

Significance
Maintaining robust circadian rhythms has been linked to longevity and metabolic health. Because these rhythms are disturbed by factors such as jet lag, shift work, and high-fat diets, there is interest in developing pharmacological control strategies to modulate circadian function. The design of therapeutic strategies is currently limited by the lack of a clear mechanistic understanding of interactions between posttranslational regulators, as efficient control of clock behavior will likely require several simultaneous modulations. Here we show fundamentally different clock responses from the manipulation of two clock regulators previously thought to act via similar mechanisms. Using mathematical modeling, we provide a mechanistic interpretation for the relationship between these two regulators, lending insight into circadian regulation and potential pharmacological control.
Keywords: identifiability analysis, gene regulation, sensitivity analysis
Abstract
Posttranslational regulation of clock proteins is an essential part of mammalian circadian rhythms, conferring sensitivity to metabolic state and offering promising targets for pharmacological control. Two such regulators, casein kinase 1 (CKI) and F-box and leucine-rich repeat protein 3 (FBXL3), modulate the stability of closely linked core clock proteins period (PER) and cryptochrome (CRY), respectively. Inhibition of either CKI or FBXL3 leads to longer periods, and their effects are independent despite targeting proteins with similar roles in clock function. A mechanistic understanding of this independence, however, has remained elusive. Our analysis of cellular circadian clock gene reporters further differentiated between the actions of CKI and FBXL3 by revealing opposite amplitude responses from each manipulation. To understand the functional relationship between the CKI-PER and FBXL3-CRY pathways, we generated robust mechanistic predictions by applying a bootstrap uncertainty analysis to multiple mathematical circadian models. Our results indicate that CKI primarily regulates the accumulating phase of the PER-CRY repressive complex by controlling the nuclear import rate, whereas FBXL3 separately regulates the duration of transcriptional repression in the nucleus. Dynamic simulations confirmed that this spatiotemporal separation is able to reproduce the independence of the two regulators in period regulation, as well as their opposite amplitude effect. As a result, this study provides further insight into the molecular clock machinery responsible for maintaining robust circadian rhythms.
Circadian rhythms are autonomous, near-24-h oscillations that coordinate daily changes in physiology and metabolism. Because circadian and metabolic regulators are tightly integrated, circadian disruptions often manifest in metabolic disease (1). Recent efforts have therefore sought to gain a mechanistic understanding of these pathways, such that the metabolic burdens imposed by a 24-h society might be mitigated. Posttranslational regulators, which play key roles in connecting circadian and metabolic processes, serve as likely targets for future therapeutics, demonstrated by the wealth of available circadian-active small molecules (2, 3).
Oscillations in circadian gene transcription are generated through a time-delayed transcription-translation negative-feedback loop. In mammals, transcription factors CLOCK and BMAL1 promote transcription of E box-containing genes Period (Per) and Cryptochrome (Cry) (Fig. 1A). PER and CRY protein products form heterodimers to accumulate in the nucleus, in which PER is stoichiometrically limiting (4), and subsequently close the negative feedback loop by inhibiting CLOCK-BMAL1–promoted gene expression. Although steady-state endpoint assays have shown the possibility of nuclear entry of CRY without PER (5–7), experiments from Per1−/−Per2−/− mice demonstrated that PER proteins are required for the timely nuclear accumulation of CRY (4). Clearance of nuclear repressors reactivates CLOCK-BMAL1, allowing the cycle to begin anew (8).
Fig. 1.
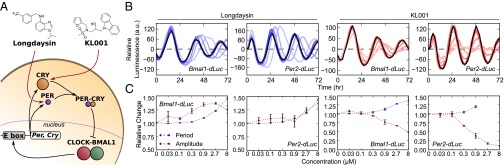
Different amplitude effect of small molecule circadian modulators targeting CKI-PER and FBXL3-CRY. (A) Schematic of the core circadian feedback loop, with the targets of small molecule modulators longdaysin (CKI inhibitor) and KL001 (inhibitor of FBXL3-dependent CRY degradation) shown. The size of each protein molecule is representative of relative concentration. (B) Detrended luminescence profiles (first 72 h, mean of two independent replications) obtained from U2OS reporter cells with increasing concentration of longdaysin and KL001. Black profiles indicate control conditions (0 μM); lighter colors indicate higher concentrations of small molecule (from 0.03 to 8 μM). (C) Relative change in period and amplitude of the results shown in B. Error bars indicate SD of two independent experiments.
The stabilities of PER and CRY are tightly regulated: PER proteins are phosphorylated by the casein kinase I family of proteins (CKIδ/ε), prompting β-transducin repeat containing protein (β-TrCP)–mediated degradation (9) and nuclear import (10). The degradation of CRY proteins is separately regulated by the SCFFBXL3 ubiquitin ligase complex (11–13). The activities of both CKI-PER and F-box and leucine-rich repeat protein 3 (FBXL3)-CRY may be further coupled to the cell’s metabolic state through AMPK signaling (14). These posttranslational regulatory mechanisms have a strong effect on period length. Increasing or decreasing CKI-dependent PER phosphorylation shortens or lengthens the period, as demonstrated by the gain-of-function mutant CKIϵtau, leading to hyperphosphorylation of PER (15, 16), and small molecule CKI inhibitors such as longdaysin (17), respectively. In contrast, increased CRY stability leads to longer periods, as shown by genetic mutations of FBXL3 (12, 13) and KL001, a small molecule inhibitor of FBXL3-dependent CRY degradation (18). Because the scale and complexity of the circadian network complicate an intuitive understanding of these relationships, mathematical models have played important roles in understanding how these manipulations affect circadian period (9, 15, 18).
Given that both CKI and FBXL3 pathways similarly regulate the stability of linked negative factors, it was thought that simultaneous perturbations to both pathways might lead to nonadditive effects: i.e., the slowest link would determine the period (19). This relationship was expected because the stabilities of both nuclear PER and CRY were thought to be important in the determination of clock kinetics (16, 18). However, both small molecule (18) and genetic experiments (19) have demonstrated the independent period effects of CKI and FBXL3 posttranslational regulations, as the inhibition of one pathway does not diminish the sensitivity of the other. This observation may be explained by a recent clarification of the canonical clock feedback circuit, where dissociated CRY was revealed as the dominant repressor of CLOCK-BMAL1–mediated E box transcription (5). This distinction helps differentiate between the roles of the otherwise similar PER and CRY proteins, in which the main role of PER in transcriptional repression is likely regulating the timing of CRY’s nuclear accumulation. Therefore, although previous mathematical models in which PER acts as a direct repressor have proposed mechanisms for CKI-dependent period lengthening (15, 20), they are likely not suitable for distinguishing between CKI-PER– and FBXL3-CRY–mediated period change.
In this study, we used human cells harboring clock gene reporters together with mathematical modeling to gain insight into the relationship between PER and CRY posttranslational regulation. Consequently, we provide a mechanism by which CKI-dependent PER phosphorylation controls the circadian period separately from the FBXL3-CRY pathway. The resulting detailed understanding of PER and CRY regulation in the core feedback loop provides a framework on which to interpret metabolic and pharmacological control of circadian rhythms.
Results and Discussion
Longdaysin and KL001 Yield Opposite Effects on the Amplitude of Circadian Reporter Expression.
To better understand both the CKI-PER and FBXL3-CRY pathways, we first studied the effects of small molecule compounds longdaysin and KL001, which cause stabilization of PER and CRY, respectively (17, 18) (Fig. 1A). We used Bmal1- and Per2-dLuciferase (dLuc) as circadian reporters, which represent different loops of the core clock mechanism and show circadian luminescence rhythms with mutually opposite phase. Time course data on circadian reporter expression under increasing concentrations of longdaysin and KL001 (18) were analyzed for period and amplitude change (Fig. 1 B and C and Fig. S1). Longdaysin caused dose-dependent increases in period and detrended amplitude to ∼50% of control values in both Bmal1- and Per2-dLuc reporter cells. In contrast, KL001 induced a simultaneous increase in period and strong reduction in amplitude. Altering the activity of CKI-PER and FBXL3-CRY is therefore differentiated by an opposite amplitude response.
To evaluate potential cross-interactions between CKI-PER and FXBL3-CRY pathways, the effect of longdaysin and KL001 on PER and CRY abundance was characterized by using PER1-LUC and CRY1-LUC reporters constitutively expressed in HEK293 cells (Fig. S2). Levels of PER1-LUC were increased only in the presence of longdaysin, whereas levels of CRY1-LUC were similarly increased only in the presence of KL001. These results suggest a lack of crossover between the two mechanisms.
Bootstrap Approach Reveals Main Period-Determining Perturbations.
We next used in silico modeling to gain insight into potential mechanisms underlying CKI-PER– and FBXL3-CRY–mediated circadian regulation. We previously described the connection between inhibition of FBXL3-dependent CRY degradation and period change (18): increasing the stability of nuclear CRY results in longer transcriptional repression and increased period length. However, although CKI has been linked to modulating PER stability and nuclear entry, it remained unclear which pathway controls the period and whether these processes are sufficient to separate the effects of CKI and FBXL3.
Although mathematical models can be used to determine whether a hypothetical mechanism is feasible, many approximations of the experimental system must be made. To generate predictions that are consistent across slight differences in model assumptions, we chose three mathematical models from the literature based on their moderate size and similar scope (18, 21, 22). The models included, at a minimum, the expression and nuclear entry mechanisms of PER and CRY. We considered the formation of the PER-CRY heterodimer as a key step in nuclear entry, which is supported by the fact that, to the best of our knowledge, all circadian models that consider both PER and CRY use this kinetic assumption (18, 21–24).
Dynamic models of genetic regulatory networks are typically comprised of a set of reaction equations and their associated kinetic parameters, which are chosen such that the model best fits the available experimental data. These kinetic parameters play a large role in determining the model’s predictions. However, the sparsity of available data and high dimensionality of the parameter sets can result in a lack of confidence in the value of any particular fit (25). In this report, we demonstrate that our predictions are independent of parameterization by using a bootstrap identifiability analysis (26). In a bootstrap method, multiple randomized in silico data sets are generated from experimental mRNA and protein concentration profiles (4). For each of these trials, a new optimal parameter set is found by minimizing the difference between the model and the data. In this fashion, we are able to determine confidence intervals in model trajectories and output predictions as a function of the data quality. The state trajectories of the resulting 2,000 parameter sets for each model are shown in Fig. 2, with reasonable agreement between models and experiment.
Fig. 2.

Time series trajectories of the 2,000 bootstrap trials for each model. Shaded regions indicate 95% confidence regions. The data were scaled to have a maximum value of 1, except for protein species, where relative values were important for clock stoichiometry.
To identify parameters associated with PER and CRY protein activity that had the most reliable effect on period, we checked for parameter changes that shifted the period in a consistent direction across bootstrap trials. This search was achieved using a first-order period sensitivity analysis, which measures the derivative of the circadian period with respect to the value of the kinetic parameter (Fig. S3). To simplify analysis, we present only those rate parameters that are associated with experimentally supported mechanisms of CKI and FBXL3 in Fig. 3. We first tested parameters associated with potential FBXL3-CRY activity (Fig. 3A) to evaluate if our method matched the experimentally verified effect of KL001 (18). Because CRY is the dominant repressor of CLOCK-BMAL1 (5), we attribute degradation rates of the PER-CRY complex to be representative of CRY clearance rates. We found that only parameters governing nuclear CRY degradation show a period lengthening effect on inhibition, whereas rates associated with cytoplasmic CRY degradation show period shortening effects. These results match with our previous assertion that period lengthening occurs via nuclear CRY stabilization (18). Experimental evidence has also indicated cytoplasmic CRY2 stabilization may lead to period shortening (27), a result consistent with our mathematical predictions.
Fig. 3.

Bootstrap predictions of circadian actions of FBXL3-CRY and CKI-PER pathways. Violin plots of the relative period sensitivity of parameters associated with potential mechanisms for FBXL3-CRY (A) and CKI-PER (B) activity, in which a box plot is superimposed above a kernel density plot to convey the distribution of sensitivities across 2,000 realizations. The whiskers used extend to the most extreme data point within 1.5× the inner quartile range. A negative or positive period sensitivity indicate that the period of oscillation will increase or decrease when that rate is inhibited, respectively. Distributions that are not different from 0 with 95% confidence are colored red and marked with an asterisk. Descriptions of the parameters shown are listed in Tables 1 and 2.
Table 1.
Descriptions of the model parameters governing FBXL3-CRY
| Model | Parameter | Description |
| Ref. 18 | MC2n | CRY2 nuclear multiplicative degradation coefficient |
| vdCn | CRY1/2 nuclear degradation rate | |
| vdC1 | CRY1 cytoplasmic degradation rate | |
| vdC2 | CRY2 cytoplasmic degradation rate | |
| Ref. 22 | dPnpCn | Phosphorylated PER-CRY complex nuclear degradation rate |
| dPnCn | PER-CRY complex nuclear degradation rate | |
| dPcpCc | Phosphorylated PER-CRY complex cytoplasmic degradation rate | |
| dPcCc | PER-CRY complex cytoplasmic degradation rate | |
| dCc | CRY cytoplasmic degradation rate | |
| Ref. 21 | V3PC | PER-CRY complex nuclear phosphorylation rate |
| vdPCN | Phosphorylated PER-CRY complex nuclear degradation rate | |
| V1C | CRY cytoplasmic degradation rate | |
| vdCC | Phosphorylated CRY cytoplasmic degradation rate |
Table 2.
Descriptions of the model parameters governing CKI-PER
| Model | Parameter | Description |
| Ref. 18 | vaC1P | PER-CRY complex nuclear entry rate |
| vdP | PER cytoplasmic degradation rate | |
| Ref. 22 | kiPcpCc | Phosphorylated PER-CRY complex nuclear entry rate |
| kfPcpCc | Phosphorylated PER-CRY complex association rate | |
| kiPcCc | PER-CRY complex nuclear entry rate | |
| kfPcCc | PER-CRY complex association rate | |
| dPcp | Phosphorylated PER cytoplasmic degradation rate | |
| dPc | PER cytoplasmic degradation rate | |
| Ref. 21 | k1 | PER-CRY complex nuclear entry rate |
| k3 | PER-CRY complex association rate | |
| V1P | PER cytoplasmic phosphorylation rate | |
| vdPC | Phosphorylated PER cytoplasmic degradation rate |
We next describe parameters potentially associated with CKI-dependent regulation of PER localization and stability (Fig. 3B). Although the concentration (and therefore stability) of cytoplasmic PER plays a role in determining the nuclear entry rate, we treat the two rates as independent by comparing the period sensitivities of kinetic constants associated with each individual step. Because PER is rate limiting in the formation of the PER-CRY complex (4), rates associated with complex formation or nuclear import were included in this analysis. Conversely, we did not include degradation rates of PER-CRY nuclear repressive complex, because CRY alone is considered the main repressor. Surprisingly, even though both processes are related, model responses to altering PER stability were different from the responses to altering nuclear import directly. Although it was previously hypothesized in models where PER acts as a direct repressor that the regulation of PER stability would determine the period (15, 20), our assumptions revealed that parameters governing PER degradation showed only nonidentifiable responses. Alternatively, inhibition of rates associated with the nuclear entry of the PER-CRY complex showed strong period lengthening effects. These results indicate that, under our current understanding of clock kinetics, the regulation of nuclear import likely plays the prominent role in CKI-dependent period regulation.
Mathematical Insights into the Different and Independent Mechanisms of PER and CRY Regulation.
Using the model and original parameter set of Hirota et al. (18) and the perturbations identified in Fig. 3, we first confirmed that inhibition of nuclear CRY degradation (vdCn) and PER-CRY nuclear import (vaC1P) reproduced the experimental period and amplitude effects of the small molecules KL001 and longdaysin, respectively (Fig. 4A, cf. Fig. 1C). Comparison of the oscillatory profiles of Per mRNA and nuclear CRY protein (Fig. 4B) revealed that inhibition of FBXL3-dependent CRY degradation caused lingering nuclear CRY to not be completely purged each cycle. This excess repressor during the accumulating phase of Per and Cry transcripts resulted in lower E box amplitudes, providing a likely explanation for the effect of KL001.
Fig. 4.

Mechanistic insight into the effects of small molecule modulators KL001 and longdaysin. (A) In silico reproductions of the circadian reporter experiments in Fig. 1 B and C, using the predictions identified in Fig. 3. (B) Comparison of the effects of KL001 and longdaysin. Parameter changes were normalized such that the period change was equal for each pair of perturbations (vdCn: 100% → 23%, vaC1P: 100% → 51%). (C) Comparison of two candidate mechanisms for CKI inhibition. Effects of increasing PER stabilization and nuclear import inhibition on the time profiles of cytoplasmic PER (Upper) and nuclear CRY (Lower). Parameter values were selected such that the amplitudes of cytoplasmic PER are equal at each level. Lighter colors indicate stronger perturbations (vdP: 100% → 22%, vaC1P: 100% → 45%); t = 0 is set to the onset of PER accumulation.
In contrast, stabilization of cytoplasmic PER (lowering the vdP parameter) resulted in reduced transcriptional amplitude with minimal period effect (Fig. S4), consistent with experimental findings from the knockdown of β-TrCP, an F box protein responsible for PER degradation (28). However, other experimental results have shown that down-regulation of β-TrCP leads to longer periods (9), suggesting that further modeling and experimental inquiry is needed on the role of β-TrCP in clock regulation. This period lengthening might be explained through β-TrCP–mediated stabilization of nuclear PER-CRY or by using alternative kinetic assumptions for the rate of PER-CRY binding.
We further compared the effect of inhibiting PER degradation with inhibiting nuclear import on the oscillatory profile of key clock proteins (Fig. 4C) to identify mechanistic differences between the two potential effects of CKI inhibition. Both perturbations increased cytoplasmic PER, suggesting the two mechanisms are difficult to distinguish experimentally. Direct stabilization of PER in the cytoplasm (lowering vdP) shortened the time delay between transcription and inactivation by accelerating the accumulation of cytoplasmic PER and nuclear PER-CRY. However, it also lengthened the repressive phase by increasing the total amount of PER-CRY that enters the nucleus. The net result of these opposing perturbations was little period change, as indicated by a faster accumulating phase and slower declining phase in nuclear CRY after vdP reduction (Fig. 4C). In contrast, inhibiting PER-CRY nuclear entry (lowering vaC1P) caused additional free protein to build in the cytoplasm, delaying nuclear accumulation and ultimately increasing the total amount of nuclear PER-CRY. Because both of these trends work to increase period length, inhibiting PER-CRY nuclear entry resulted in longer cycles. Additionally, the longer cytoplasmic time delay resulted in increased transcription, yielding slightly higher amplitudes (Fig. 4B) that closely match the experimental results of the small molecule longdaysin.
Because CKI likely regulates both stability and subcellular localization of PER in vivo, we considered the effects of simultaneously lowering both PER cytoplasmic degradation and nuclear entry rates (Fig. 5A). The loss of oscillations under extreme reduction of both parameters (Fig. 5A, shaded regions) highlights an interesting role of CKI in conferring robustness to the circadian clock: because oscillations are lost when import of the PER-CRY complex to the nucleus ceases to be rhythmic, CKI ensures lingering PER is purged from the cytoplasm by one pathway or another before E box transcription resumes. This importance has been proven experimentally, as disruption of both CKIδ and CKIϵ-mediated regulation leads to compromised circadian oscillations (29).
Fig. 5.

Independence of CKI-PER and FBXL3-CRY pathways. (A) Contour plots of period change (Upper) and Per mRNA amplitude (Lower) for simultaneous inhibition of both PER degradation (vdP) and nuclear import (vaC1P). The gray shaded region indicates loss of oscillations. (B) Period and amplitude change contour plots for varying both vdCn (CRY nuclear degradation rate) and vaC1P (PER-CRY nuclear import).
Together, inhibition of CKI by longdaysin may increase the time required before PER-CRY can enter the nucleus to repress transcription, leading to a higher amplitude and longer period. In contrast, KL001 lengthens the period by stabilizing nuclear CRY, resulting in a longer time delay before transcription resumes and lower amplitude from increased E box repression. PER regulation through CKI is therefore partitioned to the accumulating phase, controlling the speed and amount of PER-CRY complex that enters the nucleus. CRY regulation through FBXL3 is partitioned independently to the repressive phase, controlling the length of time until CLOCK-BMAL1–dependent transcription resumes (Fig. 6). This independence was reproduced in silico by the simultaneous reduction of nuclear CRY degradation and PER-CRY nuclear import (Fig. 5B). Additive independence was quantified by calculating the deviation from a theoretical surface, as described in Materials and Methods. R2 = 0.924 and 0.999 for period change and Per mRNA amplitude, respectively, indicating net changes to clock kinetics from perturbations to both rates can be closely explained by simply adding together the individual effects from each pathway.
Fig. 6.

Spatiotemporal separation underlies independence of CKI-PER and FBXL3-CRY pathways. Longdaysin, acting through CKI-PER, lengthens the accumulating phase of the circadian cycle, whereas KL001, acting through FBXL3-CRY, lengthens the declining phase.
Conclusion.
To develop efficient therapies for modulation of the circadian clock, we need to understand how clock components interact. In this study, we used circadian reporter cells and mathematical modeling to provide insight into the differences between CKI- and FBXL3-mediated clock regulation. As a result, we clarified a process by which CKI exerts control over the circadian period, demonstrated through both the hyperphosphorylating CKIϵtau mutant and small molecule CKI inhibitors, such as longdaysin. Future work will further explore the posttranslational landscape surrounding PER and CRY, including the recently discovered FBXL21, the paralog of FBXL3 that is responsible for regulating CRY stability in the cytoplasm (30, 31). In developing our predictions, we used multiple models and parameterizations to ensure our mechanisms are consistent across many in silico realizations. These results reinforce the notion that computational modeling is essential in interpreting results in systems with complicated oscillatory feedback. Additionally, in silico analyses reveal hidden design principles of biological networks, as this work highlights the importance of the CKI family of kinases in conferring robustness to the circadian cycle.
Materials and Methods
Analysis of Luminescence Profiles.
Raw luminescence data were first separated into a moving baseline and oscillatory component using a Hodrick–Prescott filter with a smoothing parameter of 1,600. Example trajectory decompositions are shown in Fig. S1. Amplitudes (Fig. 1C) were determined by taking the SD in the baseline-subtracted data. Periods were obtained by nonlinear curve fitting, in which a four-parameter (initial amplitude, decay, period, and phase) damped cosine curve was fit to the baseline-subtracted data. Periods were not shown if the relative amplitude (found by SD) fell below 25%, because noise dominated the periodic trajectory.
PER1-LUC and CRY1-LUC Assay.
HEK293 stable cell lines expressing PER1-LUC, CRY1-LUC, or LUC from a constitutive promoter (CMV promoter) were established as described previously (18). The stable cell lines (1.0 × 104 cells) were plated onto 384-well white solid-bottom plates at 50 μL/well. After 24 h, 500 nL of the compound (final 1% DMSO) was applied to the medium. After 24 h, the medium was supplemented with luciferin (final 1 mM) and Hepes-NaOH (pH 7.2; final 10 mM), and luminescence was recorded with a microplate reader (Infinite M200; Tecan).
Cost Function.
Models were fit to a cost function of experimental results. Per, Cry, Clock, and Bmal1 protein and mRNA levels were taken from ref. 4, along with profiles of CRY nuclear localization. For the model from ref. 22, additional activity profiles on Rev-Erb and Ror were obtained from CircaDB (http://bioinf.itmat.upenn.edu/circa/). To score a model trajectory, mRNA state variables were scaled independently to minimize the squared error between model and experiment, because model parameters could be adjusted to give mRNA profiles arbitrary amplitudes. For protein species, where stoichiometric interactions are important, a single scaling parameter was used for all species. Nuclear repressor species, in which only relative measurements were available, were scaled independently. Full model equations are shown in SI Text, Models 1–3.
Parameter Estimation and Bootstrap Analysis.
Bootstrap parameter estimations were performed as described previously (26), with data from ref. 4 assumed to have a normally distributed 10% relative and 5% absolute error. Because not all states in the models were measured, initial guess values for the trajectory and parameter variables were generated by optimizing the parameter sets first with a genetic algorithm approach, described in ref. 24. To help ensure bootstrap trials remained in a similar stability region of parameter space (and protect against steady-state solutions), bootstrap parameters were bound between 50% and 150% of their initial value.
Selection of Parameters for FBXL3-CRY and CKI-PER Mechanisms.
For FBXL3-CRY, parameters that determined the degradation rate of CRY (or CRY containing complexes) were considered to be the most likely candidates. Michealis–Menten degradation parameters were omitted from Fig. 3 because perturbations to such parameters are not easily attributable to changes in FBXL3 binding affinity. In the model presented in ref. 21, CRY is degraded through a series of phosphorylation events, and these parameters were considered as representative of the rate of progression toward ubiquitination of CRY. The forward phosphorylation rates of CRY and the nuclear PER-CRY complex were therefore also considered. For CKI-PER, we considered rates that determined the degradation rate and nuclear import rate of PER. Michealis–Menten parameters were not included, similar to FBXL3-CRY. With CRY being the main repressor of E box transcription (5), the degradation rates of PER-CRY complex were not considered as potential mechanisms of CKI. In the models from refs. 21 and 22, the nuclear entry of PER-CRY requires two independent steps: the formation of the PER-CRY complex and the subsequent import of the complex. Therefore, the forward reaction rates of each of these steps were included.
Numerical Experiments.
Numerical parameter inhibitions were performed by recalculating the limit cycle trajectory for each new parameter set to a tolerance of 10−8, using computational methods described previously (32). R2 values for Fig. 5B were calculated by comparing the actual results f(x, y) to idealized surfaces 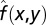 found by adding together the effects of individual perturbations to determine the degree of nonlinear effects. The idealized surface takes the form
found by adding together the effects of individual perturbations to determine the degree of nonlinear effects. The idealized surface takes the form 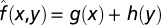 , where
, where 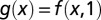 and
and 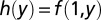 (Fig. S5). R2 calculations were restricted to the range
(Fig. S5). R2 calculations were restricted to the range 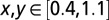 to omit bifurcation regions.
to omit bifurcation regions.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants 1R01GM096873-01 (to F.J.D.) and GM074868, MH051573, and GM085764 (to S.A.K.), and US Army Research Office Grant W911NF-09-0001 (to F.J.D.).
Footnotes
Conflict of interest statement: S.A.K. is a scientific advisory board member of Reset Therapeutics.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1323618111/-/DCSupplemental.
References
- 1.Bass J. Circadian topology of metabolism. Nature. 2012;491(7424):348–356. doi: 10.1038/nature11704. [DOI] [PubMed] [Google Scholar]
- 2.Chen Z, Yoo SH, Takahashi JS. Small molecule modifiers of circadian clocks. Cell Mol Life Sci. 2013;70(16):2985–2998. doi: 10.1007/s00018-012-1207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffett K, Burris TP. The mammalian clock and chronopharmacology. Bioorg Med Chem Lett. 2013;23(7):1929–1934. doi: 10.1016/j.bmcl.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee C, Etchegaray JP, Cagampang FR, Loudon ASI, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107(7):855–867. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- 5.Ye R, Selby CP, Ozturk N, Annayev Y, Sancar A. Biochemical analysis of the canonical model for the mammalian circadian clock. J Biol Chem. 2011;286(29):25891–25902. doi: 10.1074/jbc.M111.254680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kume K, et al. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell. 1999;98(2):193–205. doi: 10.1016/s0092-8674(00)81014-4. [DOI] [PubMed] [Google Scholar]
- 7.Yagita K, et al. Nucleocytoplasmic shuttling and mCRY-dependent inhibition of ubiquitylation of the mPER2 clock protein. EMBO J. 2002;21(6):1301–1314. doi: 10.1093/emboj/21.6.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: Implications for physiology and disease. Nat Rev Genet. 2008;9(10):764–775. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reischl S, et al. Beta-TrCP1-mediated degradation of PERIOD2 is essential for circadian dynamics. J Biol Rhythms. 2007;22(5):375–386. doi: 10.1177/0748730407303926. [DOI] [PubMed] [Google Scholar]
- 10.Takano A, Isojima Y, Nagai K. Identification of mPer1 phosphorylation sites responsible for the nuclear entry. J Biol Chem. 2004;279(31):32578–32585. doi: 10.1074/jbc.M403433200. [DOI] [PubMed] [Google Scholar]
- 11.Busino L, et al. SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science. 2007;316(5826):900–904. doi: 10.1126/science.1141194. [DOI] [PubMed] [Google Scholar]
- 12.Godinho SIH, et al. The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science. 2007;316(5826):897–900. doi: 10.1126/science.1141138. [DOI] [PubMed] [Google Scholar]
- 13.Siepka SM, et al. Circadian mutant Overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell. 2007;129(5):1011–1023. doi: 10.1016/j.cell.2007.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jordan SD, Lamia KA. AMPK at the crossroads of circadian clocks and metabolism. Mol Cell Endocrinol. 2013;366(2):163–169. doi: 10.1016/j.mce.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallego M, Eide EJ, Woolf MF, Virshup DM, Forger DB. An opposite role for tau in circadian rhythms revealed by mathematical modeling. Proc Natl Acad Sci USA. 2006;103(28):10618–10623. doi: 10.1073/pnas.0604511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meng QJ, et al. Setting clock speed in mammals: The CK1 epsilon tau mutation in mice accelerates circadian pacemakers by selectively destabilizing PERIOD proteins. Neuron. 2008;58(1):78–88. doi: 10.1016/j.neuron.2008.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirota T, et al. High-throughput chemical screen identifies a novel potent modulator of cellular circadian rhythms and reveals CKIα as a clock regulatory kinase. PLoS Biol. 2010;8(12):e1000559. doi: 10.1371/journal.pbio.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirota T, et al. Identification of small molecule activators of cryptochrome. Science. 2012;337(6098):1094–1097. doi: 10.1126/science.1223710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maywood ES, et al. Tuning the period of the mammalian circadian clock: Additive and independent effects of CK1εTau and Fbxl3Afh mutations on mouse circadian behavior and molecular pacemaking. J Neurosci. 2011;31(4):1539–1544. doi: 10.1523/JNEUROSCI.4107-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanselow K, et al. Differential effects of PER2 phosphorylation: Molecular basis for the human familial advanced sleep phase syndrome (FASPS) Genes Dev. 2006;20(19):2660–2672. doi: 10.1101/gad.397006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leloup JC, Goldbeter A. Toward a detailed computational model for the mammalian circadian clock. Proc Natl Acad Sci USA. 2003;100(12):7051–7056. doi: 10.1073/pnas.1132112100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Relógio A, et al. Tuning the mammalian circadian clock: Robust synergy of two loops. PLOS Comput Biol. 2011;7(12):e1002309. doi: 10.1371/journal.pcbi.1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forger DB, Peskin CS. A detailed predictive model of the mammalian circadian clock. Proc Natl Acad Sci USA. 2003;100(25):14806–14811. doi: 10.1073/pnas.2036281100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mirsky HP, Liu AC, Welsh DK, Kay SA, Doyle FJ., III A model of the cell-autonomous mammalian circadian clock. Proc Natl Acad Sci USA. 2009;106(27):11107–11112. doi: 10.1073/pnas.0904837106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gunawan R, Doyle FJ., III Isochron-based phase response analysis of circadian rhythms. Biophys J. 2006;91(6):2131–2141. doi: 10.1529/biophysj.105.078006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.St John PC, Doyle FJ., III Estimating confidence intervals in predicted responses for oscillatory biological models. BMC Syst Biol. 2013;7:71. doi: 10.1186/1752-0509-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurabayashi N, Hirota T, Sakai M, Sanada K, Fukada Y. DYRK1A and glycogen synthase kinase 3beta, a dual-kinase mechanism directing proteasomal degradation of CRY2 for circadian timekeeping. Mol Cell Biol. 2010;30(7):1757–1768. doi: 10.1128/MCB.01047-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohsaki K, et al. The role of beta-TrCP1 and beta-TrCP2 in circadian rhythm generation by mediating degradation of clock protein PER2. J Biochem. 2008;144(5):609–618. doi: 10.1093/jb/mvn112. [DOI] [PubMed] [Google Scholar]
- 29.Lee H, Chen R, Lee Y, Yoo S, Lee C. Essential roles of CKIdelta and CKIepsilon in the mammalian circadian clock. Proc Natl Acad Sci USA. 2009;106(50):21359–21364. doi: 10.1073/pnas.0906651106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirano A, et al. FBXL21 regulates oscillation of the circadian clock through ubiquitination and stabilization of cryptochromes. Cell. 2013;152(5):1106–1118. doi: 10.1016/j.cell.2013.01.054. [DOI] [PubMed] [Google Scholar]
- 31.Yoo SH, et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell. 2013;152(5):1091–1105. doi: 10.1016/j.cell.2013.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilkins AK, Tidor B, White J, Barton PI. Sensitivity Analysis for Oscillating Dynamical Systems. SIAM J Sci Comput. 2009;31(4):2706–2732. doi: 10.1137/070707129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.