

Significance
This study provides insights into the physiological role of Sel1L, an adaptor protein for the ubiquitin ligase Hrd1 in endoplasmic reticulum-associated degradation (ERAD). Using both animal and cell models, this study provides unequivocal evidence for an indispensable role of Sel1L in Hrd1 stabilization, mammalian ERAD, endoplasmic reticulum homeostasis, protein translation, and cellular and organismal survival. Moreover, generation of inducible knockout mouse and cell models deficient in both Sel1L and Hrd1 provides an unprecedented opportunity to elucidate the functional importance of this key branch of ERAD in vivo and to identify its physiological substrates.
Keywords: inducible ERAD-deficient models, exocrine pancreatic insufficiency, stress granule, ER dilation, ERAD tuning
Abstract
Suppressor/Enhancer of Lin-12-like (Sel1L) is an adaptor protein for the E3 ligase hydroxymethylglutaryl reductase degradation protein 1 (Hrd1) involved in endoplasmic reticulum-associated degradation (ERAD). Sel1L’s physiological importance in mammalian ERAD, however, remains to be established. Here, using the inducible Sel1L knockout mouse and cell models, we show that Sel1L is indispensable for Hrd1 stability, ER homeostasis, and survival. Acute loss of Sel1L leads to premature death in adult mice within 3 wk with profound pancreatic atrophy. Contrary to current belief, our data show that mammalian Sel1L is required for Hrd1 stability and ERAD function both in vitro and in vivo. Sel1L deficiency disturbs ER homeostasis, activates ER stress, attenuates translation, and promotes cell death. Serendipitously, using a biochemical approach coupled with mass spectrometry, we found that Sel1L deficiency causes the aggregation of both small and large ribosomal subunits. Thus, Sel1L is an indispensable component of the mammalian Hrd1 ERAD complex and ER homeostasis, which is essential for protein translation, pancreatic function, and cellular and organismal survival.
Protein misfolding and aggregation in the endoplasmic reticulum (ER) contributes significantly to the etiology and pathogenesis of many devastating diseases, including α1-antitrypsin deficiency, type-1 diabetes, Creuzfeld–Jacob disease, and cystic fibrosis (1). ER-associated degradation (ERAD) targets misfolded secretory and membrane proteins in the ER for proteasomal degradation (2–4), and the unfolded protein response (UPR) senses ER stress signals and initiates global changes in transcription and translation (5, 6). These two are the key quality-control systems in the cell to maintain ER homeostasis and adjust ER capacity in response to environmental cues. In yeast, although cells tolerate the loss of each pathway, loss of both pathways leads to synthetic lethality (7, 8), suggesting that these two pathways function in a cooperative but interdependent manner.
In mammals, the relationships between the two systems are much more complicated in part because of increased complexities within the UPR and ERAD systems. At least three major branches of UPR and five major ERAD complexes have been identified to date. Moreover, studies have suggested that different cell types in mammals have different burdens and tolerance to ER misfolded proteins, and hence different dependency and requirements for UPR and ERAD for survival. How various cell types maintain ER homeostasis remains an open and challenging question. Animal models are needed to directly address physiological significance of the ERAD and UPR in a cell type-specific manner. Several animal models defective in UPR have been characterized to date; however, studies of ERAD mouse models have been limited (9–12).
Among several key E3 ligases that have been identified so far, hydroxymethylglutaryl reductase degradation protein 1 (Hrd1) is a principle ER-resident E3 ligase and forms a complex with an ER-resident single-transmembrane protein Hrd3 in yeast or Suppressor/Enhancer of Lin-12-like (Sel1L) in mammals, responsible for the degradation of a subset of misfolded proteins in the ER (13–19). The Hrd1–Hrd3 complex was first discovered in yeast, by the Hampton group, to be responsible for the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase (13, 14) and in C. elegens by the Greenwald group through genetic interactions with Notch (20, 21). Recent studies from several groups have elegantly demonstrated that Sel1L is an integral part of the mammalian Hrd1 ERAD complex and is necessary for the ERAD process for a subset of model substrates (15–19) and endogenous substrates, including luminal hedgehog (22), transmembrane CD147 (23), and ATF6 (24). However, although Hrd3p determines the stability of Hrd1p in yeast (4, 14), knockdown of Sel1L seems to have negligible effect on the steady-state level of Hrd1 protein in cultured mammalian cells (15, 25, 26). Moreover, a recent proteomics study showed that Hrd1-mediated degradation of model substrates may proceed in a Sel1L-dependent or -independent manner, depending on substrate topology or accessibility of specific E3 ligases (15). Degradation of ER-transmembrane proteins can be Sel1L–Hrd1-independent because of functional redundancy among the ERAD complexes (27). Finally, pointing to a dispensable role of Sel1L in ER homeostasis in vivo, knockdown of Sel1L in cultured cells fails to induce UPR (28, 29) and deletion of Hrd3/Sel1 in the fly has no effect on eye size (30). Thus, how Sel1L regulates ERAD and ER homeostasis in vivo remains unclear.
Nonetheless, studies have implicated Sel1L in various cellular processes, including tumorigenesis of various cancer types (31), stem cell differentiation (32), pancreatic epithelial cell differentiation (33), and retrotranslocation of cholera toxin to the cytosol (26). Variants in the Sel1L gene have been identified in human patients with autoimmune thyroid diseases (34), in canines with progressive early-onset cerebellar ataxia (35), and in humans with Alzheimer’s disease (36). However, our ability to dissect physiological roles of Sel1L has been limited because of the embryonic lethality of the Sel1L-deficient mice (11). To circumvent this problem, we have generated and characterized inducible Sel1L-deficient mouse and cell models to permit an assessment of Sel1L function in adult animals and immortalized mouse embryonic fibroblasts (MEFs). Here our data demonstrate an indispensable role of Sel1L in mammalian ERAD and ER homeostasis and illustrate pathophysiological consequences associated with acute Sel1L deficiency.
Results
Premature Lethality of Sel1LIKO Mice.
Sel1L is ubiquitously distributed in many tissues, with the strongest expression in the pancreas (Fig. 1A). To study the role of Sel1L in vivo, we generated tamoxifen-inducible knockout mice (IKO) f/f;ERCre+ (Sel1LIKO) with f/f;ERCre− (WT) littermates as a control cohort (Fig. 1 B and C). Mice were born at an expected Mendelian ratio (Fig. S1A) and grew normally in the first 16-wk of life following weaning at 3 wk of age (Fig. S1B). To acutely induce Sel1L deficiency, adult mice were injected daily with tamoxifen for 3 d (from day 0 to day 2). Day 0 was defined as the day with the first tamoxifen injection. Following tamoxifen injection, Sel1LIKO animals progressively lost body weight starting around day 8. The mice became runted and moribund with reduced body temperature and died within 2–3 wk (Fig. 1 D–F), despite higher food intake, normal blood glucose levels, energy expenditure, and physical activity (Fig. 1 G and H and Fig. S1C).
Fig. 1.
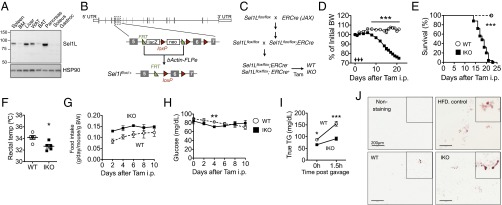
Early lethality and nutrient malabsorption in Sel1LIKO mice. (A) Western blot analysis of Sel1L in different tissues from WT mice. Gastroc, gastrocnemius; BM, bone marrow; WAT/BAT, white/brown adipose tissue; HSP90, a loading control. (B) Diagram illustrating the generation of Sel1Lflox/+ animals. Gray boxes, exons. The loxP sites flank the exon 6. (C) Diagram illustrating the generation of Sel1Lflox/flox;ERCre mice. ERCre, estrogen-receptor-controlled Cre. (D) Body weight change after three daily injections of tamoxifen in adult mice. Arrows point to three consecutive tamoxifen injection. WT, n = 20; IKO, n = 29. (E) Surviving curve of WT and IKO mice after tamoxifen injection. WT n = 16; IKO n = 25. ***P < 0.001 by the log-rank (Mantel–Cox) test. (F) Rectal body temperature at day 13 (n = 5). (G) Food intake after tamoxifen injection (n = 5). (H) Blood glucose levels (ad libitum) after tamoxifen injection (n = 5). (I) Serum Triglyceride (TG) levels before and at 1.5 h postlipid gavage in WT and IKO mice. n = 5 each. (J) Oil-red O staining of fecal smear from WT and IKO mice. Nonstained and stained fecal smear slides from 2 mo high-fat diet (HFD) -fed mice were used as negative and positive controls, respectively. Representative pictures of four mice each group shown. Data are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 by Student t test, except for E. Representative data of at least two experiments shown.
The observations that Sel1LIKO mice lost body weight despite higher food intake and normal metabolic parameters prompted the speculation that Sel1LIKO mice may suffer from maldigestion and malabsorption. To directly test this theory, we gave mice an oral bolus of lipid. Indeed, serum triglyceride level was significantly lower in Sel1LIKO mice than that of WT mice (Fig. 1I). Moreover, fecal fat contents of Sel1LIKO mice were higher than those of WT littermates (Fig. 1J). Both evidence points to the diagnosis of maldigestion and malabsorption. Thus, acute Sel1L deficiency leads to early lethality and nutrient malabsorption in adult mice.
Pancreatic Atrophy in the Pancreas of Sel1LIKO Mice.
At day 8, visual and histological examinations revealed no apparent abnormalities in tissues such as small intestine, lung, liver, heart, and skeletal muscle (Fig. S2). However, that was not the case for the pancreas. Unlike the pancreas of WT mice, which had a firm consistency and uniform yellow opaque color, the pancreas of Sel1LIKO mice was diffusely dark red, soft, and weighed about half of that of the WT mice, a change consistent with severe pancreatic atrophy (Fig. 2 A and B). Histologically, dramatic morphological degenerative changes were noted in the exocrine pancreas of Sel1LIKO mice, including dramatic reduction of eosinophilic cytoplasmic secretory zymogen granules, increased basophilia-marked anisokaryosis, and binucleated cells (Fig. 2C). A few lymphocytes and neutrophils were frequently detected in the interstitium of the exocrine pancreas of Sel1LIKO mice, a change interpreted as mild pancreatitis (Fig. S3A). In line with reduced eosinophilic staining in the H&E-stained tissue sections, the protein levels of two major components of acinar cell zymogen granules, pancreatic α-amylase and lipase, were significantly reduced in the pancreas of Sel1LIKO mice (Fig. 2D). The reduction of pancreatic α-amylase was further confirmed by immunohistochemistry staining of pancreatic tissue (Fig. 2E). Accordingly, enzymatic activities of pancreatic lipase and α-amylase were reduced by over 60–80% in the pancreas of Sel1LIKO mice (Fig. 2F). The mRNA levels of α-amylase and pancreatic lipase were down-regulated as well (Fig. 2G). Of note, Sel1L deletion was limited to the exocrine pancreas (Fig. 2E). This observation provided an explanation for the normal function of the endocrine pancreas in terms of blood glucose (Fig. 1H) and insulin/glucagon levels in the circulation and pancreatic islets (Fig. S3 B and C). Thus, Sel1LIKO mice exhibit pancreatic atrophy with defects in nutrient digestion and absorption.
Fig. 2.
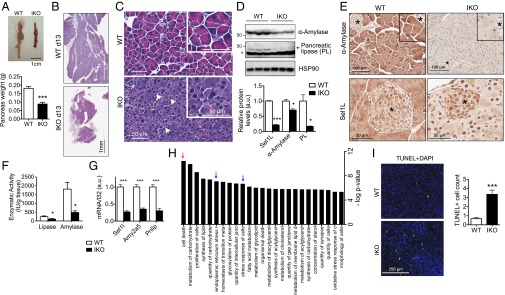
Exocrine pancreatic insufficiency and increased cell death in the pancreas of Sel1LIKO mice. Experiments were performed at day 13. (A) Picture of WT and IKO pancreas from male mice. Each picture represents five mice each group. (B) H&E images showing pancreatic atrophy in IKO mice. (C) H&E images showing reduced eosinophilic zymogen staining, different nuclei sizes (arrowheads), and binucleated cells. (D) Western blot analysis of various pancreatic enzymes with quantitation shown below. Asterisk indicates the nonspecific band. Each lane represents one mouse. (E) Immunohistochemistry of amylase and Sel1L. n = 3–5 each group. Asterisk denotes an islet. Note Sel1L deletion is largely limited in the exocrine pancreas. (F) Enzymatic activities in WT and IKO pancreas. n = 5 each group. (G) qPCR analysis of genes in the pancreas. n = 5 each. Amy2a5, amylase 2a5; Pnlip, pancreatic lipase. (H) Ingenuity analysis showing top 25 significantly changed functional annotations between WT and IKO pancreas (day 13, n = 4 mice each) with a threshold of fold change >1.5 or <−1.5 and q value <0.01. Red arrow, cell death; blue arrows, stress responses. (I) Confocal images of TUNEL staining (green) with nuclei stained with DAPI. Positive/negative controls shown in Fig. S5A. Quantitation of TUNEL-positive cells in 40 random views under 20× magnification shown on the right. Data are mean ± SEM. *P < 0.05, ***P < 0.001 by Student t test. Representative data of at least two experiments shown.
The phenotypes of the Sel1LIKO pancreas and mice indeed resemble the condition of exocrine pancreatic insufficiency, a disease often seen in mammals, including humans, dogs, and cats (37). Exocrine pancreatic insufficiency is associated with a reduction in pancreatic enzymes and hence nutrient maldigestion and malabsorption.
Increased Cell Death in the Pancreas of Sel1LIKO Mice.
To identify signaling pathways that were affected by acute Sel1L deficiency, we performed a nonbiased whole-genome microarray analysis of the pancreas at day 13 (Fig. S4A). Strikingly, over 2,000 genes were altered in the pancreas of Sel1LIKO mice, with a fold-change of >1.5 or <−1.5 and q-value <0.01. Among these genes, 1,195 genes were up-regulated by >1.5 and 1,000 genes were down-regulated more than 1.5-fold. The top 20 genes that were either up-regulated or down-regulated in Sel1LIKO samples are shown in Fig. S4B. Functional pathway analysis of Sel1L-responsive transcripts revealed that Sel1L deficiency facilitated expression of a host of biological processes including cell death, ER stress response, and lipid and carbohydrate metabolism (Fig. 2H). Indeed, ingenuity analysis further revealed that, in addition to XBP1s, key regulators of lipid metabolism, including members of peroxisome proliferator-activated receptor family (PPARα, PPARγ, and PPARβ/δ) and sterol regulatory element binding transcription factor 2 were activated in the pancreas of Sel1LIKO mice (Fig. S4C).
TUNEL staining showed a three- to fourfold increase of TUNEL-positive cells in the exocrine pancreas of Sel1LIKO mice (Fig. 2I and Fig. S5A), confirming increased cell death. Accordingly, the anti-apoptotic protein Bcl-2 was significantly reduced (Fig. S5B). Histological assessment of H&E-stained pancreatic tissues of Sel1LIKO mice revealed pyknotic nuclei with eosinophilic cytoplasm (Fig. S5C), a finding consistent with increased cell death. Although disruption of ER homeostasis is linked with the activation of autophagy in some cell types (38, 39), Sel1L deficiency was not associated with increase of autophagy in pancreas as measured by LC3B cleavage (Fig. S5B). Expression of a proliferation marker Ki-67 was significantly reduced in the exocrine pancreas of Sel1LIKO mice compared with that in WT mice (Fig. S5D). Thus, in line with pancreatic atrophy, Sel1L deficiency promotes cell death and pancreatic atrophy in the exocrine pancreas.
Characterization of the Sel1L–Hrd1 ERAD Complex in the Exocrine Pancreas of Sel1LIKO Mice.
Although recent studies have shown a dispensable role of Sel1L in Hrd1 stability and ER homeostasis (see introductory remarks), our microarray data suggested that ER stress response was induced by Sel1L deficiency (Fig. 2H). Because Sel1L is a known Hrd1 cofactor, we first determined the impact of Sel1L deficiency on Hrd1 and ERAD. Taking advantage of an inducible model, we analyzed dynamic cellular response to Sel1L loss. Following tamoxifen injection, Sel1L protein level was greatly abolished at day 4. Remarkably, Hrd1 protein levels decreased gradually, reaching 30% at day 4 and 20% at day 8 (Fig. 3 A and B). The decrease of Hrd1 protein levels was not a result of transcriptional suppression because Hrd1 mRNA was actually increased (Fig. 3C). The concurrent reduction of Sel1L and Hrd1 protein levels in the pancreas of Sel1LIKO mice was further confirmed using immunohistochemistry (Fig. 3D). Similar observation of Sel1L-regulated Hrd1 stability was seen in other tissues of Sel1LIKO mice, including the kidney and ileum (Fig. 3E). Indeed, Hrd1 and Sel1L protein levels exhibited a linear correlation in various Sel1L-deficient tissues and MEFs (Fig. 3F). Taking these data together, we conclude that Sel1L regulates Hrd1 protein stability in vivo and that we have generated an inducible mouse model deficient in both Sel1L and Hrd1.
Fig. 3.
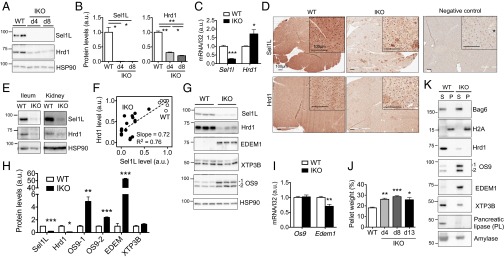
Sel1L is indispensable for the stability of Sel1L-Hrd1 complex in vivo. Pancreas was harvested at days 4, 8, and 13. (A) Western blot analysis of Sel1L and Hrd1 in WT and IKO pancreas at day 4 and 8, with quantitation shown in B upon being normalized to the loading control HSP90. (C) qPCR analysis of Sel1L and Hrd1 genes at day 13; n = 5. (D) Immunohistochemical staining of Sel1L and Hrd1 in pancreas at day 13; n = 3 each. (E) Western blot analysis of Sel1L and Hrd1 in the ileum and kidney of IKO mice. (F) Linear regression analysis between Hrd1 and Sel1L protein levels in various WT or Sel1L-deficient tissues and cell lines, including pancreas, gut, kidney, and MEFs. See SI Materials and Methods for details. Each dot represents one sample (n = 25). The slope of the regression line and the square of the correlation coefficient (R2) are shown. (G) Western blot analysis of Sel1L-associated factors (EDEM1, OS9, and XTP3B) in WT and IKO pancreas at day 13 with quantitation shown in H. (I) qPCR analysis of Edem1 and Os9 in the pancreas at day 13. (J) Percentage of Nonidet P-40 insoluble pellet weight in total tissue weight at the indicated times following tamoxifen injection; n = 2–6. WT samples were pooled from two samples of day 8 and four samples of day 13. (K) Western blot analysis of various proteins in the NP40P and NP40S fractions of pancreas at day 13 using lysis buffer containing 0.5% Nonidet P-40. The distribution of Bag6 and H2A marks the soluble (S) and insoluble (P) fractions, respectively. Representative data of three samples each shown. Data are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 by Student t test. Representative data of two experiments shown.
Hrd3/Sel1L–Hrd1 belongs to a larger macromolecular complex involved in ERAD, including ER degradation enhancer, mannosidase alpha-like 1 (EDEM1), amplified in osteosarcoma 9 (OS9), and endoplasmic reticulum lectin 1 (XTP3B) (16, 40–45). We next examined how Sel1L deficiency affects their protein levels. Strikingly, protein levels of EDEM1, OS9.1, and OS9.2 were elevated by over 50-, 5-, and 2-fold, respectively, in the pancreas at day 13, but not XTP3B (Fig. 3 G and H). Elevated EDEM1 and OS9 protein levels were not a result of transcriptional regulation because their mRNA levels were not increased in the pancreas of Sel1LIKO mice (Fig. 3I). Thus, Sel1L–Hrd1 deficiency causes the accumulation of EDEM1 and both OS9 isoforms, but not XTP3B, in pancreas.
Providing further support that Sel1L deficiency leads to ERAD defects, the amount of detergent-insoluble fraction of the pancreas of Sel1LIKO mice was significantly increased upon the loss of Sel1L (Fig. 3J). Detergent-insoluble fraction represents protein aggregates formed from misfolded proteins (46), as shown by the distribution of soluble cytosolic protein BAG6 and aggregation-prone histone protein H2A (47) (Fig. 3K). Of note, the alteration of protein levels of Hrd1 and other Sel1L-associated factors, as well as some digestion enzymes, was not because of their redistribution between the soluble and insoluble fractions of the pancreas (Fig. 3K). Thus, these data support the notion that Sel1L deficiency causes the accumulation of misfolded proteins in the ER. Taking these data together, we find that Sel1L is an indispensable component of the Hrd1 ERAD complex and mammalian ERAD.
UPR Activation in the Exocrine Pancreas Following the Loss of Sel1L.
We next determined the extent of UPR activation and the dynamic UPR response in the exocrine pancreas of Sel1LIKO mice at days 4, 8, and 13. Phosphorylation of UPR sensor PKR-like ER kinase (PERK) was significantly elevated at all time points (Fig. 4 A and B). Phosphorylation of its downstream target eukaryotic translation initiation factor 2a (eIF2α) at serine 51 increased at day 4 and continued to increase with time (Fig. 4 A–C). Nuclear accumulation of Ddit3, DNA-damage inducible transcript 3 (CHOP) was increased in Sel1LIKO pancreas (Fig. 4D). Providing further support to the UPR activation, Xbp1 mRNA splicing by another UPR sensor inositol-requiring enzyme 1a (IRE1α) was doubled in the pancreas of Sel1LIKO mice (Fig. 4E). mRNA levels of a subset of ER chaperones were elevated (Fig. 4F). At the protein level, Grp58 protein was up-regulated in the pancreas of Sel1LIKO mice, whereas others, such as Grp78, Calnexin, and Pdia1, were not (Fig. 4 G and H). Thus, our data demonstrate that Sel1L is essential for the maintenance of ER homeostasis, and that acute Sel1L deficiency induces persistent UPR, which may account for increased cell death and pancreatic atrophy.
Fig. 4.
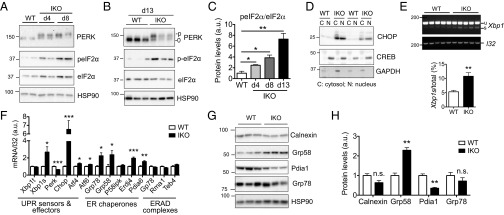
Sel1L is required for ER homeostasis in the exocrine pancreas. Pancreas were harvested at the indicated times. (A and B) Western blot analysis of PERK activation in WT and IKO pancreas at days 4 and 8 (A) and 13 (B). HSP90, loading control. (C) Quantitation of A and B. (D) Western blot analysis of CHOP in cytosolic (C) and nuclear (N) fractions of the pancreas. CREB and GAPDH represent nuclear and cytosolic makers, respectively. (E) RT-PCR analysis of Xbp1 splicing with the quantitation of the ratio of Xbp1s to total Xbp1 mRNA shown below. (F) qPCR analysis of UPR and ERAD-related genes. (G) Western blot analysis of various chaperones in pancreas at day 13 with quantitation shown in H. Data are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 by Student t test. n = 3 mice each group except RT-PCR where n = 5. Representative data of two experiments shown.
Dilated ER and Smaller Zymogen Granules in the Exocrine Pancreas of Sel1LIKO Mice.
We next delineated the consequence of Sel1L deficiency at the organelle and molecular levels using transmission electron microscopy (TEM). The central lumen of the acinus was characterized by the presence of apical cell microvilli (Fig. 5A). Strikingly, rather than being long and thin sheet-like densely packed cisternae as in WT cells, the ER in the acinar cells of Sel1LIKO mice was extensively swollen and fragmented (Fig. 5A). The alterations of ER morphology in the pancreas of Sel1LIKO mice were distinct from those previously reported in the exocrine pancreas of XBP1−/− neonates (48), PERK−/− neonates (49), and Grp78+/− mice (50), likely because of different status of ERAD and UPR. Moreover, secretory zymogen granules that were highly prominent in the exocrine pancreas of WT mice were significantly reduced in size in the pancreas of Sel1LIKO mice compared with that in WT mice (Fig. 5 B and C). Thus, acute Sel1L deficiency leads to massively dilated and fragmented ER in the exocrine pancreas with smaller zymogen granules.
Fig. 5.
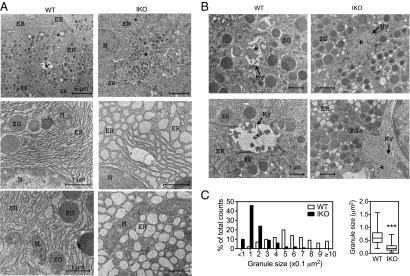
Dilated ER and smaller zymogen granules in the exocrine pancreas of Sel1LIKO mice. Pancreas was harvested at day 12. (A) Representative TEM images of exocrine pancreas of WT and IKO mice showing a profound dilation of the ER. (B) Representative TEM images of exocrine pancreas of WT and IKO mice showing smaller secretory granules. Asterisks indicate the acinar lumen; N, nucleus; M, mitochondrion; ZG, zymogen granules; ER, endoplasmic reticulum; and Mv, Microvillus. Scale bar shown. (C) Quantitation of granule sizes in 100 granules each genotype in histogram (Left) and Whiskers plot (Right). In the Whiskers plot, the medium, the 25th to 75th percentiles and the minimum to maximum range of granule sizes are shown. n = 3 mice each group. Data are mean ± SEM. ***P < 0.001 by Student t test.
Reduced Polysome Association and Increased Ribosomal Aggregation in the Exocrine Pancreas of Sel1LIKO Mice.
Dilated ER lumen and increased eIF2α phosphorylation suggested an altered translation rates in Sel1LIKO pancreas. Indeed, translation was reduced in the pancreas of Sel1LIKO mice, as demonstrated by reduced phosphorylation of p70 S6 ribosomal kinase (S6K), a downstream target of AKT and mammalian target of rapamycin (mTOR) (30) (Fig. 6A). Polysome profiling using the kidneys of Sel1LIKO mice revealed increased 80S monosomal peak along with a dramatic reduction in the number of polysomes (Fig. 6B). The ratio of polysomes (P) to monosomes (M) reduced by over 50% in the Sel1LIKO tissue. Multiple attempts to detect polysomes in the pancreas failed, likely because of the presence of large amount of pancreatic RNase. Thus, these data indicate that acute loss of Sel1L reduces polysome association, and hence attenuates global translation.
Fig. 6.
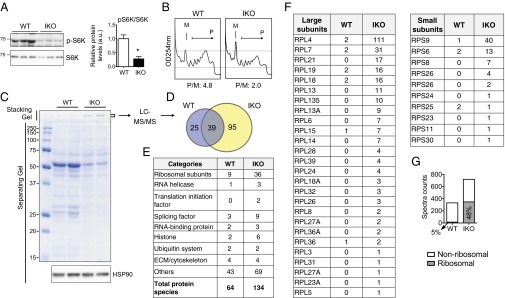
Reduced translation and increased ribosomal aggregation in the exocrine pancreas of Sel1LIKO mice. (A) Western blot analysis of (p)-S6K in of pancreas at day 13. Quantitation shown on the right. Data are mean ± SEM. *P < 0.05 by Student t test. (B) Polysome profiling analysis of the kidneys of WT and IKO mice at day 13. M, monosomes; P, polysomes; P/M, ratio of polysomes to monosomes based on quantitation of area under curve of each fraction. (C) Coomassie blue-stained SDS/PAGE gel of WT and IKO pancreatic lysates (loaded at an equal tissue weight) at day 13. Note stronger signals in the stacking gel of the IKO samples, which were sliced and subjected to LC-MS/MS analysis (D–G). HSP90, a loading control. (D) Venn diagram showing the overlap of hits in WT and IKO samples identified by the LC-MS/MS analysis. Complete list of hits shown in Table S1. (E) Functional categories of the hits. (F) Spectra counts of ribosomal subunits. RPL and RPS, 60S and 40S ribosomal subunits, respectively. (G) Diagram showing spectral counts of ribosomal and nonribosomal proteins. The percent of counts from ribosomal proteins were 5% in WT (15 of 332) and 48% in IKO (348 of 720), which accounted for the increased total spectral counts in IKO sample.
Translation attenuation was very obvious when the same amount of total lysates (per gram of tissue) prepared in 0.5% mild detergent Nonidet P-40 were separated on SDS/PAGE (Fig. 6C). Intriguingly, we noticed a strong band in the stacking gel of Sel1LIKO samples, which likely represents soluble aggregates (Fig. 6C). LC-MS/MS analysis of the band identified a total of 134 proteins in Sel1LIKO vs. 64 in WT samples, with 39 overlapping hits (Fig. 6D). Lists of total hits with spectra counts are shown in Table S1. Strikingly, there were 36 distinct ribosomal proteins present in Sel1LIKO samples (vs. 9 in WT), which included 10 small and 26 large ribosomal subunits (vs. 3 and 6 in WT, respectively) (Fig. 6 E and F). The percent of spectral counts contributed by ribosomal proteins was nearly 50% in Sel1LIKO sample vs. 5% in the WT sample, which accounted for the increase of total spectral counts in Sel1LIKO samples (Fig. 6G). In addition, eIF3A, a core stress granule constituent (51), as well as several helicases and splicing factors were detected (Fig. 6E and Table S1). The presence of ribosomal subunits as well as other RNA processing components likely marks the formation of stress granules in the pancreas of Sel1LIKO mice, which indeed is often associated with eIF2α phosphorylation and translational arrest (51). Thus, acute Sel1L deficiency reduces translation and induces ribosomal aggregation, likely in the form of stress granules.
A Cell-Culture Model of Acute Sel1L Deficiency.
To further establish the cell-autonomous effect of Sel1L and to allow a temporal regulation of Sel1L function in vitro, we generated f/f;ERCre+ immortalized MEFs, termed Sel1LIKO MEFs. The f/f;ERCre− MEFs treated with the tamoxifen analog 4-hydroxytamoxifen (4-OHT) or f/f;ERCre+ MEFs treated with vehicle were used interchangeably as controls. Starting at the first passage (p1) following 4-OHT treatment, Sel1LIKO MEFs exhibited decreased growth and cell viability, and at p2 growth and viability of Sel1LIKO MEFs were greatly attenuated (Fig. 7 A and B). Both Sel1L and Hrd1 protein levels were progressively reduced with passage of f/f;ERCre+ MEFs treated with 4-OHT (Fig. 7C), further supporting the notion that Sel1L is required for Hrd1 stability. Degradation of a known substrate null Hong Kong variant of α1-antitrypsin (A1ATNHK)-GFP was attenuated in p1 Sel1LIKO MEFs (Fig. 7D). Moreover, ER stress was activated upon the loss of Sel1L, as illustrated by a fourfold increase of Xbp1 mRNA splicing, increased Brefeldin A (BFA)-marked ER/Golgi mass and eIF2α phosphorylation (Fig. 7 E–G). Conversely, translation in Sel1LIKO MEFs was attenuated as shown by phosphorylation of ribosomal S6 protein (Fig. 7G) and polysome profiling (Fig. 7H). It is worth pointing out that the effect of Sel1L deficiency on translation is modest in comparison with that of WT MEFs treated with 300 nM thapsigargin (Tg, an ER stress inducer) for 2 h. Thus, acute loss of Sel1L in vitro alters ER homeostasis and negatively affects cell growth and translation, thus representing a great model to investigate the function and physiological substrates of the Sel1L–Hrd1 ERAD complex as well as primary cellular response to the accumulation of misfolded proteins.
Fig. 7.

Sel1L is indispensable for Hrd1 stability, cellular growth, and ER homeostasis in vitro. (A) Microscopic images of f/f;ERCre− and f/f;ERCre+ MEFs at day 4 of different passages (p). p0, vehicle (ethanol) treated; p1/2, 4-OHT treated. Quantitation of cell number from one experiment (representative of two) shown on the right. (B) Cell viability analysis as measured by CCK-8 of MEFs at day 4 of each passage. (C) Western blot analysis of Sel1L and Hrd1 in f/f;ERCre+ MEFs at day 4 of each passages. Quantitation from one representative experiment of at least three repeats shown below the gel. (D) Flow cytometric analysis of transfected A1ATNHK-GFP in p0 or p1 Sel1LIKO MEFs. GFP+ cells from each passage were gated and overlaid in the histogram (n = 3). Representative microscopic images shown on the right. Scale bar, 50 μm. (E) Xbp-1 mRNA splicing of MEFs at day 4. Quantitation of the percent of Xbp1s in total Xbp1 mRNA shown on the right. (F) Flow cytometric analysis of the ER/Golgi volume in MEFs stained with BFA-Bodipy at each passage. Quantitation of mean fluorescence intensity shown on the right. (G) Western blot analysis of eIF2α and S6 phosphorylation in MEFs at day 4 of each passage. HSP90, a loading control. Quantitation from three experiments shown on the right. (H) Polysome profiling of f/f;ERCre+ MEFs at p0 and p1. Tg (300 nM, 2 h)-treated p0 MEFs were included as a positive control for ER stress. M, monosomes; P, polysomes; P/M, ratio of polysomes to monosomes based on the quantitation of area under curve of respective fraction. Data are mean ± SEM. *P < 0.05, **P < 0.01, ***. P < 0.01 compared with WT. Representative data of at least two experiments shown.
Discussion
Our data herein demonstrate that Sel1L has an indispensable role in mammalian ERAD and ER homeostasis. Acute Sel1L deletion results in a dramatic down-regulation of Hrd1 and progressive activation of the UPR, which leads to massive dilation and fragmentation of the ER and promotes cell death. Moreover, our data further show that acute Sel1L deficiency reduces translation efficiency and induces ribosomal aggregation. Consistently, the effect of Sel1L on Hrd1 stability, ER stress, translation, and cell death were observed in vitro in Sel1LIKO MEFs. Further pointing to its physiological importance, Sel1LIKO adult mice exhibit pancreatic atrophy and die within 3 wk. Thus, Sel1L is indispensable for Hrd1 stability, ERAD, and ER homeostasis, which is essential for pancreas secretory function and survival of adult animals.
Hrd3/Sel1L is a highly conserved ER-resident type 1 single-transmembrane protein responsible for the degradation of a subset of misfolded ER proteins (13, 14, 17, 19). The physiological importance of Sel1L in mammalian ERAD has yet to be demonstrated. Our data herein show that Sel1L is indispensable for mammalian ERAD and ER homeostasis. Like Hrd3 in yeast, Sel1L controls the stability of Hrd1 in multiple tissues as well as MEFs. Acute loss of Sel1L results in a reduction of overall Hrd1-Sel1L ERAD activity and concurrent induction of UPR and dilation of ER in the pancreas and MEFs. The discrepancies between our and other recent studies on Sel1L-mediated regulation of Hrd1 stability (15, 25, 26) are likely because of the difference in cell types or residual Sel1L levels in previous studies. Thus, our data show that the effect of Sel1L on Hrd1 stability and the importance of Sel1L in ERAD are evolutionarily conserved from yeast to mammals. Because of significant down-regulation of Hrd1 in Sel1L-deficient mice, our mouse model is indeed deficient for both Sel1L and Hrd1. Therefore, the Sel1LIKO mouse model provides a unique model system to further delineate the function of Sel1L and Sel1L-Hrd1 ERAD in vivo.
It is well known that Hrd3/Sel1L–Hrd1 belongs to a larger macromolecular complex involved in ERAD, including EDEM1, OS9, and XTP3B (16, 40–45). These cofactors play an important role in the initial recognition and recruitment of substrate to the Hrd3/Sel1L–Hrd1 complex. Our data show that Sel1L–Hrd1 deficiency in the pancreas increases the protein levels of endogenous OS9 and EDEM1, but not XTP3B. These changes are uncoupled from the transcriptional regulation, suggesting that OS9 and EDEM1 may be degraded through the Sel1L–Hrd1 ERAD complex, likely together with misfolded proteins. Indeed, ERAD-mediated degradation of OS9 in the transfected cell culture system has been recently reported (23). Given the importance of EDEM1 and OS9 in the substrate recognition and recruitment, we propose that Hrd1-mediated degradation of EDEM1 and OS9 may serve as an important self-regulatory mechanism to prevent the overactivation of ERAD, which may represent one form of “ERAD tuning” (29).
Why is the pancreas so sensitive to Sel1L dysfunction? Across all tissues, the pancreas is the most active in protein synthesis, with over 90% of the newly synthesized proteins being targeted to the secretory pathway (52). In response to food intake, acinar cells secrete a large amount of pancreatic juice containing electrolytes and a variety of digestive enzymes, including pancreatic lipase and α-amylase (53, 54). It is thus well established that ER homeostasis is critical for pancreatic function. Animal models with defects in UPR such as X-box binding protein 1 (Xbp1)- (48) and Perk-deficient mice (49) all exhibit profound defects in the exocrine pancreas. Interestingly, for reasons that are currently unclear, IRE1α-deficient adult mice exhibit only mild defects in exocrine pancreas (55). We showed recently that 2-h refeeding following overnight fasting causes UPR activation in pancreas (56). The Hrd1–Sel1L ERAD complex may play an important role in the clearance of misfolded proteins in the ER and, hence is indispensable for the reset of ER homeostasis during refeeding. Loss of the Sel1L–Hrd1 complex alters ER homeostasis and leads to persistent UPR. This UPR may induce the expression of ER chaperones and reduce translation, thereby resetting ER homeostasis. However, as the accumulation of a subset of misfolded proteins continues, it overwhelms the UPR system, which causes ribosomal aggregation and ultimately leads to cell death in the pancreas. In yeast, accumulation of misfolded protein within the ER lumen causes persistent UPR activation, leading to oxidative stress and ultimately cell death (57). How other mammalian cell types respond to acute Sel1L deficiency remains an intriguing question. Further investigation and comparisons of tissue and cell type-specific role of Sel1L will likely provide key insights into the physiological role of ERAD.
Sel1L function in vivo is likely to be mediated through the E3 ligase Hrd1. Sel1L, however, may have Hrd1-independent functions in other cellular processes (15). Cellular Sel1L is five- to sevenfold more abundant than Hrd1 (58). As Sel1L and Hrd1 exist in a 1:1 stoichiometric complex in ERAD (14, 18), Sel1L may exist in an Hrd1-independent complex. Indeed, a recent study showed that Sel1L may form a Sel1L–LC3-I complex to deliver certain proteins involved in ERAD, such as OS9 for lysosomal degradation, thereby controlling the efficacy of ERAD (29). Our microarray showed that Sel1L deficiency alters the expression of over 2,000 genes in the pancreas. Although many of these genes are linked to UPR and cell death, others are related to nutrient metabolism, including carbohydrate and lipids. Whereas lipid metabolism may be correlated with XBP1s activation in Sel1LIKO cells (59), how carbohydrate metabolism is altered by Sel1L deficiency remains unclear. Thus, more studies are required to decipher whether Sel1L exerts an Hrd1-independent function in vivo. One of the foreseeable challenges, however, is to distinguish a novel function of Sel1L from its effect on ERAD and ER homeostasis.
Our data herein show that acute loss of Sel1L increases eIF2α phosphorylation and reduces polysome-mediated translation. Serendipitously, our biochemical fractionation of soluble pancreatic aggregates, coupled with LC-MS/MS, reveals many ribosome subunits, both small and large, in the soluble aggregates of Sel1L-deficient pancreas. Strikingly, several RNA binding proteins and initiation factors including helicases, splicing factors, and eIF3A are present in the soluble aggregates as well, leading to the speculation that Sel1L deficiency may lead to the formation of stress granules in the pancreas. Stress granules are often associated with translational arrest induced by eIF2α phosphorylation and contain nontranslating mRNAs, translation initiation components, and many additional proteins modulating mRNA metabolism (51). Phosphorylation of eIF2α prevents the delivery of initiator methionine to the 40S preinitiation complex, which stalls translation initiation. The stalled preinitiation complex, together with mRNAs and other RNA binding proteins, forms a highly dynamic cytosolic structure known as stress granules, which not only protect mRNAs but also make them readily available upon the relief of the stress (51). Although stress granules are often associated with eIF2α phosphorylation, whether it occurs in vivo in response to physiological ER stress remains unknown. Thus, our data suggest that stress granules may form in response to ERAD deficiency and UPR activation in vivo.
As stress granules are highly dynamic and contain weakly aggregated particles, biochemical fractionation of stress granules has been challenging (60). To date, contents of stress granules have only been defined using immunostaining. Thus, our serendipitous finding may open the door for future biochemical purification of stress granules and further characterization of their contents. In addition, because stress granules are thought to contain only small ribosomal subunits (51), the identification of many large ribosomal subunits in the soluble aggregates of the Sel1L-deficient pancreas is interesting. We speculate that, similar to the small subunits, these large subunits may aggregate as well when the small units are not available, and become available when the stress is relieved. Indeed, studies have shown that the composition of stress granules is stress-specific (51, 61). Thus, aggregation of small and large ribosomal subunits may be associated with Sel1L deficiency-induced stress granules. The nature of this event deserves further investigation.
In summary, our study establishes the physiological significance of Sel1L and the Hrd1–Sel1L ERAD complex in vivo. Sel1L is indispensable for Hrd1 stability and ERAD function, and likely together with Hrd1, Sel1L regulates ER homeostasis, UPR, and translation. The inducible Sel1L animal and cell models are invaluable tools to further delineate tissue- and cell-type–specific role of Sel1L and Sel1L–Hrd1 ERAD in vivo, to identify its physiological substrate, and lastly, to characterize cellular response to constitutive UPR in vivo.
Materials and Methods
Mice.
The Sel1ltm1e(KOMP)Wtsi (Sel1Lflox/+) ES cells on the C57BL/6N background were purchased from the KOMP Repository Project (ID CSD44577, University of California, Davis, www.komp.org/geneinfo.php?project=44577) (62). Exon 6 of the Sel1L gene was flanked with two loxP sites (floxed). The lacZ-neo cassette was deleted by crossing the animals onto the βActin-FLPe deleter mice on the C57BL/6J background (JAX 003800). The resulting Sel1Lflox/+ animals were intercrossed to generate Sel1Lflox/flox (f/f) mice. The Sel1Lflox/flox mice were then crossed with actin-promoter driven estrogen receptor-Cre fusion protein transgenic (ERCre) mice on the C57BL/6J background (JAX 004781). The last cross generated f/f;ERCre+ and the control cohort f/f;ERCre− littermates at 1:1 ratio. Mice were fed on low-fat diet consisted of 13% fat, 67% carbohydrate, and 20% protein (Harlan Teklad 2914). Genotyping primer sequences are listed in Table S2. WT B6 mice were purchased from JAX and bred in our mouse facility. All animal procedures have been approved by the Cornell Institutional Animal Care and Use Committee (#2007–0051).
Tamoxifen-Mediated Sel1L Deletion and Survival Analysis.
Tamoxifen (Sigma T5648) was dissolved in sunflower oil at 5 mg/mL. Age-matched littermates at the age of 8–20 wk were injected intraperitoneally with tamoxifen at the volume of 5 μL/g body weight daily for three consecutive days. Body weights were monitored daily. For humane reasons and guidelines from Institutional Animal Care and Use Committee, when body weight drops below 80% of the starting body weight, mice were announced “dead” and euthanized.
TEM.
Mice at day 12 after tamoxifen injection were killed at 9:00–10:00 AM ad libitum. The pancreas was immediately sliced into 1- to 2-mm3 pieces and fixed, stained, dehydrated, and embedded in Poly/bed 812 (Polysciences). Fixation and embedding processes were carried out at the Cornell Center for Materials Research Core Facility. Embedded samples were cut with Leica Ultracut Ultramicrotome system and images were taken using JEM-1400 TEM on a fee-for-service basis at the Electron Microscopy and Histology Core Facility at Weill Cornell Medical College.
Histological Analysis and Immunohistochemistry Staining.
Tissues were fixed in 10% (vol/vol) neutralized formalin, and processed by the Cornell Histology Core Facility for paraffin embedding, sectioning, and H&E staining on a fee-for-service basis. Tissue section examination was performed by our collaborating pathologist in this study (G.E.D.). For immunohistochemistry staining (IHC), paraffin-embedded pancreatic sections were rehydrated, boiled in 1 mM EDTA for antigen retrieval, and stained with Histostain kit and DAB substrate from Invitrogen. Antibodies used for IHC were: α-Amylase (1:200), Sel1L (1:200), and Ki-67 (1:50) from Abcam, and Hrd1 (rabbit, 1:200) from Novus Biologicals. Staining of insulin and glucagon was performed by the Cornell Histology Core Facility. H&E and IHC sections were scanned using the Aperio Scanscope and pictures were taken at various magnifications.
RNA Extraction, RT-PCR for Xbp1 mRNA Splicing, and Quantitative PCR.
To extract RNA from the pancreas, mice were anesthetized and pancreas tissues were perfused locally with RNAlater reagent (Qiagen). Tissues were then excised and soaked in RNAlater on ice for stabilization. Total RNA was extracted using TRIzol and RNA miniprep kit (Sigma RTN350) with DNaseI digestion (Roche). RNA quality was determined by measuring the OD260/280 and visualized on an agarose gel. RT-PCR was performed as previously described (63). Percent of Xbp1 mRNA splicing defined as the ratio of Xbp1s level to total Xbp1 (Xbp1u + Xbp1s) levels was quantitated using the ImageLab software. Reactions without cDNA were included as negative controls to ensure the specificity. All quantitative PCR (qPCR) data were normalized to ribosomal l32 gene in the corresponding sample. qPCR primer sequences are listed in Table S2. Xbp1 mRNA splicing in Sel1LIKO MEFs was analyzed as described above.
Microarray and Data Analysis.
RNA quality and concentration was examined using the RNA 6000 Nano kit on the Agilent 2100 bioanalyzer (Fig. S4A). The cDNA array of pancreatic RNA was performed as previously described (64). A fold-change of 1.5 and q value < 0.01 were set as cutoff for differential regulation and analyzed by Ingenuity analysis. The microarray datasets have been submitted to Gene Expression Omnibus GSE52929.
Polysome Profiling of Tissues and Cells.
Sel1LIKO MEFs were treated with vehicle or 4-OHT for 3 d, and harvested at 80% confluence from 10 10-cm plates. Cells were treated with vehicle or Tg (300 nM) for 2 h and 0.1 mg/mL cycloheximide (CHX) was added at the last 3 min of treatment. The plates were washed with ice-cold PBS containing 0.1 mg/mL CHX, and cells were scraped in 1 mL PBS. After centrifugation at 500 × g, cell pellet was lysed in 1 mL lysis buffer [20 mM Tris pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 250 µg/mL CHX, 1% Triton-X100, 100 µg/mL heparin, 120 U/mL RNAseOUT (Invitrogen) and 0.5% protease inhibitor mixture (Sigma)] and homogenized with Dounce grinder. Lysate was incubated on ice for 10 min and centrifuged at 12,000 × g for 10 min, and 800 μL of the supernatant was layered onto a 12 mL 15–45% (wt/vol) sucrose gradient prepared using the Gradient Master (BioComp Instruments) with the polysome buffer (20 mM Tris pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT). After centrifugation at 32,000 rpm for 2.5 h in a SW40 rotor (LE-80K, Beckman-Coulter, Pasadena, CA), the gradients were fractioned at 1.5 mL/min with an automated fractionation system (ISCO), which continuously monitored absorbance values at 254 nm. For kidneys, mice on day 9 were anesthetized and the bottom half of the left kidney was removed, homogenized with Dounce grinder in 1 mL lysis buffer (with 200 U/mL RNAseOUT). Lysate was cleared by centrifugation and fractioned with 15–45% sucrose gradients as described above. Our multiple attempts using pancreas for polysome profiling failed likely because of the large amount of tissue RNases.
Nonidet P-40 Fractionation for Pancreas Tissue.
Frozen pancreas tissue from WT and Sel1LIKO mice at day 13 (∼15 mg) was weighed and homogenized in Nonidet P-40 lysis buffer (50 mM Tris⋅HCl pH 8.0, 0.5% Nonidet P-40, 150 mM NaCl, 5 mM MgCl2) supplied with protease inhibitor (Sigma). The lysate volume was normalized by tissue weight at 500 µL per 10 mg tissue. The lysate was centrifuged at 12,000 × g for 10 min and the supernatant was collected as NP40S fraction. The pellet weight was measured and presented as the percent of the total tissue weight. The pellet was then resuspended in 1× SDS sample buffer (50 mM Tris⋅Cl, 2% (wt/vol) SDS, 0.29 M 2-mecaptoethanol, 10% (vol/vol) glycerol, 0.01% bromophenyl blue) with the volume normalized to initial tissue weight, heated at 95 °C for 30 min and collected as the NP40P fraction. The NP40S and NP40P fractions were subsequently analyzed by Western blot or Coomassie Brilliant blue staining (see below).
Mass Spectrometry Analysis of Soluble Protein Aggregates in Pancreas.
The NP40S fraction was mixed with 5× SDS sample buffer. Samples were heated at 65 °C for 5 min and 20 µL was loaded onto 12% SDS/PAGE. Gel was incubated with Coomassie Brilliant blue (0.5 g Coomassie Brilliant blue R-250 in 450 mL methanol, 450 mL Milli-Q water, and 100 mL acetic acid) with gentle rocking for 30 min at room temperature, followed by two washes with Milli-Q water 5 min each. Gel was destained in destaining solution [45% (vol/vol) methanol, 45% Milli-Q water, 10% acetic acid] that was changed every 30 min until bands could be clearly visualized. Protein bands in the stacking gels were considered as soluble protein aggregates, which were excised and subjected to an in-gel trypsin digestion. The resultant peptide mixtures were pressure-loaded onto a C18 reverse-phase capillary column and analyzed by online nanoflow LC-MS/MS on an Agilent 1200 quaternary HPLC system (Agilent) connected to an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific) using a 2-h gradient. MS data were searched with ProLuCID on IP2 (Integrated Proteomics Applications) against a mouse UniProt database, and filtered using DTASelect2 with a 5 ppm Δ mass cutoff of the peptide masses and a false-positive rate below 1%.
Generation and Characterization of Sel1LIKO MEFs.
MEFs were generated from day 13.5 embryos of mating between f/f;ERCre+ and f/f;ERCre− mice. Briefly, head and liver from embryos at day 13.5 were carefully removed (saved for genotyping), and the remaining part was minced and digested with 2 mL of trypsin at 37 °C for 10 min, vigorously pipetted into single-cell suspension, and cultured in a 10-cm dish. After two passages, MEFs were immortalized by transduction with the pBabe-SV40 retroviral system (gifts from Robert Weiss, Cornell University, Ithaca, NY) and selected with puromycin, as previously described (63). To induce Sel1L deletion, f/f;ERCre+ or control f/f;ERCre− (WT) MEFs were treated with 400 nM 4-OHT (Sigma H7904, dissolved in 100% ethanol). Every 4 d, cells were trypsinized, counted and plated at 2 × 105 cells per 10-cm dish as p1 and p2; p0 refers to parental cells without 4-OHT treatment. Cell viability assays were carried out using the Cell Counting Kit-8 (CCK-8) per supplier’s protocol in 96-well plates (Dojindo). BFA-Bodipy staining of MEFs was performed as previously described (65). For transfection with the model substrate A1ATNHK-GFP, f/f;ERCre+ MEF cells in six-well plates were transfected with plasmids encoding A1ATNHK-GFP using Lipofectamine 2000 per supplier’s protocol (Invitrogen). Six hours later, transfection medium was replaced with fresh culture medium containing either vehicle or 4-OHT for 3 d before analysis. Flow cytometric analysis was performed using BD FACSCalibur and gated on a GFP+ population. Cells were also examined using the inverted Nikon Eclipse Ti microscopy (Nikon).
Statistical Analysis.
Results are expressed as mean ± SEM unless indicated otherwise. Comparisons between groups were made by unpaired two-tailed Student t test, where P < 0.05 was considered as statistically significant. Survival curves were compared by the log-rank (Mantel–Cox) test. All experiments were repeated at least two to three times, or performed with several independent biological samples, and representative data are shown.
Supplementary Material
Acknowledgments
We thank Drs. Yihong Ye, Robert Weiss, and Marc Montminy for reagents, Yihong Ye for suggestions and sharing the protocols, Xiangwei Gao for help with polysome profiling analysis, Changyou Lin for the use of scanscope, John Grazul, and Lee Cohen-Gould for TEM, Haibo Sha for sharing the mice, Hana Kim for the multiplex analysis of the insulin and glucagon, Cindy Wang for the assistance with genotyping and Western blot, and other members of L.Q. laboratory for comments and suggestions. The Cornell Center for Materials Research for provided assistance in the processing of transmission electron microscopy samples (National Science Foundation DMR-1120296). This work was supported by National Institutes of Health Grants CA168997 (to J.W.L.), R21AI085332 (to G.E.D.), and P41 GM103533 (to J.R.Y.); the Netherlands Nutrigenomics Centre (S.K.); Chinese National Science Foundation Grant 31371391 (to Q.L.); and Grant R01DK082582 and American Diabetes Association 1-12-CD-04 (to L.Q.). S.S. is an International Student Research Fellow of the Howard Hughes Medical Institute (59107338). L.Q. is the recipient of the Junior Faculty and Career Development Awards from the American Diabetes Association.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE52929).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1318114111/-/DCSupplemental.
References
- 1.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Olzmann JA, Kopito RR, Christianson JC. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb Perspect Biol. 2013;5(9) doi: 10.1101/cshperspect.a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brodsky JL. Cleaning up: ER-associated degradation to the rescue. Cell. 2012;151(6):1163–1167. doi: 10.1016/j.cell.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hampton RY, Sommer T. Finding the will and the way of ERAD substrate retrotranslocation. Curr Opin Cell Biol. 2012;24(4):460–466. doi: 10.1016/j.ceb.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 5.Walter P, Ron D. The unfolded protein response: From stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 6.Hetz C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 7.Travers KJ, et al. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101(3):249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 8.Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol. 2000;2(7):379–384. doi: 10.1038/35017001. [DOI] [PubMed] [Google Scholar]
- 9.Dougan SK, et al. Derlin-2-deficient mice reveal an essential role for protein dislocation in chondrocytes. Mol Cell Biol. 2011;31(6):1145–1159. doi: 10.1128/MCB.00967-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yagishita N, et al. Essential role of synoviolin in embryogenesis. J Biol Chem. 2005;280(9):7909–7916. doi: 10.1074/jbc.M410863200. [DOI] [PubMed] [Google Scholar]
- 11.Francisco AB, et al. Deficiency of suppressor enhancer Lin12 1 like (SEL1L) in mice leads to systemic endoplasmic reticulum stress and embryonic lethality. J Biol Chem. 2010;285(18):13694–13703. doi: 10.1074/jbc.M109.085340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eura Y, et al. Derlin-1 deficiency is embryonic lethal, Derlin-3 deficiency appears normal, and Herp deficiency is intolerant to glucose load and ischemia in mice. PLoS ONE. 2012;7(3):e34298. doi: 10.1371/journal.pone.0034298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hampton RY, Gardner RG, Rine J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1996;7(12):2029–2044. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardner RG, et al. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol. 2000;151(1):69–82. doi: 10.1083/jcb.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christianson JC, et al. Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol. 2012;14(1):93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christianson JC, Shaler TA, Tyler RE, Kopito RR. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10(3):272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mueller B, Lilley BN, Ploegh HL. SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J Cell Biol. 2006;175(2):261–270. doi: 10.1083/jcb.200605196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lilley BN, Ploegh HL. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc Natl Acad Sci USA. 2005;102(40):14296–14301. doi: 10.1073/pnas.0505014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mueller B, Klemm EJ, Spooner E, Claessen JH, Ploegh HL. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc Natl Acad Sci USA. 2008;105(34):12325–12330. doi: 10.1073/pnas.0805371105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grant B, Greenwald I. The Caenorhabditis elegans sel-1 gene, a negative regulator of lin-12 and glp-1, encodes a predicted extracellular protein. Genetics. 1996;143:237–247. doi: 10.1093/genetics/143.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sundaram M, Greenwald I. Suppressors of a lin-12 hypomorph define genes that interact with both lin-12 and glp-1 in Caenorhabditis elegans. Genetics. 1993;135:765–783. doi: 10.1093/genetics/135.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, et al. Processing and turnover of the Hedgehog protein in the endoplasmic reticulum. J Cell Biol. 2011;192(5):825–838. doi: 10.1083/jcb.201008090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tyler RE, et al. Unassembled CD147 is an endogenous endoplasmic reticulum-associated degradation substrate. Mol Biol Cell. 2012;23(24):4668–4678. doi: 10.1091/mbc.E12-06-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horimoto S, et al. The unfolded protein response transducer ATF6 represents a novel transmembrane-type endoplasmic reticulum-associated degradation substrate requiring both mannose trimming and SEL1L protein. J Biol Chem. 2013;288(44):31517–31527. doi: 10.1074/jbc.M113.476010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iida Y, et al. SEL1L protein critically determines the stability of the HRD1-SEL1L endoplasmic reticulum-associated degradation (ERAD) complex to optimize the degradation kinetics of ERAD substrates. J Biol Chem. 2011;286(19):16929–16939. doi: 10.1074/jbc.M110.215871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams JM, Inoue T, Banks L, Tsai B. The ERdj5-Sel1L complex facilitates cholera toxin retrotranslocation. Mol Biol Cell. 2013;24(6):785–795. doi: 10.1091/mbc.E12-07-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernasconi R, Galli C, Calanca V, Nakajima T, Molinari M. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J Cell Biol. 2010;188(2):223–235. doi: 10.1083/jcb.200910042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cattaneo M, et al. SEL1L and HRD1 are involved in the degradation of unassembled secretory Ig-mu chains. J Cell Physiol. 2008;215(3):794–802. doi: 10.1002/jcp.21364. [DOI] [PubMed] [Google Scholar]
- 29.Bernasconi R, et al. Role of the SEL1L:LC3-I complex as an ERAD tuning receptor in the mammalian ER. Mol Cell. 2012;46(6):809–819. doi: 10.1016/j.molcel.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 30.Zhang W, et al. ER stress potentiates insulin resistance through PERK-mediated FOXO phosphorylation. Genes Dev. 2013;27(4):441–449. doi: 10.1101/gad.201731.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Biunno I, et al. SEL1L a multifaceted protein playing a role in tumor progression. J Cell Physiol. 2006;208(1):23–38. doi: 10.1002/jcp.20574. [DOI] [PubMed] [Google Scholar]
- 32.Cardano M, et al. mSEL-1L (Suppressor/enhancer Lin12-like) protein levels influence murine neural stem cell self-renewal and lineage commitment. J Biol Chem. 2011;286(21):18708–18719. doi: 10.1074/jbc.M110.210740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S, Francisco AB, Munroe RJ, Schimenti JC, Long Q. SEL1L deficiency impairs growth and differentiation of pancreatic epithelial cells. BMC Dev Biol. 2010;10:19. doi: 10.1186/1471-213X-10-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ban Y, et al. SEL1L microsatellite polymorphism in Japanese patients with autoimmune thyroid diseases. Thyroid. 2001;11(4):335–338. doi: 10.1089/10507250152039064. [DOI] [PubMed] [Google Scholar]
- 35.Kyöstilä K, et al. A SEL1L mutation links a canine progressive early-onset cerebellar ataxia to the endoplasmic reticulum-associated protein degradation (ERAD) machinery. PLoS Genet. 2012;8(6):e1002759. doi: 10.1371/journal.pgen.1002759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saltini G, et al. A novel polymorphism in SEL1L confers susceptibility to Alzheimer’s disease. Neurosci Lett. 2006;398(1–2):53–58. doi: 10.1016/j.neulet.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 37.Batt RM. Exocrine pancreatic insufficiency. Vet Clin North Am Small Anim Pract. 1993;23(3):595–608. doi: 10.1016/s0195-5616(93)50308-x. [DOI] [PubMed] [Google Scholar]
- 38.Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4(12):e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281(40):30299–30304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hosokawa N, et al. A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2001;2(5):415–422. doi: 10.1093/embo-reports/kve084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oda Y, Hosokawa N, Wada I, Nagata K. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science. 2003;299(5611):1394–1397. doi: 10.1126/science.1079181. [DOI] [PubMed] [Google Scholar]
- 42.Cormier JH, Tamura T, Sunryd JC, Hebert DN. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol Cell. 2009;34(5):627–633. doi: 10.1016/j.molcel.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hosokawa N, Kamiya Y, Kamiya D, Kato K, Nagata K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J Biol Chem. 2009;284(25):17061–17068. doi: 10.1074/jbc.M809725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126(2):349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- 45.Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126(2):361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- 46.Sakakibara A, Furuse M, Saitou M, Ando-Akatsuka Y, Tsukita S. Possible involvement of phosphorylation of occludin in tight junction formation. J Cell Biol. 1997;137(6):1393–1401. doi: 10.1083/jcb.137.6.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Q, et al. A ubiquitin ligase-associated chaperone holdase maintains polypeptides in soluble states for proteasome degradation. Mol Cell. 2011;42(6):758–770. doi: 10.1016/j.molcel.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005;24(24):4368–4380. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harding HP, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7(6):1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 50.Ye R, et al. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes. 2010;59(1):6–16. doi: 10.2337/db09-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kedersha N, Ivanov P, Anderson P. Stress granules and cell signaling: more than just a passing phase? Trends Biochem Sci. 2013;38(10):494–506. doi: 10.1016/j.tibs.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scheele GA, Kern HF. Cellular compartmentation, protein processing and secretion in the exocrine pancreas. In: Go VL, et al., editors. The Pancreas: Biology, Pathobiology and Disease. NY: Raven Press; 1993. pp. 121–150. [Google Scholar]
- 53.Novak I. Purinergic receptors in the endocrine and exocrine pancreas. Purinergic Signal. 2008;4(3):237–253. doi: 10.1007/s11302-007-9087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Case RM. Synthesis, intracellular transport and discharge of exportable proteins in the pancreatic acinar cell and other cells. Biol Rev Camb Philos Soc. 1978;53(2):211–354. doi: 10.1111/j.1469-185x.1978.tb01437.x. [DOI] [PubMed] [Google Scholar]
- 55.Iwawaki T, Akai R, Kohno K. IRE1α disruption causes histological abnormality of exocrine tissues, increase of blood glucose level, and decrease of serum immunoglobulin level. PLoS ONE. 2010;5(9):e13052. doi: 10.1371/journal.pone.0013052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang L, et al. A Phos-tag-based approach reveals the extent of physiological endoplasmic reticulum stress. PLoS ONE. 2010;5(7):e11621. doi: 10.1371/journal.pone.0011621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haynes CM, Titus EA, Cooper AA. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol Cell. 2004;15(5):767–776. doi: 10.1016/j.molcel.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 58.Kislinger T, et al. Global survey of organ and organelle protein expression in mouse: Combined proteomic and transcriptomic profiling. Cell. 2006;125(1):173–186. doi: 10.1016/j.cell.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 59.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320(5882):1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Souquere S, et al. Unravelling the ultrastructure of stress granules and associated P-bodies in human cells. J Cell Sci. 2009;122(Pt 20):3619–3626. doi: 10.1242/jcs.054437. [DOI] [PubMed] [Google Scholar]
- 61.Buchan JR, Parker R. Eukaryotic stress granules: The ins and outs of translation. Mol Cell. 2009;36(6):932–941. doi: 10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skarnes WC, et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474(7351):337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sha H, et al. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 2009;9(6):556–564. doi: 10.1016/j.cmet.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun S, Xia S, Ji Y, Kersten S, Qi L. The ATP-P2X7 signaling axis is dispensable for obesity-associated inflammasome activation in adipose tissue. Diabetes. 2012;61(6):1471–1478. doi: 10.2337/db11-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.He Y, et al. Nonmuscle myosin IIB links cytoskeleton to IRE1α signaling during ER stress. Dev Cell. 2012;23(6):1141–1152. doi: 10.1016/j.devcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.