen J, Price-troska T, Secreto C, Zent CS, Kay N. Axl Receptor Tyrosine Kinase Signaling Pathway and the p53 Tumor Suppressor Protein Exist In A Novel Regulatory Loop In B-Cell Chronic Lymphocytic Leukemia Cells. ASH Annual Meeting Abstracts. 2011 Nov 18;118(21):799. [Google Scholar]
- 84.Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, Croce CM, et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984 Sep 6–11;311(5981):29–33. doi: 10.1038/311029a0. Research Support, U.S. Gov’t, P.H.S. [DOI] [PubMed] [Google Scholar]
- 85.Liu X, Yao W, Newton RC, Scherle PA. Targeting the c-MET signaling pathway for cancer therapy. Expert opinion on investigational drugs. 2008 Jul;17(7):997–1011. doi: 10.1517/13543784.17.7.997. Review. [DOI] [PubMed] [Google Scholar]
- 86.Eksioglu-Demiralp E, Akdeniz T, Bayik M. Aberrant expression of c-met and HGF/c-met pathway provides survival advantage in B-chronic lymphocytic leukemia. Cytometry Part B, Clinical cytometry. 2011 Jan;80(1):1–7. doi: 10.1002/cyto.b.20553. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 87.Giordano S, Di Renzo MF, Narsimhan RP, Cooper CS, Rosa C, Comoglio PM. Biosynthesis of the protein encoded by the c-met proto-oncogene. Oncogene. 1989 Nov;4(11):1383–8. Research Support, Non-U.S. Gov’t. [PubMed] [Google Scholar]
- 88.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nature reviews Molecular cell biology. 2003 Dec;4(12):915–25. doi: 10.1038/nrm1261. Review. [DOI] [PubMed] [Google Scholar]
- 89.Christensen JG, Burrows J, Salgia R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005 Jul 8;225(1):1–26. doi: 10.1016/j.canlet.2004.09.044. Review. [DOI] [PubMed] [Google Scholar]
- 90.Jiang WG, Martin TA, Parr C, Davies G, Matsumoto K, Nakamura T. Hepatocyte growth factor, its receptor, and their potential value in cancer therapies. Crit Rev Oncol Hematol. 2005 Jan;53(1):35–69. doi: 10.1016/j.critrevonc.2004.09.004. Review. [DOI] [PubMed] [Google Scholar]
- 91.Gherardi E, Youles ME, Miguel RN, Blundell TL, Iamele L, Gough J, et al. Functional map and domain structure of MET, the product of the c-met protooncogene and receptor for hepatocyte growth factor/scatter factor. Proc Natl Acad Sci U S A. 2003 Oct 14;100(21):12039–44. doi: 10.1073/pnas.2034936100. In Vitro Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ponzetto C, Bardelli A, Zhen Z, Maina F, dalla Zonca P, Giordano S, et al. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell. 1994 Apr 22;77(2):261–71. doi: 10.1016/0092-8674(94)90318-2. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 93.Uehara Y, Minowa O, Mori C, Shiota K, Kuno J, Noda T, et al. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature. 1995 Feb 23;373(6516):702–5. doi: 10.1038/373702a0. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 94.Sugiura K, Taketani S, Yoshimura T, Nishino T, Nishino N, Fujisawa J, et al. Effect of hepatocyte growth factor on long term hematopoiesis of human progenitor cells in transgenic-severe combined immunodeficiency mice. Cytokine. 2007 Mar;37(3):218–26. doi: 10.1016/j.cyto.2007.04.001. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 95.Chan PC, Chen SY, Chen CH, Chen HC. Crosstalk between hepatocyte growth factor and integrin signaling pathways. J Biomed Sci. 2006 Mar;13(2):215–23. doi: 10.1007/s11373-005-9061-7. Research Support, Non-U.S. Gov’t Review. [DOI] [PubMed] [Google Scholar]
- 96.Johnson DE, Williams LT. Structural and functional diversity in the FGF receptor multigene family. Adv Cancer Res. 1993;60:1–41. doi: 10.1016/s0065-230x(08)60821-0. Comparative Study Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. Review. [DOI] [PubMed] [Google Scholar]
- 97.Moroni E, Dell’Era P, Rusnati M, Presta M. Fibroblast growth factors and their receptors in hematopoiesis and hematological tumors. J Hematother Stem Cell Res. 2002 Feb;11(1):19–32. doi: 10.1089/152581602753448513. Research Support, Non-U.S. Gov’t Review. [DOI] [PubMed] [Google Scholar]
- 98.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nature reviews Cancer. 2010 Feb;10(2):116–29. doi: 10.1038/nrc2780. Review. [DOI] [PubMed] [Google Scholar]
- 99.Turner N, Lambros MB, Horlings HM, Pearson A, Sharpe R, Natrajan R, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010 Apr 8;29(14):2013–23. doi: 10.1038/onc.2009.489. Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ramos AH, Dutt A, Mermel C, Perner S, Cho J, Lafargue CJ, et al. Amplification of chromosomal segment 4q12 in non-small cell lung cancer. Cancer biology & therapy. 2009 Nov;8(21):2042–50. doi: 10.4161/cbt.8.21.9764. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Elbauomy Elsheikh S, Green AR, Lambros MB, Turner NC, Grainge MJ, Powe D, et al. FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast cancer research : BCR. 2007;9(2):R23. doi: 10.1186/bcr1665. Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kunii K, Davis L, Gorenstein J, Hatch H, Yashiro M, Di Bacco A, et al. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res. 2008 Apr 1;68(7):2340–8. doi: 10.1158/0008-5472.CAN-07-5229. [DOI] [PubMed] [Google Scholar]
- 103.Takeda M, Arao T, Yokote H, Komatsu T, Yanagihara K, Sasaki H, et al. AZD2171 shows potent antitumor activity against gastric cancer over-expressing fibroblast growth factor receptor 2/keratinocyte growth factor receptor. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007 May 15;13(10):3051–7. doi: 10.1158/1078-0432.CCR-06-2743. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 104.Malgeri U, Baldini L, Perfetti V, Fabris S, Vignarelli MC, Colombo G, et al. Detection of t(4;14)(p16.3;q32) chromosomal translocation in multiple myeloma by reverse transcription-polymerase chain reaction analysis of IGH-MMSET fusion transcripts. Cancer Res. 2000 Aug 1;60(15):4058–61. Comparative Study Research Support, Non-U.S. Gov’t. [PubMed] [Google Scholar]
- 105.Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998 Nov 1;92(9):3025–34. Research Support, U.S. Gov’t, P.H.S. [PubMed] [Google Scholar]
- 106.Chang H, Qi XY, Samiee S, Yi QL, Chen C, Trudel S, et al. Genetic risk identifies multiple myeloma patients who do not benefit from autologous stem cell transplantation. Bone Marrow Transplant. 2005 Nov;36(9):793–6. doi: 10.1038/sj.bmt.1705131. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 107.Squires M, Ward G, Saxty G, Berdini V, Cleasby A, King P, et al. Potent, selective inhibitors of fibroblast growth factor receptor define fibroblast growth factor dependence in preclinical cancer models. Molecular cancer therapeutics. 2011 Sep;10(9):1542–52. doi: 10.1158/1535-7163.MCT-11-0426. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 108.Taylor JGt, Cheuk AT, Tsang PS, Chung JY, Song YK, Desai K, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. The Journal of clinical investigation. 2009 Nov;119(11):3395–407. doi: 10.1172/JCI39703. Research Support, N.I.H., Extramural. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang JK, Gao G, Goldfarb M. Fibroblast growth factor receptors have different signaling and mitogenic potentials. Mol Cell Biol. 1994 Jan;14(1):181–8. doi: 10.1128/mcb.14.1.181. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000 Sep;7(3):165–97. doi: 10.1677/erc.0.0070165. Review. [DOI] [PubMed] [Google Scholar]
- 111.Menzel T, Rahman Z, Calleja E, White K, Wilson EL, Wieder R, et al. Elevated intracellular level of basic fibroblast growth factor correlates with stage of chronic lymphocytic leukemia and is associated with resistance to fludarabine. Blood. 1996 Feb 1;87(3):1056–63. [PubMed] [Google Scholar]
- 112.Krejci P, Dvorakova D, Krahulcova E, Pachernik J, Mayer J, Hampl A, et al. FGF-2 abnormalities in B cell chronic lymphocytic and chronic myeloid leukemias. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2001 Feb;15(2):228–37. doi: 10.1038/sj.leu.2402012. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 113.Faderl S, Do KA, Johnson MM, Keating M, O’Brien S, Jilani I, et al. Angiogenic factors may have a different prognostic role in adult acute lymphoblastic leukemia. Blood. 2005 Dec 15;106(13):4303–7. doi: 10.1182/blood-2005-03-1010. [DOI] [PubMed] [Google Scholar]
- 114.Bairey O, Zimra Y, Shaklai M, Rabizadeh E. Bcl-2 expression correlates positively with serum basic fibroblast growth factor (bFGF) and negatively with cellular vascular endothelial growth factor (VEGF) in patients with chronic lymphocytic leukaemia. Br J Haematol. 2001 May;113(2):400–6. doi: 10.1046/j.1365-2141.2001.02731.x. [DOI] [PubMed] [Google Scholar]
- 115.Konig A, Menzel T, Lynen S, Wrazel L, Rosen A, Al-Katib A, et al. Basic fibroblast growth factor (bFGF) upregulates the expression of bcl-2 in B cell chronic lymphocytic leukemia cell lines resulting in delaying apoptosis. Leukemia. 1997 Feb;11(2):258–65. doi: 10.1038/sj.leu.2400556. [DOI] [PubMed] [Google Scholar]
- 116.Murakami M, Nguyen LT, Hatanaka K, Schachterle W, Chen PY, Zhuang ZW, et al. FGF-dependent regulation of VEGF receptor 2 expression in mice. The Journal of clinical investigation. 2011 Jul;121(7):2668–78. doi: 10.1172/JCI44762. Research Support, N.I.H., Extramural. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rosenwald A, Alizadeh AA, Widhopf G, Simon R, Davis RE, Yu X, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. The Journal of experimental medicine. 2001 Dec 3;194(11):1639–47. doi: 10.1084/jem.194.11.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Green JL, Kuntz SG, Sternberg PW. Ror receptor tyrosine kinases: orphans no more. Trends Cell Biol. 2008 Nov;18(11):536–44. doi: 10.1016/j.tcb.2008.08.006. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Masiakowski P, Carroll RD. A novel family of cell surface receptors with tyrosine kinase-like domain. J Biol Chem. 1992 Dec 25;267(36):26181–90. Comparative Study. [PubMed] [Google Scholar]
- 120.Matsuda T, Nomi M, Ikeya M, Kani S, Oishi I, Terashima T, et al. Expression of the receptor tyrosine kinase genes, Ror1 and Ror2, during mouse development. Mech Dev. 2001 Jul;105(1–2):153–6. doi: 10.1016/s0925-4773(01)00383-5. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 121.Oldridge M, Fortuna AM, Maringa M, Propping P, Mansour S, Pollitt C, et al. Dominant mutations in ROR2, encoding an orphan receptor tyrosine kinase, cause brachydactyly type B. Nat Genet. 2000 Mar;24(3):275–8. doi: 10.1038/73495. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 122.van Bokhoven H, Celli J, Kayserili H, van Beusekom E, Balci S, Brussel W, et al. Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nat Genet. 2000 Aug;25(4):423–6. doi: 10.1038/78113. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 123.Afzal AR, Rajab A, Fenske CD, Oldridge M, Elanko N, Ternes-Pereira E, et al. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat Genet. 2000 Aug;25(4):419–22. doi: 10.1038/78107. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 124.Ermakov S, Malkin I, Keter M, Kobyliansky E, Livshits G. Family-based association study of ROR2 polymorphisms with an array of radiographic hand bone strength phenotypes. Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2007 Dec;18(12):1683–92. doi: 10.1007/s00198-007-0401-5. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 125.Klein U, Tu Y, Stolovitzky GA, Mattioli M, Cattoretti G, Husson H, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. The Journal of experimental medicine. 2001 Dec 3;194(11):1625–38. doi: 10.1084/jem.194.11.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yoda A, Oishi I, Minami Y. Expression and function of the Ror-family receptor tyrosine kinases during development: lessons from genetic analyses of nematodes, mice, and humans. J Recept Signal Transduct Res. 2003 Feb;23(1):1–15. doi: 10.1081/rrs-120018757. Research Support, Non-U.S. Gov’t Review. [DOI] [PubMed] [Google Scholar]
- 127.Daneshmanesh AH, Mikaelsson E, Jeddi-Tehrani M, Bayat AA, Ghods R, Ostadkarampour M, et al. Ror1, a cell surface receptor tyrosine kinase is expressed in chronic lymphocytic leukemia and may serve as a putative target for therapy. International journal of cancer Journal international du cancer. 2008 Sep 1;123(5):1190–5. doi: 10.1002/ijc.23587. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 128.Reddy UR, Phatak S, Pleasure D. Human neural tissues express a truncated Ror1 receptor tyrosine kinase, lacking both extracellular and transmembrane domains. Oncogene. 1996 Oct 3;13(7):1555–9. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. [PubMed] [Google Scholar]
- 129.Lowell CA. Src-family kinases: rheostats of immune cell signaling. Mol Immunol. 2004 Jul;41(6–7):631–43. doi: 10.1016/j.molimm.2004.04.010. Review. [DOI] [PubMed] [Google Scholar]
- 130.Bromann PA, Korkaya H, Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004 Oct 18;23(48):7957–68. doi: 10.1038/sj.onc.1208079. Review. [DOI] [PubMed] [Google Scholar]
- 131.Ishizawar R, Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell. 2004 Sep;6(3):209–14. doi: 10.1016/j.ccr.2004.09.001. Review. [DOI] [PubMed] [Google Scholar]
- 132.Lu Y, Yu Q, Liu JH, Zhang J, Wang H, Koul D, et al. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J Biol Chem. 2003 Oct 10;278(41):40057–66. doi: 10.1074/jbc.M303621200. In Vitro Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. [DOI] [PubMed] [Google Scholar]
- 133.Belsches AP, Haskell MD, Parsons SJ. Role of c-Src tyrosine kinase in EGF-induced mitogenesis. Frontiers in bioscience : a journal and virtual library. 1997 Oct 15;2:d501–18. doi: 10.2741/a208. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. Review. [DOI] [PubMed] [Google Scholar]
- 134.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002 May 31;296(5573):1655–7. doi: 10.1126/science.296.5573.1655. Review. [DOI] [PubMed] [Google Scholar]
- 135.Guarino M. Src signaling in cancer invasion. J Cell Physiol. 2010 Apr;223(1):14–26. doi: 10.1002/jcp.22011. Review. [DOI] [PubMed] [Google Scholar]
- 136.Lai SY, Johnson FM. Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies: implications for future therapeutic approaches. Drug Resist Updat. 2010 Jun;13(3):67–78. doi: 10.1016/j.drup.2010.04.001. Review. [DOI] [PubMed] [Google Scholar]
- 137.Sen B, Johnson FM. Regulation of SRC family kinases in human cancers. J Signal Transduct. 2011;2011:865819. doi: 10.1155/2011/865819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Contri A, Brunati AM, Trentin L, Cabrelle A, Miorin M, Cesaro L, et al. Chronic lymphocytic leukemia B cells contain anomalous Lyn tyrosine kinase, a putative contribution to defective apoptosis. J Clin Invest. 2005 Feb;115(2):369–78. doi: 10.1172/JCI22094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kulathu Y, Hobeika E, Turchinovich G, Reth M. The kinase Syk as an adaptor controlling sustained calcium signalling and B-cell development. The EMBO journal. 2008 May 7;27(9):1333–44. doi: 10.1038/emboj.2008.62. Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Turner M, Schweighoffer E, Colucci F, Di Santo JP, Tybulewicz VL. Tyrosine kinase SYK: essential functions for immunoreceptor signalling. Immunol Today. 2000 Mar;21(3):148–54. doi: 10.1016/s0167-5699(99)01574-1. Review. [DOI] [PubMed] [Google Scholar]
- 141.Kuno Y, Abe A, Emi N, Iida M, Yokozawa T, Towatari M, et al. Constitutive kinase activation of the TEL-Syk fusion gene in myelodysplastic syndrome with t(9;12)(q22;p12) Blood. 2001 Feb 15;97(4):1050–5. doi: 10.1182/blood.v97.4.1050. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 142.Streubel B, Vinatzer U, Willheim M, Raderer M, Chott A. Novel t(5;9)(q33;q22) fuses ITK to SYK in unspecified peripheral T-cell lymphoma. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2006 Feb;20(2):313–8. doi: 10.1038/sj.leu.2404045. [DOI] [PubMed] [Google Scholar]
- 143.Buchner M, Fuchs S, Prinz G, Pfeifer D, Bartholome K, Burger M, et al. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009 Jul 1;69(13):5424–32. doi: 10.1158/0008-5472.CAN-08-4252. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 144.Baudot AD, Jeandel PY, Mouska X, Maurer U, Tartare-Deckert S, Raynaud SD, et al. The tyrosine kinase Syk regulates the survival of chronic lymphocytic leukemia B cells through PKCdelta and proteasome-dependent regulation of Mcl-1 expression. Oncogene. 2009 Sep 17;28(37):3261–73. doi: 10.1038/onc.2009.179. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 145.Antic D, Jovanovic MP, Fekete MD, Cokic V. Assessment of bone marrow microvessel density in chronic lymphocytic leukemia. Appl Immunohistochem Mol Morphol. 2010 Jul;18(4):353–6. doi: 10.1097/PAI.0b013e3181d18ae2. [DOI] [PubMed] [Google Scholar]
- 146.Frater JL, Kay NE, Goolsby CL, Crawford SE, Dewald GW, Peterson LC. Dysregulated angiogenesis in B-chronic lymphocytic leukemia: morphologic, immunohistochemical, and flow cytometric evidence. Diagn Pathol. 2008;3:16. doi: 10.1186/1746-1596-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gora-Tybor J, Jamroziak K, Szmigielska-Kaplon A, Krawczynska A, Lech-Maranda E, Wierzbowska A, et al. Evaluation of circulating endothelial cells as noninvasive marker of angiogenesis in patients with chronic lymphocytic leukemia. Leuk Lymphoma. 2009 Jan;50(1):62–7. doi: 10.1080/10428190802549883. [DOI] [PubMed] [Google Scholar]
- 148.Martinelli S, Maffei R, Castelli I, Santachiara R, Zucchini P, Fontana M, et al. Increased expression of angiopoietin-2 characterizes early B-cell chronic lymphocytic leukemia with poor prognosis. Leuk Res. 2008 Apr;32(4):593–7. doi: 10.1016/j.leukres.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 149.Quiney C, Billard C, Mirshahi P, Fourneron JD, Kolb JP. Hyperforin inhibits MMP-9 secretion by B-CLL cells and microtubule formation by endothelial cells. Leukemia. 2006 Apr;20(4):583–9. doi: 10.1038/sj.leu.2404134. [DOI] [PubMed] [Google Scholar]
- 150.Smolej L, Andrys C, Krejsek J, Belada DZ, Zak P, Siroky O, et al. Basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) are elevated in peripheral blood plasma of patients with chronic lymphocytic leukemia and decrease after intensive fludarabine-based treatment. Vnitr Lek. 2007 Nov;53(11):1171–6. [PubMed] [Google Scholar]
- 151.Paesler J, Gehrke I, Gandhirajan RK, Filipovich A, Hertweck M, Erdfelder F, et al. The vascular endothelial growth factor receptor tyrosine kinase inhibitors vatalanib and pazopanib potently induce apoptosis in chronic lymphocytic leukemia cells in vitro and in vivo. Clin Cancer Res. 2010 Jul 1;16(13):3390–8. doi: 10.1158/1078-0432.CCR-10-0232. [DOI] [PubMed] [Google Scholar]
- 152.Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010 Apr 1;115(13):2578–85. doi: 10.1182/blood-2009-08-236471. Clinical Trial, Phase I Clinical Trial, Phase II Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Weinblatt ME, Kavanaugh A, Genovese MC, Musser TK, Grossbard EB, Magilavy DB. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. The New England journal of medicine. 2010 Sep 30;363(14):1303–12. doi: 10.1056/NEJMoa1000500. Clinical Trial, Phase II Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 154.Hantschel O, Rix U, Schmidt U, Burckstummer T, Kneidinger M, Schutze G, et al. The Btk tyrosine kinase is a major target of the Bcr-Abl inhibitor dasatinib. Proc Natl Acad Sci U S A. 2007 Aug 14;104(33):13283–8. doi: 10.1073/pnas.0702654104. Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wiestner A. Emerging role of kinase targeted strategies in chronic lymphocytic leukemia. Blood. 2012 Aug 8; doi: 10.1182/blood-2012-05-423194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Amrein PC, Attar EC, Takvorian T, Hochberg EP, Ballen KK, Leahy KM, et al. Phase II study of dasatinib in relapsed or refractory chronic lymphocytic leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011 May 1;17(9):2977–86. doi: 10.1158/1078-0432.CCR-10-2879. Clinical Trial, Phase II Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010 Feb 15;70(4):1544–54. doi: 10.1158/0008-5472.CAN-09-2997. [DOI] [PubMed] [Google Scholar]