

Significance
Defects in spermatogenesis, many of which are unexplained, underlie the infertility problems of ∼20% of couples. Although specific roles for the p53 family members in female fertility have been described, their involvement in spermatogenesis is largely unexpected. Using gene-targeted mice, we have demonstrated that deficiency of TAp73, but not p53 or ∆Np73, leads to male infertility caused by severely impaired germ cell differentiation and maturation to viable sperms in the testes. Importantly, our work has established that TAp73, but not p53, regulates many genes involved in spermatogenesis. Thus, our results provide previously unidentified in vivo evidence that TAp73 is a “guardian” of male germ cells and may point toward a novel avenue for the diagnosis and management of male infertility.
Keywords: ADAM17, MMP13, Serpin, Timp
Abstract
The generation of viable sperm proceeds through a series of coordinated steps, including germ cell self-renewal, meiotic recombination, and terminal differentiation into functional spermatozoa. The p53 family of transcription factors, including p53, p63, and p73, are critical for many physiological processes, including female fertility, but little is known about their functions in spermatogenesis. Here, we report that deficiency of the TAp73 isoform, but not p53 or ΔNp73, results in male infertility because of severe impairment of spermatogenesis. Mice lacking TAp73 exhibited increased DNA damage and cell death in spermatogonia, disorganized apical ectoplasmic specialization, malformed spermatids, and marked hyperspermia. We demonstrated that TAp73 regulates the mRNA levels of crucial genes involved in germ stem/progenitor cells (CDKN2B), spermatid maturation/spermiogenesis (metalloproteinase and serine proteinase inhibitors), and steroidogenesis (CYP21A2 and progesterone receptor). These alterations of testicular histology and gene expression patterns were specific to TAp73 null mice and not features of mice lacking p53. Our work provides previously unidentified in vivo evidence that TAp73 has a unique role in spermatogenesis that ensures the maintenance of mitotic cells and normal spermiogenesis. These results may have implications for the diagnosis and management of human male infertility.
In Western countries, ∼20% of couples experience infertility, and half of these cases are attributed to defective spermatogenesis in the male. Spermatogenesis occurs in the testes as a sequence of coordinated steps (1). Spermatogonial germ cells located on the basement membrane of the seminiferous tubules undergo either self-renewal or mitotic division to generate primary spermatocytes. Primary spermatocytes undergo a first meiotic division to generate secondary spermatocytes, which then enter a second meiotic division to yield four haploid, round-shaped spermatids that each develop and elongate through the process of spermiogenesis into mature spermatozoa anchored near the central lumen of the seminiferous tubules. During spermiogenesis, an acrosome is formed at one pole of the spermatid to form the sperm head, whereas the flagellum extends from the opposite pole and, subsequently, elongates into the sperm tail. Mature spermatozoa are released into the lumens of the seminiferous tubules and travel to the epididymides, where fully functional sperm are stored.
Spermatogenesis is regulated by stromal cells and steroid hormones. Two main somatic cell types supporting spermatogenesis are Sertoli cells and Leydig cells. On the basement membrane, germ cells associate with Sertoli cells to obtain the nutrients, hormones, and other molecules required for sperm maturation. Adjacent Sertoli cells are joined by tight junctions to form the basolateral blood–testes barrier (BTB), which prevents large molecules from passing from the blood into the seminiferous tubules. Secondary spermatocytes eventually transit across the BTB and cluster at a more apical region near the seminiferous tubule lumen called the apical ectoplasmic specialization (ES). The apical ES is an actin-based junction that anchors elongating spermatids to Sertoli cells. The apical ES is important for spermiogenesis because it prevents spermatids and spermatozoa from entering the seminiferous tubule lumen until they are fully differentiated (2).
The Leydig cells are stromal cells that are located between the seminiferous tubules and secrete androgen steroid hormones. Among these are the gonadotropins, which regulate spermatogenesis and so are important for male fertility. Luteinizing hormone stimulates Leydig cells to secrete androgens, including testosterone. This testosterone combines with follicle-stimulating hormone to stimulate Sertoli cells to carry out their spermatogenesis-promoting functions. Thus, Sertoli cells and Leydig cells, as well as the integrity of the BTB and apical ES, are critical for efficient spermatogenesis (2, 3).
Meiotic recombination is a complex process, and a large number of genes involved in DNA double-strand break formation/repair, DNA recombination, cell cycle, and aneuploidy participate in spermatogenesis. These processes involve the p53 family of transcriptional factors, namely p53, p63, and p73, with their isoforms and distinct functions. For example, each of the Trp63 and Trp73 genes can be transcribed from two alternative promoters to produce two major isoforms, only one of which contains the transcription transactivation (TA) domain. For p73, these isoforms are designated TAp73 (contains the TA domain) and ∆Np73 (lacks the TA domain) (4). In general, p53, TAp63, and TAp73 activate distinct target genes, whereas ΔNp63 and ΔNp73 may act as dominant-negative inhibitors by heterodimerizing with the TA isoforms or by selectively blocking the responsive elements. Structurally, p53, p63, and p73 are highly homologous in sequence and possess a DNA-binding domain (DBD) and an oligomerization domain (OD), in addition to the TA domain. Because of this similarity, their transcriptional profiles partially overlap in the regulation of several cellular processes (4). However, each p53 family member exhibits distinctive functions. For instance, TAp73 (i) ensures mitotic progression and the accuracy of chromosomal segregation by regulating the spindle assembly checkpoint (5, 6), (ii) fine tunes the activation of the DNA damage response (7), (iii) sustains stem cell renewal (8, 9), and (iv) regulates responses to oxidative stress by influencing mitochondrial respiration (10).
A role for the p53 family members in the maintenance of maternal fertility has been reported (11), but little is known about their functions, if any, in male fertility. Our previous work showed that, unlike p53 KO and ∆Np73 KO mice (7, 12), most TAp73 KO mice showed male infertility (5), implying a specific requirement for TAp73 in male germ cells. Here, we provide in vivo evidence of TAp73 in spermatogenesis.
Results
Loss of TAp73 in Mice Results in Severe Male Infertility Influenced by Genetic Background and Age.
We previously reported that almost all TAp73 KO male mice (9/10) were infertile (5). To determine whether TAp73 is required in germ cells, we crossed wild-type (WT) C57BL6 females to WT or TAp73 KO male mice of the C57B6/sv129Ola mixed genetic background, or TAp73 KO male mice representing F1, F6, or F9 backcrosses of these mice into the C57BL6 background. The percentage of females plugged was similar for WT and TAp73 KO male mice regardless of genetic background (WT, 90–100%; TAp73 KO, 80–100%), suggesting that the neuronal phenotypes (5), including pheromonal defects (13), previously described in TAp73 KO mice have no effect on mating capacity (Table S1). However, fewer TAp73 KO mice (0–50%, depending on age) were able to father litters (Table S1), and the average litter size for TAp73 KO males was 40–73% smaller than WT controls (Table S1). Male TAp73 KO mice of a pure C57BL6 (F9 backcross) had a less severe defect in fertility (30–50%) than male TAp73 KO mice of a mixed F1 or partially pure C57BL6 (F6) background (0–40%). These findings indicate that the effect of TAp73 loss on fertility depends, at least in part, on genetic background and that TAp73 is important, but not absolutely required, for the fertility because a subset of the TAp73 KO mice were fertile. Control examinations of p53 KO and ΔNp73 KO mice (F9, C57BL6) (Table S1) confirmed previous reports that these mutants show normal male fertility (7, 12). Interestingly, the infertility of TAp73 KO males became more severe with age regardless of genetic background. Among C57BL6 (F6) mice, about half (6/10) of 7-wk-old TAp73 KO males were infertile, whereas 100% of older (11- and 16-wk-old) TAp73 KO males were infertile (Table S1). Thus, male infertility associated with loss of TAp73 increases in an age-dependent fashion, consistent with the overall acceleration of aging previously described in TAp73 KO mice (10).
Loss of TAp73 Impairs Spermatogenesis.
To gain insight into the mechanism underlying TAp73 KO male infertility, we initially compared the testes of WT and TAp73 KO males. Regardless of genetic background, TAp73 KO testes weighed significantly less than WT testes (Fig. 1A) and exhibited a dramatic decrease in sperm number (Fig. 1B). Residual sperm of TAp73 KO mice showed reduced motility compared with controls (Table S1), suggesting that they were not functional. When we examined the histology of seminiferous tubules and epididymides in the mutant testes, very few sperm were detected in TAp73 KO epididymides (Fig. 1C). Although the number of seminiferous tubules in the testes was comparable among 11-wk-old WT, TAp73 KO, and ΔNp73 KO mice, seminiferous tubules lacking elongated spermatids or mature spermatozoa were observed only in TAp73 KO testes (Fig. 1D and Fig. S1). The number of Leydig cells between seminiferous tubules was also decreased in mutant testes (Fig. 1D, arrowheads). These results confirm that male infertility is associated only with loss of the TAp73, and is primarily due to an absence of mature sperm.
Fig. 1.
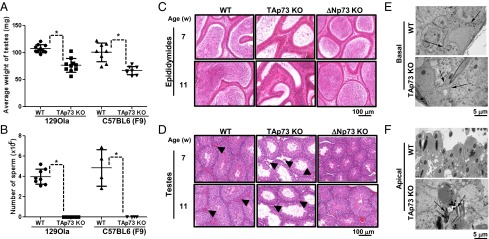
Testes of TAp73 KO mice exhibit altered germ cell numbers, germ cell localization, testicular histology, and ultrastructure. (A and B) The average weights of testes (A) and the number of mature sperm isolated from epididymides (B) were measured in 36-wk-old WT (n = 10) and TAp73 KO (n = 10) mice of the 129Ola background and in 16-wk-old WT (n = 8) and TAp73 KO (n = 8) mice of the C57BL6 (F9) background. Data points are values for individual mice. The horizontal line is the group mean ± SD (*P < 0.02; unpaired Student t test). (C and D) H&E-stained histological sections of epididymides (C) and testes (D) of WT, TAp73 KO [C57BL6 (F9)], and ΔNp73 KO (control) mice of 7 or 11 wk of age. Arrowheads show Leydig cells. (E and F) Electron micrographs of testes from 36-wk-old WT and TAp73 KO mice. (E) Normal basal ES (Basal) of BTB (arrows) surrounding the lower lateral membranes of Sertoli cells in WT and TAp73 KO mice. (F) In contrast to orderly apical ES (apical) in WT testes, TAp73 KO testes show disorganized apical ES populated by malformed spermatids (arrows).
Further histopathological assessment of TAp73 KO testes revealed multifocal/segmental seminiferous tubular degeneration, along with hypospermia (Fig. S2). Most TAp73 KO seminiferous tubules were characterized by an epithelium comprising Sertoli cells and one or two layers of germ cells/spermatocytes, but some lacked germ cells, as well as maturing spermatocytes. Although synchronous maturation was apparent in some tubules, it was limited to early-stage round spermatids (Fig. S2A), with only a few interspersed elongating spermatids and mature spermatozoa being present (Fig. S2B). Scattered apoptotic germ cells were detected in the epithelial layer (Fig. S2C). These histological alterations were not observed in the ΔNp73 KO testes (Fig. S2D). Consistent with our litter size data, the histological alterations observed in TAp73 KO testes were more marked in aged (36-wk-old) males. Compared with WT controls, these mutants showed marked reduction in germ cells within the seminiferous tubules and almost no cells in tubule lumens (Fig. S2 E and F).
We next subjected TAp73 KO testes to ultrastructural examination by electron microscopy. The regions of the TAp73 KO seminiferous tubules devoid of spermatids were lined by a thin, stratified epithelium comprised of morphologically normal cells. Areas containing spermatids were much less common but exhibited a much deeper epithelium. The basal ES of the mutant BTB appeared normal and surrounded the lower lateral membranes of the Sertoli cells (Fig. 1E). However, we were intrigued to observe that the apical ES layers of the epithelium were disorganized and populated by numerous malformed spermatids (Fig. 1F). Many of these spermatids contained several intact and/or fragmented tails. These abnormalities did not affect the structure of constituent axonemes but were consistent with a lack of normal motility. There were very few Leydig cells present between the seminiferous tubules of TAp73 KO testes (Fig. 1D, arrowheads).
Taken together, our histological and ultrastructural analyses indicate that TAp73 KO male infertility is attributable to failed spermatogenesis. Moreover, the profile and timing of the histological abnormalities we observed are consistent with the pattern of impaired fertility in TAp73 KO males.
Loss of TAp73 Decreases Proliferation and Increases DNA Damage and Apoptosis of Germ Cells.
To gain further insights into TAp73-mediated regulation of spermatogenesis, we performed immunohistochemical analysis of TAp73 to detail its cellular localization within the testes. TAp73 is expressed in most testicular cells, with particularly high levels in the cytoplasm of spermatogonia and nuclei of spermatids, raising the possibility that TAp73 plays roles in particularly spermatogonia and spermatids (Fig. 2A). To gain cellular insight into the mechanism by which TAp73 promotes male fertility, we performed histochemical examinations of markers of spermatid (protamine 1), proliferation (Ki67), apoptosis (TUNEL), and DNA damage (γH2AX) in testes from WT and TAp73 KO littermates. Protamine 1 staining was observed in spermatids in WT, but not the TAp73 KO, testes, indicating that spermatids are reduced in the TAp73 KO testes (Fig. 2B). Whereas normal spermatogonial germ cells are capable of Ki67+ proliferation, differentiated germ cells are not. Ki67 staining was significantly reduced in TAp73 KO testes compared with controls, indicating that loss of TAp73 impairs spermatogonial proliferation (Fig. 2 C and D). In addition, both TUNEL staining (Fig. 2 E and F) and γH2AX staining (Fig. 2 G and H) were enhanced in TAp73 KO testes compared with controls. These data suggest that the impaired male fertility in TAp73 KO mice arises, at least in part, from defects at both the pre- and postmeiotic stages of spermatogenesis.
Fig. 2.

Fewer spermatids, reduced spermatogonial proliferation, and increased apoptosis and DNA damage in TAp73 KO testes. (A–C) Representative immunohistochemical analysis to detect TAp73 (A), protamine 1 (B), and Ki67+ (C) in testes of 36-wk-old WT and TAp73 KO mice. Arrowheads in A show positive staining in cytoplasm of spermatogonia. (D) Quantitation of the data in C expressed as percentage of Ki67+ cells. Data shown are the means ± SD (n = 5). (E) Representative TUNEL assay detecting apoptosis in testes of 36-wk-old WT (n = 4) and TAp73 KO (n = 5) mice. (F) Quantitation of the data in E expressed as percentage of TUNEL+ cells. Data shown are the means ± SD. (G) Representative immunohistochemical analysis to detect the DNA damage marker γH2AX in testes of 16-wk-old WT (n = 5) and TAp73 KO (n = 5) mice. (H) Quantitation of the data in G expressed as percentage of γH2AX+ cells. Data shown are the mean ± SD. P values were determined according to unpaired Student t test.
Loss of TAp73 Decreases Serum Progesterone.
Steroid hormones regulate spermatogenesis, and reduced testosterone is associated with male infertility (3). Because Leydig cells are essential sources of steroid hormones during spermatogenesis, and we had found reduced numbers of Leydig cells in TAp73 KO testes, we measured serum hormone levels of 36-wk-old TAp73 KO mice and littermate controls. TAp73 deficiency did not have a significant effect on serum levels of most steroid hormones, including testosterone and cholesterol (Fig. 3 D–I and Fig. S3 A and C). However, there was a statistically significant reduction in progesterone in the serum of TAp73 KO male mice (Fig. S3B). Serum levels of triglyceride were also decreased by loss of TAp73 (Fig. S3F). We performed quantitative RT-PCR (qPCR) to analyze mRNA levels of numerous steroidogenesis-related genes (Fig. S4A) and steroid hormone receptors (Fig. S4B) in extracts of whole testes and found a significant increase in the expression of the CYP21A2, which encodes 21-hydroxylase (Fig. S4A), and progesterone receptor genes (Fig. S4B) in the TAp73 KO, but not p53 KO, testes (Figs. S3D and S4C). Because 21-hydroxylase catalyzes the hydroxylation of carbon in progesterone to generate 11-deoxycorticosterone, an up-regulation of 21-hydroxylase expression could be responsible for the decreased serum progesterone in TAp73 KO mice.
Fig. 3.

TAp73 plays a unique role in regulating the expression of genes involved in spermatogenesis. (A–C) qPCR determinations of levels of the indicated mRNAs related to markers of differentiated germ cells (A), serine proteinase inhibitors (B), and ADAM, MPPs, and MPP inhibitors (C) in testes of 36-wk-old WT and TAp73 KO mice. Data were analyzed as in A. (D) qPCR determinations of levels of the indicated mRNAs in testes of 18-wk-old WT (n = 3) and p53 KO (n = 3) mice. Data were analyzed as in A.
The Infertility in TAp73KO Male Mice Is Not Attributable to Impaired Responses to Oxidative Stress.
The increase in cell death and DNA damage in TAP73 KO testes prompted us to consider whether the mutants might have defective stress responses. We previously reported that, in the absence of TAp73, decreased expression of the mitochondrial gene cytochrome c oxidase 4 subunit 1 (Cox4i1) leads to defective mitochondrial function and consequent accumulation of oxidative damage and senescence markers in tissues of aged TAp73 KO mice (10). We therefore investigated whether TAp73 KO testes suffered from a similar defect in oxidative metabolism. As expected, TAp73 KO testes showed an increase in the expression of the senescence marker CDKN2B/p16 and a significant decrease in Cox4il (Fig. S5A). However, when we performed immunohistochemical analysis to detect 8-hydroxy-2′deoxyguanosine (a marker of oxidative DNA damage), we could not find any difference between WT and TAp73 KO testes (Fig. S5B). Moreover, comparative qPCR analysis of mRNA levels in WT and TAp73 KO testes revealed that the mutant showed no up-regulation of oxidative stress response genes, including NRF2, NQO1, HO-1, GCLC, and GCLM (Fig. S5A).
To examine oxidative stress responses in vivo, 3-wk-old WT and TAp73 KO littermates were supplied with drinking water containing (or not) the antioxidant N-acetyl-l-cysteine (NAC). After 13 wk, all TAp73 KO mice (5/5) that had been given normal water exhibited the expected infertility (Table S2). Interestingly, all TAp73 KO mice (4/4) that had been provided with water containing 10 mg/mL NAC for 13 wk were also infertile (Table S2). Thus, although other tissues of TAp73 KO mice exhibit oxidative damage, and the expression of Cox4il is reduced in TAp73 KO testes, this tissue sustained no significant oxidative damage in the absence of TAp73. We conclude that oxidative stress does not play a major role in the infertility of TAp73 KO males.
Loss of TAp73 Alters Critical mRNA Expression Patterns During Spermatogenesis.
To molecularly characterize the defect in spermatogenesis in TAp73 KO testes, we performed qPCR to detect the expression of markers specific for each cell type involved in this process. Expression levels of CDH1 and Plzf mRNAs, which are markers of spermatogonia, were slightly but not significantly reduced in TAp73 KO testes (Fig. 3A). However, more severe decreases were observed for mRNAs of Dmc1 and Id2, which are markers of spermatocytes, and Pgk2 and Prm1, which are spermatid markers (Fig. 3A). These data are consistent with our histological and ultrastructural findings and indicate that loss of TAp73 affects gene transcription required for spermatogenesis.
Microarray analysis using samples from both human subjects and infertile mouse models suggested that a number of genes, including those related to proteolysis, are important for male fertility (14). Also, because cell adhesion/migration proteins, metalloproteinases, and serine proteinase inhibitors are all important for spermatogenesis and spermatid maturation (spermiogenesis) (2, 15, 16), we used qPCR to compare the mRNA levels of several genes encoding these proteins in WT and TAp73 KO testes. In keeping with the results of the microarray analysis (14), S100 calcium-binding protein A10 (S100A10) was up-regulated in TAp73 KO but not p53 KO testes (Fig. S6). No changes in the expression of genes involved in cell adhesion were observed (Fig. S7), but the mRNAs of several serine proteinase inhibitors, including SPINK2 and SPINT1, were significantly down-regulated in TAp73 KO testes (Fig. 3B). Strikingly, no such suppression of serine proteinase inhibitors was observed in parallel examinations of p53 KO mice (Fig. 3D). An even greater reduction was observed in the mRNA levels of several metalloproteinase genes, including several members of the a disintegrin and metalloproteinase (ADAM) family and numerous matrix metalloproteinases (MMPs) (Fig. 3C). In contrast, mRNAs for metalloproteinase inhibitors, including TIMP1 and TIMP4, were significantly up-regulated in the absence of TAp73 (Fig. 3C). Of note, these changes were specific for the TAp73 isoform, because the vast majority of ADAM, MMP, TIMP, and SERPIN molecules were largely unaffected by loss of p53 (Fig. 3D).
ADAM17 and MMP13 Were Direct-Target Genes of TAp73.
To confirm whether TAp73 directly regulates expression levels of ADAM, MMP, TIMP, and SERPIN, we initially used the SAOS-2 TAp73β, ΔNp73, and p53 tet-on inducible cell lines (Fig. 4A). Doxycycline-induced expressions of TAp73β, not ΔNp73 or p53, increased mRNAs levels of MMP13, SPINK2, and SPINT1 and decreased mRNA levels of CYP21A2 and TIMP4. We searched for potential p53 family binding sites using MatInspector and identified consensus binding sites (p53BS) in the promoter sequences of ADAM17 and MMP13 genes (Fig. 4B). ChIP analysis showed that overexpressed TAp73β binds not only to p21 and MDM2 (positive controls) but also to one of p53-binding sites (BS2) of ADAM17 and MMP13 (Fig. 4C).
Fig. 4.

MMP13 and ADAM17 are previously unidentified TAp73 target genes. (A) HA tag TAp73β, ΔNp73, and p53 Saos-2 tet-on cells were exposed to 2 μM doxycycline for 16 h, followed by qPCR determinations of the indicated mRNAs. Data shown are the means ± SD (n = 3). P values were determined according to unpaired Student t test. (B) Bioinformatics analysis of human ADAM17 and MMP13 promoters using the MatInspector program has been analyzed for putative p53 binding sites (p53BS). (C) ChIP assay was performed using nuclear extracts from HA-TAp73β–overexpressing Saos-2 cells. Protein–chromatin complexes were immunoprecipitated with anti-HA antibody or control IgG. PCR was performed with primers designed against promoter region predicted or validated p53-binding sites of indicated genes. MDM2- and p21-responsive elements were used as positive controls.
Discussion
TAp73 is the only p53 family member linked thus far to male fertility. In this study, we used TAp73 KO mice to show that (i) TAp73 is required for efficient spermatogenesis and particularly for the maintenance of spermatogonia, the differentiation of matured spermatids; and (ii) TAp73 controls the expression of genes involved in germ cell senescence, spermiogenesis, and steroidogenesis. Hence, TAp73 is a central controller of male germ cell differentiation and a “guardian of male fertility.”
Despite the high degree of structural homology among the p53, TAp63, and TAp73 proteins, the phenotypes of p53 KO, p63 KO, ∆Np73, and TAp73 KO mice are clearly different, suggesting that each isoform of each family member can have unique functions. For instance, whereas p53 KO mice develop normally (17), p73 KO mice show neurological and immunological defects (13). In our study, we identified decreased serum progesterone in TAp73 KO males, as well as increased DNA damage and cell death in TAp73 KO spermatogonia and severely impaired spermiogenesis. Unlike p53, which is expressed primarily in spermatocytes but not in spermatogonia, Leydig, or Sertoli cells (18), we showed that TAp73 is expressed in all testicular cells, with particularly high levels in spermatogonia and spermatids (Fig. 2A), which is largely consistent with previous reports (19, 20). These findings could partially explain why we observed extensive DNA damage and apoptosis of spermatogonia (Fig. 2 E–H), as well as defective spermatid maturation, in testes of TAp73 KO mice but not in testes of p53 KO mice. The increased DNA damage in spermatogonia could be attributable to defects in its ability to regulate its target genes and bind to 53BP1, as we previously reported (7). In contrast to role of TAp73 in etoposide-induced apoptosis in GC2 cells (a murine spermatocyte cell line) (20), TAp73 plays antiapoptotic roles in spermatogonia, suggesting that the roles of TAp73 in apoptosis may be context-dependent.
Testosterone is a steroid hormones that is synthesized by Leydig cells and stimulate Sertoli cells to support spermatogenesis (21). Because we observed reduced numbers of Leydig cells in the TAp73 KO testes, we measured serum hormone and metabolite levels (Fig. S3). We found that levels of progesterone, but not testosterone, were decreased in TAp73 KO mice. Concomitantly mRNA levels of CYP21A2, which encodes 21-hydroxylase, were up-regulated in the testes of these mutants (Fig. S4A); 21-hydroxylase catalyzes conversion of progesterone to 11-deoxycorticosterone, triggering aldosterone synthesis (22). Thus, an increase in 21-hydroxylase activity could accelerate progesterone conversion and, at least partially, account for the lower progesterone levels observed in TAp73 KO mice. However, testosterone also can be synthesized from androstenediol by 3β-hydroxysteroid dehydrogenase in a progesterone-independent manner, and the lower progesterone levels observed were not associated with lower testosterone levels in the TAp73 KO. Although progesterone influences sperm function within female reproductive tract (i.e., acrosome reaction), there is no evidence of a crucial role for progesterone, nor for triglyceride, in spermatogenesis. Interestingly, we also observed that progesterone receptor (PR), but not androgen receptor, was up-regulated in TAp73 KO testes (Fig. S4B). The PR knockout mice show increased sperm production (23), suggesting that the up-regulation of PR in the TAp73 KO testes could be associated with defective spermatogenesis.
In this study, we found that loss of TAp73 (but not p53) affected the expression of numerous genes reportedly involved in stem cell senescence (CDKN2B), spermiogenesis (MMPs, ADAMs, SERPINs and TIMPs), and steroidogenesis (CYP21A2 and progesterone receptor). Importantly, gene expression profiles using human clinical samples supports the importance of SERPINs and TIMPs in male fertility (14). These deficits likely make a major contribution to the infertility of TAp73 KO males and establish that TAp73 has a specific role in gene expression associated with male fertility.
Our gene expression data may explain the ultrastructural anomalies of TAp73 KO testes. The apical ES layers were disorganized in TAp73 KO males. Cells of the apical ES are located between elongating/elongated spermatids and Sertoli cells and anchor the spermatids until they can complete their maturation. In TAp73 KO testes, we observed a disorganized apical ES populated by malformed spermatids. Although the apical ES is an actin-based adhesion junction, other adhesion junctions are composed of other proteins (2). For instance, the laminin α3, β3, and γ3 chains are localized with spermatids and form adhesion junctions with α6β1-integrin expressed by Sertoli cells at the apical ES. Laminin is an MMP substrate, and laminin must be cleaved by these enzymes to generate biologically active peptides that mediate cell adhesion or migration (2). We found that MMPs were deregulated in the absence of TAp73, which may explain the disorganized apical ES and malformed spermatids in TAp73 KO males. In more detail, ChIP analysis showed that MMP13 and ADAM17 are previously unidentified TAp73 target genes, although TAp73 affects mouse ADAM17 expression in testes but not human ADAM17 in Saos-2 cells. This result might be attributable to the lack of critical transcriptional cofactors expressed specifically in testis. On the other hand, p73 regulates MMP13 expression positively in Saos-2 cells (Fig. 4A) but negatively in mouse testes (Fig. 3). In contrast to the human gene, the first 1 Kb of mouse MMP13 promoter does not include any p53 family binding sites, according to MatInspector software (Fig. S8). However, analysis showed that p73 might still be able to directly regulate mouse MMP13, binding a predicted binding site localized after the transcriptional start site. This dissimilarity in human and mouse MMP13 might underlie differential control exerted by p73 in our human cell lines and mouse testes.
Another gene group affected by loss of TAp73 was the Serpin family of serine proteinase inhibitors. Serpins have multiple functions, including tissue remodeling and maintenance of fertility. For instance, SERPINEA5 KO mice show a severe defect in spermatogenesis because of disorganization of BTB (24), whereas SPINK2 mutant mice exhibit increased germ cell apoptosis and impaired fertility (25). We believe that reduction of Serpin in TAp73 KO testes could contribute to spermatogenesis defects. How TAp73 uniquely regulates a number of spermatogenesis-related genes should be elucidated. In conclusion, our findings reveal an important role for TAp73 in supporting male fertility that is not shared by other p53 family members or even other p73 isoforms. p53 and p63 are proposed to be guardians of the genome and female germ cells, respectively (11, 26, 27). Here, we propose TAp73 is “a guardian of male germ cells” regulating primarily mitotic proliferation and spermiogenesis, offering previously unidentified molecular mechanisms to understand the etiology of human male infertility.
Materials and Methods
Fertility, sperm numbers and motility, histology, immunohistochemistry, NAC administration, electron microscopy, cell lines, ChIP, and measurement of metabolites in serum are described in SI Materials and Methods.
Mice.
The generation of TAp73, ΔNp73, and p53 KO mice has been described previously (5, 7, 28). All breedings were approved by the University Health Network Animal Care Committee (identification no. AUP985).
Real-Time qPCR.
All procedures have been described previously (29). The primer sequences used for real-time RT-PCR are listed in Tables S3 and S4.
Supplementary Material
Acknowledgments
We thank all members of the T.W.M. laboratory, particularly Ms. Irene Ng, for assistance. We also thank Dr. Eleonora Candi (University of Rome Tor Vergata) and Dr. Gerald M. Cohen and Dr. Richard A. Knight [Medical Research Council (MRC) Toxicology Unit] for constructive suggestions, Maria Guerra Martin and Tim Smith for preparation of samples for electron microscopy and the support of the staffs of the genotyping facility and the animal resource center at Princess Margaret Hospital, and Dr. Mary Saunders for scientific editing of the manuscript. This work was supported by Canadian Institutes of Health Research Grant RN 0000078522 (to T.W.M.), Associazione Italiana per la Ricerca sul Cancro (AIRC) Grants 2011-IG11955 and AIRC 5xmile (#9979) (to. G.M.), and the MRC.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1323416111/-/DCSupplemental.
References
- 1.Hess RA, Renato de Franca L. Spermatogenesis and cycle of the seminiferous epithelium. Adv Exp Med Biol. 2008;636:1–15. doi: 10.1007/978-0-387-09597-4_1. [DOI] [PubMed] [Google Scholar]
- 2.Cheng CY, Mruk DD. A local autocrine axis in the testes that regulates spermatogenesis. Nat Rev Endocrinol. 2010;6(7):380–395. doi: 10.1038/nrendo.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verhoeven G, Willems A, Denolet E, Swinnen JV, De Gendt K. Androgens and spermatogenesis: Lessons from transgenic mouse models. Philos Trans R Soc Lond B Biol Sci. 2010;365(1546):1537–1556. doi: 10.1098/rstb.2009.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melino G, De Laurenzi V, Vousden KH. p73: Friend or foe in tumorigenesis. Nat Rev Cancer. 2002;2(8):605–615. doi: 10.1038/nrc861. [DOI] [PubMed] [Google Scholar]
- 5.Tomasini R, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008;22(19):2677–2691. doi: 10.1101/gad.1695308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomasini R, et al. TAp73 regulates the spindle assembly checkpoint by modulating BubR1 activity. Proc Natl Acad Sci USA. 2009;106(3):797–802. doi: 10.1073/pnas.0812096106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilhelm MT, et al. Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev. 2010;24(6):549–560. doi: 10.1101/gad.1873910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agostini M, et al. p73 regulates maintenance of neural stem cell. Biochem Biophys Res Commun. 2010;403(1):13–17. doi: 10.1016/j.bbrc.2010.10.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujitani M, et al. TAp73 acts via the bHLH Hey2 to promote long-term maintenance of neural precursors. Curr Biol. 2010;20(22):2058–2065. doi: 10.1016/j.cub.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 10.Rufini A, et al. TAp73 depletion accelerates aging through metabolic dysregulation. Genes Dev. 2012;26(18):2009–2014. doi: 10.1101/gad.197640.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. The p53 family: Guardians of maternal reproduction. Nat Rev Mol Cell Biol. 2011;12(4):259–265. doi: 10.1038/nrm3086. [DOI] [PubMed] [Google Scholar]
- 12.Hu W, Feng Z, Teresky AK, Levine AJ. p53 regulates maternal reproduction through LIF. Nature. 2007;450(7170):721–724. doi: 10.1038/nature05993. [DOI] [PubMed] [Google Scholar]
- 13.Yang A, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404(6773):99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- 14.Rockett JC, Patrizio P, Schmid JE, Hecht NB, Dix DJ. Gene expression patterns associated with infertility in humans and rodent models. Mutat Res. 2004;549(1-2):225–240. doi: 10.1016/j.mrfmmm.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 15.Moreno RD, Urriola-Muñoz P, Lagos-Cabré R. The emerging role of matrix metalloproteases of the ADAM family in male germ cell apoptosis. Spermatogenesis. 2011;1(3):195–208. doi: 10.4161/spmg.1.3.17894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao X, Mruk DD, Cheng CY. Intercellular adhesion molecules (ICAMs) and spermatogenesis. Hum Reprod Update. 2013;19(2):167–186. doi: 10.1093/humupd/dms049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donehower LA, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356(6366):215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 18.Beumer TL, et al. The role of the tumor suppressor p53 in spermatogenesis. Cell Death Differ. 1998;5(8):669–677. doi: 10.1038/sj.cdd.4400396. [DOI] [PubMed] [Google Scholar]
- 19.Hamer G, Gademan IS, Kal HB, de Rooij DG. Role for c-Abl and p73 in the radiation response of male germ cells. Oncogene. 2001;20(32):4298–4304. doi: 10.1038/sj.onc.1204568. [DOI] [PubMed] [Google Scholar]
- 20.Codelia VA, Cisterna M, Alvarez AR, Moreno RD. p73 participates in male germ cells apoptosis induced by etoposide. Mol Hum Reprod. 2010;16(10):734–742. doi: 10.1093/molehr/gaq045. [DOI] [PubMed] [Google Scholar]
- 21.Walker WH. Testosterone signaling and the regulation of spermatogenesis. Spermatogenesis. 2011;1(2):116–120. doi: 10.4161/spmg.1.2.16956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flück CE, Pandey AV. Clinical and biochemical consequences of p450 oxidoreductase deficiency. Endocr Dev. 2011;20:63–79. doi: 10.1159/000321221. [DOI] [PubMed] [Google Scholar]
- 23.Lue Y, et al. Functional role of progestin and the progesterone receptor in the suppression of spermatogenesis in rodents. Andrology. 2013;1(2):308–317. doi: 10.1111/j.2047-2927.2012.00047.x. [DOI] [PubMed] [Google Scholar]
- 24.Uhrin P, et al. Disruption of the protein C inhibitor gene results in impaired spermatogenesis and male infertility. J Clin Invest. 2000;106(12):1531–1539. doi: 10.1172/JCI10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee B, et al. Impaired spermatogenesis and fertility in mice carrying a mutation in the Spink2 gene expressed predominantly in testes. J Biol Chem. 2011;286(33):29108–29117. doi: 10.1074/jbc.M111.244905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358(6381):15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 27.Suh EK, et al. p63 protects the female germ line during meiotic arrest. Nature. 2006;444(7119):624–628. doi: 10.1038/nature05337. [DOI] [PubMed] [Google Scholar]
- 28.Jacks T, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4(1):1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 29.Inoue S, et al. Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by preventing c-Myc/Miz1-mediated down-regulation of p21 and p15. Genes Dev. 2013;27(10):1101–1114. doi: 10.1101/gad.214577.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.