

Abstract
Retinoblastoma (Rb) protein is a tumor suppressor that is dysregulated in a majority of human cancers. Rb functions to inhibit cell cycle progression in part by directly disabling the E2F family of cell cycle-promoting transcription factors. Because the de novo synthesis of multiple glutamine-derived anabolic precursors is required for cell cycle progression, we hypothesized that Rb also may directly regulate proteins involved in glutamine metabolism. We examined glutamine metabolism in mouse embryonic fibroblasts (MEFs) isolated from mice that have triple knock-outs (TKO) of all three Rb family members (Rb-1, Rbl1, and Rbl2) and found that loss of global Rb function caused a marked increase in 13C-glutamine uptake and incorporation into glutamate and TCA cycle intermediates in part via upregulated expression of the glutamine transporter ASCT2 and the activity of glutaminase 1 (GLS1). The Rb-controlled transcription factor E2F-3 altered glutamine uptake by direct regulation of ASCT2 mRNA and protein expression, and E2F-3 was observed to associate with the ASCT2 promoter. We next examined the functional consequences of the observed increase in glutamine uptake and utilization and found that glutamine exposure potently increased oxygen consumption whereas glutamine deprivation selectively decreased ATP concentration in the Rb TKO MEFs but not the WT MEFs. In addition, TKO MEFs exhibited elevated production of glutathione from exogenous glutamine, and had increased expression of gamma-glutamylcysteine ligase relative to WT MEFs. Importantly, this metabolic shift towards glutamine utilization was required for the proliferation of Rb TKO MEFs but not for the proliferation of the WT MEFs. Last, addition of the TCA cycle intermediate α-ketoglutarate to the Rb TKO MEFs reversed the inhibitory effects of glutamine deprivation on ATP, GSH levels, and viability. Taken together, these studies demonstrate that the Rb/E2F cascade directly regulates a major energetic and anabolic pathway that is required for neoplastic growth.
Keywords: Retinoblastoma Protein, Metabolism, Glutamine, Cancer, Tumor Suppressor
Introduction
Neoplastic cells are characterized by un-controlled proliferation which depends on their ability to increase cellular metabolism in order to produce the ATP and reducing power to drive the synthesis of macromolecules required for cell division (1). Dysregulated metabolism has been extensively studied for several decades, with the primary focus directed at the capacity of tumor cells to activate aerobic glycolysis (2–5). This metabolic alteration ultimately leads to increased glycolytic flux to lactate and a decrease in glucose carbon availability for the TCA cycle and other downstream biochemical processes. Accordingly, neoplastic cells require nutrient sources such as glutamine to replenish the anaplerotic carbon in the TCA cycle necessary to maintain their rapid cell division (6–8). In tumor cells, glutamine carbon is utilized for mitochondrial TCA cycling, glutathione synthesis, and fatty acid production (6, 9–12). Importantly, glutamine restriction or inhibitors of glutaminolytic enzymes decrease cell proliferation both in vitro and in vivo (7, 13–15). Although both the proto-oncogene MYC and the tumor suppressor p53 have been found to contribute to the control of glutamine uptake and glutaminolysis, the precise regulation of glutamine metabolism in human cancers is not fully understood (6, 16–20). The tumor suppressor, retinoblastoma protein (Rb), is a master regulator of several transcription factors involved in cell cycle regulation, yet the involvement of Rb pocket proteins in glutamine metabolism is unknown.
The Rb family of transcription factors (Rb-1, Rbl1, and Rbl2) has multiple inter-related target genes that converge on cell cycle regulators. Loss of Rb family function as a result of mutation or inactivating hyperphosphorylation by deregulated cyclin dependent kinases is a hallmark of all tumors, and understanding the full spectrum of Rb’s functions may enable the identification of proteins that can be targeted for the development of anti-cancer agents. The Rb family is classically believed to interact with the E2F transcription factors to regulate cell cycle progression. The Rb family members have over-lapping and compensatory functions and, as a result, genomic deletion of all three Rb family members (triple knockout, TKO) is required for the loss of cell cycle control in fibroblasts (21, 22). Beyond its role in cell cycle control, the Rb family regulates cell senescence in culture and cell contact inhibition and the TKO MEFs are thus immortal and lack cell contact inhibition (21, 22). Furthermore, Rb/E2F has been shown to have expanding roles including potentially linking cell proliferation to certain biochemical pathways (reviewed in (23)). Given that a dysfunctional Rb cascade promotes loss of proliferative control, we hypothesized that the Rb pathway also may directly regulate the uptake and conversion of glutamine into anabolic precursors that are required for neoplastic cell growth and survival.
Herein, we report that Rb family-deficient fibroblasts increase glutamine consumption to sustain mitochondrial function through anaplerosis and to maintain elevated intracellular glutathione. Importantly, elevated glutamine uptake is mediated through direct activity of specific E2F family members on glutaminolytic enzymes, and enhanced glutaminolysis is required for the growth and survival of Rb-dysfunctional cells. These data expand our understanding of the mechanisms by which tumor cells manipulate nutrient pathways to promote enhanced proliferation, and support the targeting of glutamine metabolism in Rb-deficient tumors as a potential therapeutic strategy.
Results
Rb-family Deletion Increases Glutamine Uptake and Conversion to Glutamate In Part Via Expression of the Glutamine Transporter ASCT2 and GLS1 Activity
Rb TKO cells contain genetic deletions of the three Rb family members (Rb1, Rbl1, and Rbl2) and exhibit several classic hallmarks of neoplastic cells including enhanced proliferation and immortalization (22). Initially, we examined basal 14C-glutamine uptake of Rb WT, Rb-1 null (Rb-1−/−), and Rb TKO mouse embryonic fibroblast (MEF) cells. Whereas Rb-1−/− cells exhibited a 4-fold increase in glutamine uptake, Rb TKO cells consumed 6-fold more 14C-glutamine relative to Rb WT MEFs (Fig. 1a). To confirm specificity of Rb-1 function in regulating glutamine uptake, Rb-1 was reconstituted in both the Rb-1−/− and TKO MEFs (Fig. 1b & Fig. S1a). As illustrated in Figure 1b, Rb-1 expression significantly suppressed glutamine uptake in both cell types with uptake in the Rb-1−/− cells relatively reduced to levels observed in the WT cells. These data indicate that although loss of Rb-1 function is sufficient to cause a marked increase in glutamine uptake in MEFs, Rbl1 and/or Rbl2 either can contribute to the suppression of glutamine uptake or can compensate for the loss of Rb-1 expression. Importantly, the entire Rb family can be suppressed by common mutations in human cancers (e.g. p16INK4A (24, 25)) and we thus elected to continue our studies on the metabolic effects of Rb dysfunction by directly comparing the Rb WT and the fully Rb-deficient TKO cells. To determine whether the observed increase in glutamine uptake was a regulated process, we examined mRNA expression of several glutamine transporters. The glutamine transporters ASCT2 and SLC38A5 previously have been identified to be elevated in tumor tissues and their expression has been demonstrated to be transcriptionally regulated by specific oncogenes (9, 26). In the Rb TKO cells, only ASCT2, and not SLC38A5, exhibited elevated mRNA levels compared to Rb WT cells and ASCT2 protein expression was found to be increased 6-fold in the Rb TKO cells (Fig. 1c). Consistent with a functional role of Rb-1 in regulating glutamine uptake, re-expression of Rb-1 also resulted in a marked decrease in ASCT2 expression (Fig. S1b). Examination of glutaminase (GLS) isoforms, which convert glutamine to glutamate, revealed that GLS1 mRNA is solely expressed in the MEF cells, but genetic deletion of the Rb family yielded no increase in GLS1 transcript levels (Fig. 1d). However, protein analysis revealed an increase in GLS1 expression (Fig. 1d), suggesting both transcriptional and post-transcriptional regulation of glutaminolytic enzymes by the Rb pathway. Given the increased GLS1 expression in Rb TKO cells, we expected that these cells would convert a greater portion of glutamine to glutamate and in doing so generate higher levels of ammonia, which is another product of GLS1 activity. As demonstrated in Figure 1e, NH4+ excretion was significantly higher in TKO compared to WT MEFs. To confirm these results and to identify specific glutamine-metabolic pathways within both WT and TKO MEFs, we conducted isotopomer analysis using [13C5,15N2]-glutamine by 2D-NMR. As shown in Figure 1f, TKO MEFs converted a relatively greater amount of extracellular glutamine to glutamate compared to WT MEFs. Together, these results demonstrate for the first time that genetic deletion of Rb family causes a direct induction of glutamine uptake and utilization by modulating specific enzymes involved in the glutaminolytic pathway.
Figure 1. Deletion of the Rb family Enhances Glutamine Uptake and Conversion to Glutamate Via Elevated ASCT2 Expression and GLS1 Activity.
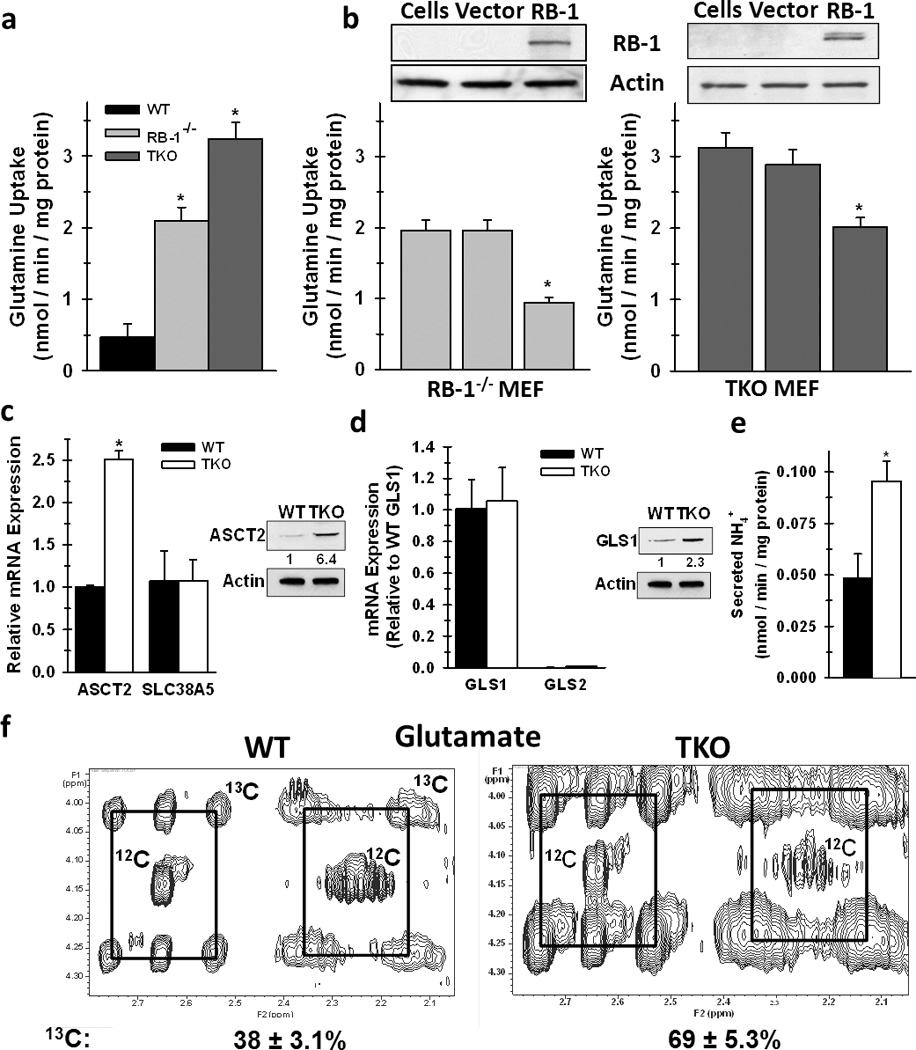
a. Glutamine uptake in WT, Rb-1−/− and TKO cells was assayed by 14C-glutamine labeling. Data are represented as nmol / min / mg protein. Mean ± s.d. from triplicate measurements from three independent cell preparations. * p < 0.005. b. Glutamine uptake was determined in Rb-1−/− and TKO cells following reconstitution of Rb-1 expression. Data are represented as nmol / min / mg protein. Mean ± s.d. from triplicate measurements from three independent cell preparations. * p < 0.01. c. mRNA expression of ASCT2 and SLC38A5 between WT and TKO MEFs. For the ASCT2 and SLC38A5 comparisons, data are represented as relative mRNA with WT levels set to 1. Protein expression of ASCT2 between WT and TKO MEFs is represented as relative protein expression with WT levels set to 1. For protein expression, shown is a representative image from three separate experiments. * p < 0.01. d. For GLS, mRNA levels are relative to expression of WT GLS1 set to 1. For mRNA, shown is mean ± s.d. from three independent RNA preparations. Protein expression of GLS1 between WT and TKO MEFs is represented as relative protein expression with WT levels set to 1. For protein expression, shown is a representative image from three separate experiments. * p < 0.01. e. Secreted ammonia levels were determined in the medium from WT and TKO cells. Data are represented as nmol / min / mg protein, and shown are mean ± s.d. from triplicate measurements from two independent experiments. * p < 0.005. f. Incorporation of glutamine carbon into glutamate was determined by stable isotope labeling of WT and TKO cells with [13C5,15N2]-glutamine, as described in the methods. Shown are representative 2-D NMR spectra of peaks representative of glutamate. The boxes connect the 13C satellite peaks of glutamate and the central peak denoted 12C is from glutamate that contains no 13C.
Rb-Family Deletion Causes Elevated Anaplerosis of Glutamine Carbon and Increased Mitochondrial Function
To determine whether glutamine carbons enter the TCA cycle after conversion to glutamate, we next analyzed 13C incorporation into aspartate, which is generated from 13C-labeled oxaloacetate via activity of aspartate aminotransferase. We observed 63 ± 4.2% of aspartate carbon originated from glutamine in Rb TKO cells, whereas only 29 ± 2.4% was labeled in Rb WT cells (Fig. 2a). However, no labeling of lactate from glutamine was observed in the Rb TKO cells or in the medium (data not shown) suggesting that malic enzyme activity is low in these cells and glutamine carbon is preferentially metabolized to replenish TCA intermediates.
Figure 2. Loss of the Rb Pathway Increases TCA Anaplerosis of Glutamine Carbon and Stimulates Mitochondrial Metabolism.
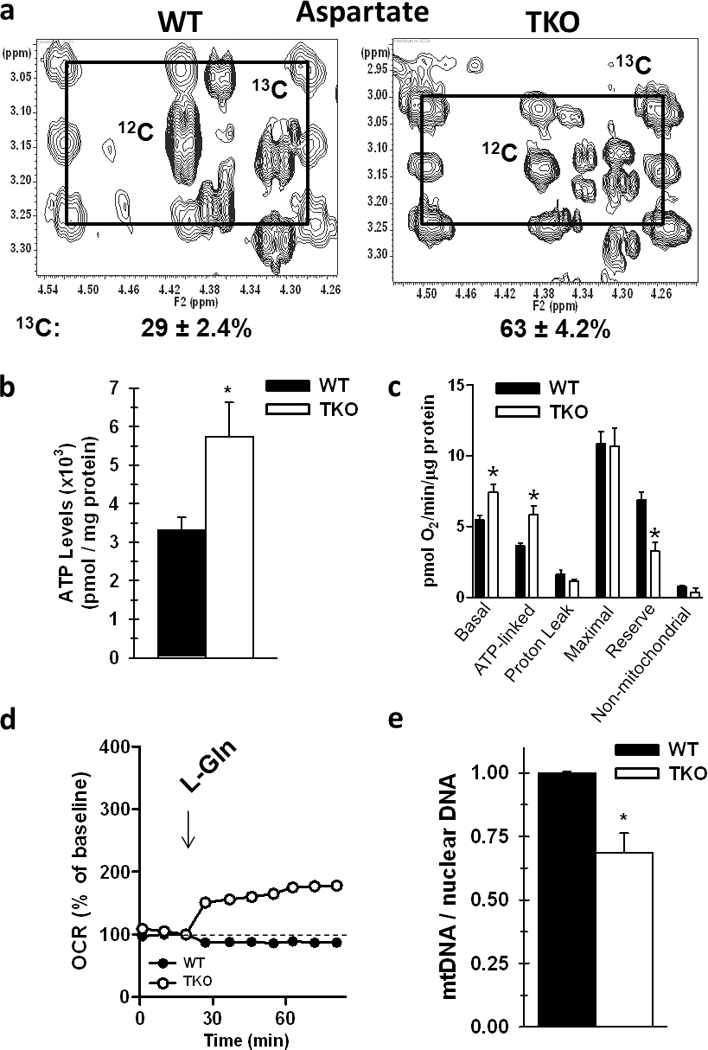
a. Incorporation of glutamine carbon into aspartate was determined by stable isotopomer labeling of WT and TKO cells with [13C5,15N2]-glutamine, as described in the Methods and in Figure 1. Shown are representative 2-D NMR spectra of TOCSY cross-peaks corresponding to aspartate C2-C3 and 13C satellites. The pattern indicates a mixture of fully labeled aspartate via the first turn of the Krebs cycle, and aspartate that shows the second turn of the Krebs cycle after condensation of OAA with unlabeled acetyl CoA from unlabeled glucose, as in Figure 1. b. Steady-state ATP levels in WT versus TKO cells. Data are represented as pmol / mg protein, and shown is the mean ± s.d. from duplicate measurements from three independent experiments. * p < 0.01. c. O2 consumption in WT and TKO cells was measured on Seahorse XF 24 analyzer and the oxygen consumption rate (OCR) was normalized to protein content and data is presented as pmol / min / µg protein. Shown is mean ± s.e.m. from five replicates from two separate experiments. ATP linked, maximal (reserve), and non-mitochondrial O2 fractions were determined by addition of oligomycin, FCCP, and AA/rotenone, respectively. * p < 0.01. d. O2 consumption was measured after addition of exogenous glutamine to WT and TKO cells. Shown is the % baseline oxygen consumption (set at 100%) as a function of time from 5 replicates from two independent experiments. e. Relative amounts of mitochondrial DNA to nuclear DNA between WT and TKO cells was determined by real-time PCR using primers specific for either mitochondrial or nuclear specific genes. Shown is the ratio of mtDNA / nuclear DNA with WT set to 1 from four separate cell preparations. * p < 0.005.
We also measured steady-state ATP levels and the rate of oxygen consumption by Rb WT and Rb TKO cells. Deletion of the Rb family resulted in 1.8-fold higher level of intracellular ATP (Fig. 2b), and an increase in the basal O2 consumption rate [OCR] (Fig. 2c). Furthermore, this increase in OCR was directly coupled to ATP production, with no differences in mitochondrial proton leak in the Rb TKO cells compared with WT cells (Fig. 2c). These measurements do not distinguish between the contribution of glutamine oxidation for mitochondrial function from oxidation of other carbon precursors such as glucose or fatty-acids. For this, we determined the baseline OCR in both cell types cultured in medium deprived of both glucose and glutamine and then measured the OCR response to the addition of exogenous glutamine (4 mM final concentration). Unlike WT MEFs, Rb TKO cells were able to enhance their O2 consumption with the addition of glutamine, indicating that cells devoid of Rb function can increase their mitochondrial metabolism by using glutamine carbon through TCA anaplerosis (Fig. 2d). Lastly, it is important to note that the increase in oxygen consumption and ATP levels observed in the Rb TKO cells could be a result of a greater number of mitochondria compared to the Rb WT cells. However, mitochondrial genome analysis revealed that Rb TKO cells contain significantly less mitochondria relative to WT cells (Fig. 2e). Together, these data suggest that the mitochondria in the TKO cells are more active, which is presumably attributable to the increase in the availability and capacity for glutamine metabolism.
Rb-Family Deficiency Results in Enhanced Production of Reactive Oxygen Species and Decreased Ratio of Reduced Glutathione
Since Rb TKO cells exhibited elevated mitochondrial function relative to Rb WT cells, we postulated that the production of reactive oxygen species (ROS) such as superoxide and hydrogen peroxide would be significantly increased in the Rb-depleted cells. Based on DCF fluorescence, a higher percentage of TKO cells (60.3%) were positive for ROS formation compared to WT (13.5%) (Fig. 3a), and the mean fluorescence intensity was elevated in the Rb TKO cells (Fig. 3b). Furthermore, the rate of ROS production also was increased in the TKO cells (Fig. 3b). To determine mitochondrial ROS production within these cells, both Rb WT and Rb TKO cells were labeled with MitoSOX, which is an ROS indicator that is specifically targeted to the mitochondria (27). As illustrated in Figure 3c, Rb TKO cells generated significantly more mitochondrial ROS compared to Rb WT cells.
Figure 3. Rb-Family Deficient Cells Have Elevated ROS Levels, Increased Total Glutathione, and a Lower Ratio of GSH to Total Glutathione.
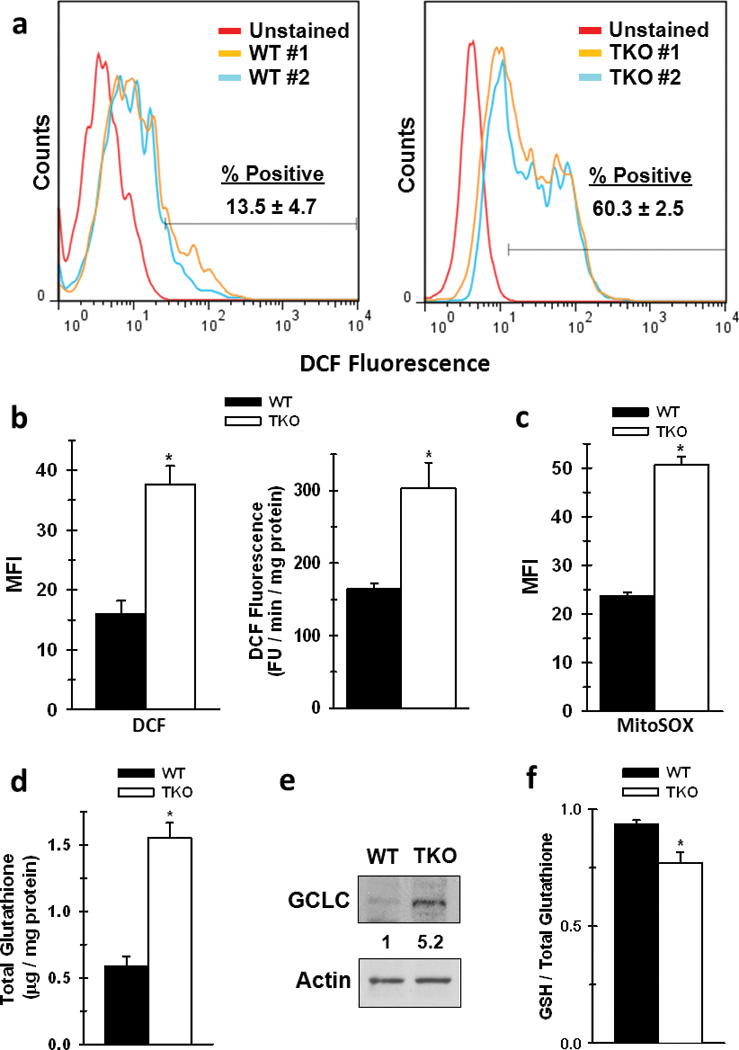
a. Representative flow cytometry histograms of WT or TKO measuring DCF fluorescence. % positive cells were determined as number of stained cells beyond that of the unstained population. b. Mean fluorescent intensity (MFI) between WT and TKO cells were determined by FlowJo software. Data are presented as mean ± s.d. from duplicate measurements from two separate experiments. * p < 0.01. Rate of ROS production was determined in whole WT or TKO cells by measurement of DCF fluorescence as a function of time. Data are represented as relative FU / min / mg protein, and shown as the mean ± s.d. of 5 replicates from three independent experiments. * p < 0.01. c. MFI of WT and TKO MEFs stained with the mitochondrial specific ROS indicator MitoSOX. Data are presented as mean ± s.d. of three replicates from two separate experiments. d. Total glutathione was measured as described in the Materials and Methods. Data are represented as µg / mg protein between WT and TKO cells. Shown is the mean ± s.d. from three replicates from two separate experiments. * p < 0.005 e. Protein expression of gamma glutamyl-cysteine ligase (GCLC). Shown is a representative image from three independent cell lysate preparations, and ratio of expression was determined by densitometry analysis with WT expression set to 1. f. The ratio of GSH / Total glutathione was measured as described in the Materials and Methods. Data are represented as the ratio of GSH / total glutathione between WT and TKO cells. Shown is the mean ± s.d. from three replicates from two separate experiments. p < 0.01.
In addition to being a precursor for TCA anaplerosis, glutamine is the major carbon source for glutathione biosynthesis in multiple cell systems (9, 10). To determine whether Rb suppresses glutathione production, we initially measured total glutathione levels, and found that Rb TKO cells have significantly elevated glutathione compared to WT cells (Fig. 3d). This was further confirmed by metabolomics analysis with 13C-glutamine, whereby Rb TKO cells exhibited an increase in labeled glutamine incorporation into GSH (Fig. S2a&b). We also found that this increase is likely driven in part by elevated expression of gamma-glutamylcysteine ligase (GCLC) in the Rb TKO cells, which is the rate-limiting step in glutathione synthesis (Fig. 3e). However, an increase in GSSG formation within the TKO cells resulted in a decrease in the levels of reduced glutathione (GSH) to total glutathione in the Rb TKO cells (Fig 3f). Together, these data suggest that Rb-deficient cells not only increase glutamine metabolism to provide the anaplerotic carbon for enhanced mitochondrial function, but also the synthesis of glutathione.
Growth and Survival of Rb-dysfunctional Cells is Dependent on Glutamine Metabolism
Our results demonstrate that loss of Rb function enhanced glutamine metabolism for both mitochondrial metabolism and glutathione production, two pathways necessary for the growth and survival of neoplastic cells. In order to determine the relative dependence of Rb WT versus Rb TKO cells on glutamine, we next cultured the cells in the absence of glutamine and quantified the viable cell number over 48 hours. We found that the growth of Rb WT cells was relatively unaffected by glutamine withdrawal even after 48 hours, whereas Rb TKO cells were unable to survive and grow in the absence of glutamine (Fig. 4a). In addition, while siRNA-mediated suppression of either ASCT2 or GLS1 reduced glutamine uptake and NH4+ secretion in both the WT and Rb TKO MEFs, only the growth of the Rb TKO cells was negatively affected (Fig. S3). These results further highlight the potential utility of small molecule antagonists of these enzymes as anti-cancer agents in Rb-deficient tumors. Interestingly, suppression of GLS1 resulted in a significant reduction in glutamine uptake in these cells. While the mechanism by which GLS1 suppression is able to decrease glutamine uptake is not well defined, inhibition of primary metabolizing enzymes within other biochemical pathways have previously been observed to inhibit nutrient uptake (for example, effects of inhibition of HK2 and choline kinase on glucose and choline transport, respectively) (28, 29). Furthermore, in support of the requirement of glutamine for maintaining the energetic requirements for elevated proliferation, glutamine withdrawal also resulted in a rapid decrease in steady-state ATP levels (3 hrs.), while ATP levels were unchanged until 48 hours in Rb WT cells under the same conditions (Fig. 4b). We then supplemented the medium with cell permeable α-ketoglutarate (α-kg), a TCA cycle intermediate, and observed a significant rescue in ATP levels as well as cytotoxicity caused by glutamine withdrawal in the Rb TKO cells (Fig. 5a&b). Furthermore, consistent with glutamine being the primary carbon source for glutathione biosynthesis and the observation that Rb TKO cells increase glutamine carbon incorporation into GSH (Figure S2), glutamine depletion significantly decreased GSH levels and resulted in an overall increase in cellular ROS production in the Rb TKO cells (Fig 5c–e). Similar observations have been reported in which small molecule inhibition of GLS1 resulted in lower GSH levels and a concomitant increase in ROS levels in lymphoma cells (10). However, addition of α-kg decreased ROS levels and resulted in partial rescue of GSH levels in the Rb TKO cells (Fig 5c–e). Combined, our data indicate that deletion of the Rb pathway directly stimulates glutamine utilization in order to provide the energy and macromolecular precursors necessary for enhanced cell division and survival.
Figure 4. Loss of Rb Function Renders Cells Dependent on Glutamine for Cell Survival and Growth and for Maintenance of Cellular Steady-State ATP Levels.
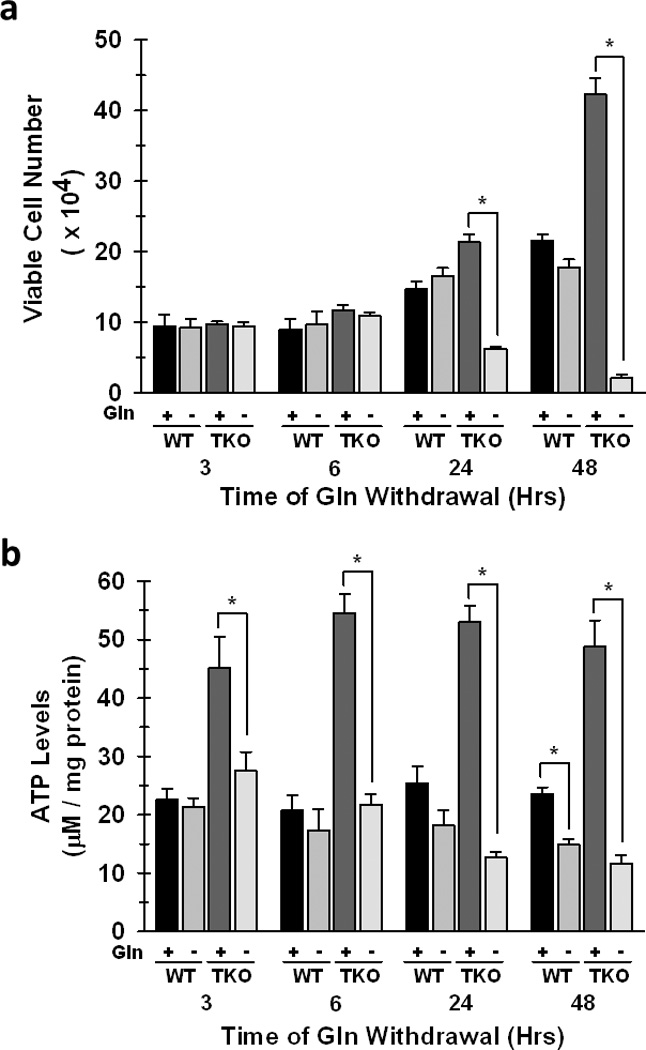
WT and TKO cells were cultured in glutamine-free medium for the indicated time periods. a. Viable cell number was determined by hemocytometer enumeration after trypan blue staining. Data are represented as mean viable cell number ± s.d. from triplicate measurements from three independent experiments. * p < 0.005 b. Steady-state ATP was determined between WT and TKO cells as described in the Materials and Methods. Shown are mean ± s.d. of triplicate readings from two different experiments. * p < 0.01.
Figure 5. Alpha-Ketoglutarate Rescues ATP Production, GSH Levels, and Cell Survival During Glutamine Withdrawal in Cells Lacking the Rb Family.
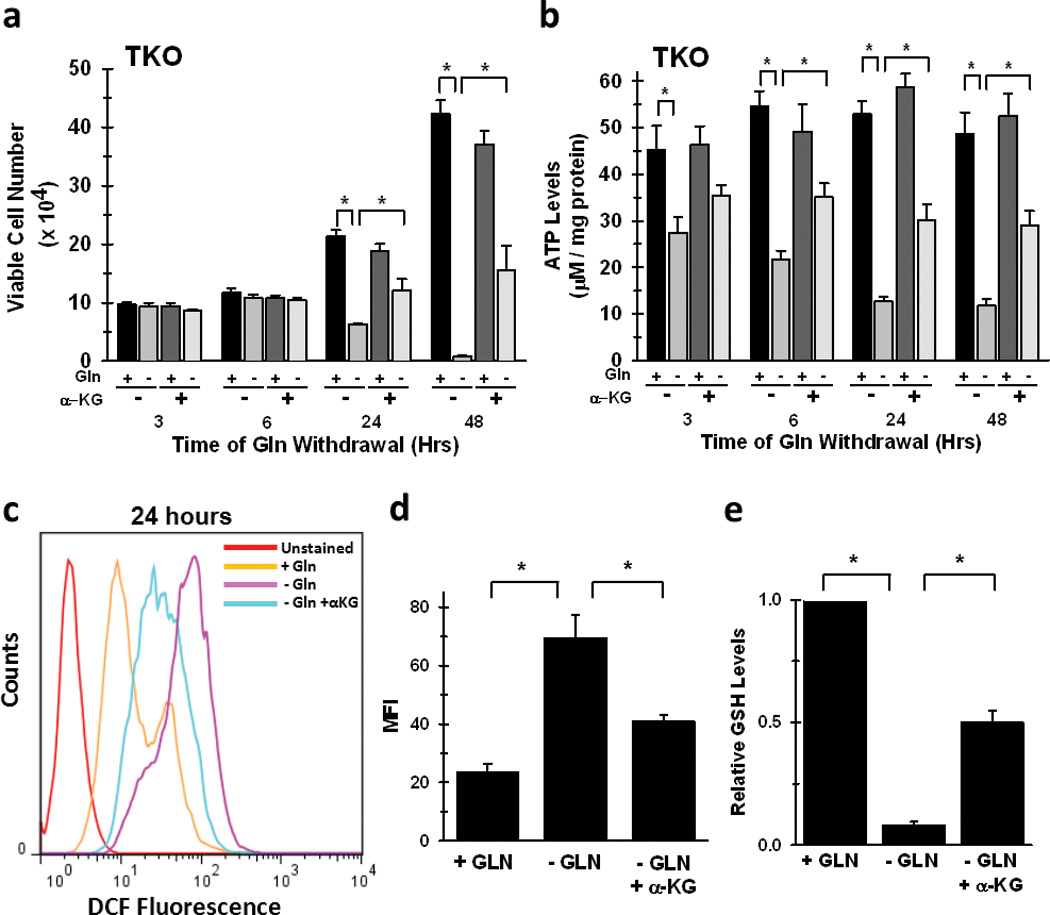
TKO cells were cultured in glutamine-free medium for the indicated time periods with or without addition of 7mM dimethyl-α-ketoglutarate. a. Viable cell number was determined by hemocytometer enumeration after trypan blue staining. Data are represented as mean viable cell number ± s.d. from triplicate measurements from three independent experiments. * p < 0.005. b. Steady-state ATP was determined between WT and TKO cells as described in the Materials and Methods. Data are represented as µM / mg protein, and shown are mean ± s.d. of triplicate readings from two different experiments. * p < 0.01. c. Representative flow cytometry histograms of TKO MEFs cultured in complete medium, medium lacking glutamine, or medium lacking glutamine with the addition of α-KG for twenty-four hours. d. Mean fluorescence intensity in TKO MEFs cultured in the absence or presence of glutamine or α-KG. Data are presented as the mean ± s.d. from triplicate readings from two separate experiments. * p < 0.01. e. GSH levels in TKO MEFs after culture in indicated medium conditions for twenty-four hours. Data are represented as relative GSH levels between the different conditions with + GLN sample set to 1, and shown are the mean ± s.d. from triplicate measurements from two separate experiments. * p < 0.005.
E2F Family Members Regulate Elevated Glutamine Metabolism in the Absence of Rb Function
The Rb family is known to negatively regulate gene expression through direct interaction with E2F family members, and Rb functions to suppress the activity of E2F-1, 2, and -3 to transcriptionally activate downstream target genes (24). To determine if these E2F members were altered in the absence of the Rb family, we examined the level of expression of each member between Rb WT and Rb TKO cells. Both E2F-1 and E2F-2 mRNA (data not shown) and protein expression were significantly elevated in the TKO cells, whereas E2F-3 levels exhibited little change (Fig. 6a). Since our analysis of the Rb WT and Rb TKO cells examined the effects of Rb dysfunction on glutaminolysis, we sought to determine whether any of these E2F family members played a role in mediating these alterations in the Rb TKO cells. We transfected the cells with E2F-1, -2, and -3 siRNA and measured the expression of ASCT2 and GLS1. We found that only E2F-3 knock-down significantly decreased ASCT2 expression, whereas there was no effect on GLS1 levels by siRNA against any E2F member (Fig. 6b). These results are not surprising since ASCT2, but not GLS1, was found to be transcriptionally regulated in the absence of Rb (Fig. 1b). Interestingly, inhibition of the expression of E2F members revealed differential effects on glutamine consumption in Rb TKO cells, in which E2F-2 and E2F-3 decreased uptake by 20 and 35%, respectively, while E2F-1 yielded no significant difference (Fig. 6d). However, E2F-1 and E2F-3 siRNA reduced cell proliferation, while E2F-2 suppression did not significantly alter Rb TKO cell growth (Fig. 6c). These differential effects on glutamine uptake and cell growth by E2F proteins suggest that their regulatory involvement in multiple cellular pathways, be it cell cycle or metabolic control or both, cumulatively determine the extent of their influence on cell proliferation. Next, chromatin immunoprecipitation analysis was performed to assess the potential for direct regulation of E2Fs on ASCT2 gene expression. Using antibodies specific for each E2F family member, E2F-3 was observed to significantly associate with the proximal ASCT2 promoter (Fig. 6e). ChIP analysis of E2F-1,-2, and -3 binding to a known target gene promoter, cdc2 (30), was performed to assess specificity of E2F association to the ASCT2 promoter. As illustrated in Figure 6e, under these conditions, all three E2F proteins associated with the cdc2 promoter, with E2F-1 and E2F-3 exhibiting the strongest interaction. In addition, as a negative control, none of the E2F family members were found to interact with the GAPDH gene promoter (Fig. 6e), as has been previously demonstrated for E2F-1 in the Rb TKO cells (31). These results correlate with our protein and glutamine uptake analysis in that E2F-3 suppression alone decreased ASCT2 expression and had a significant effect on glutamine consumption. Taken together, these results indicate that the Rb pathway regulates glutamine metabolism for mitochondrial function and glutathione synthesis partially through direct control of glutamine uptake via specific E2F mediated transcription of target genes, such as ASCT2, and partly through unknown alternative regulatory mechanisms (i.e. post-transcriptional – GLS1) of proteins involved in glutaminolysis (Fig. 7).
Figure 6. E2F Family Members Contribute to Enhanced Glutamine Metabolism in Cells with Non-Functional Rb Through Direct Regulation of Glutaminolytic Enzyme Expression.
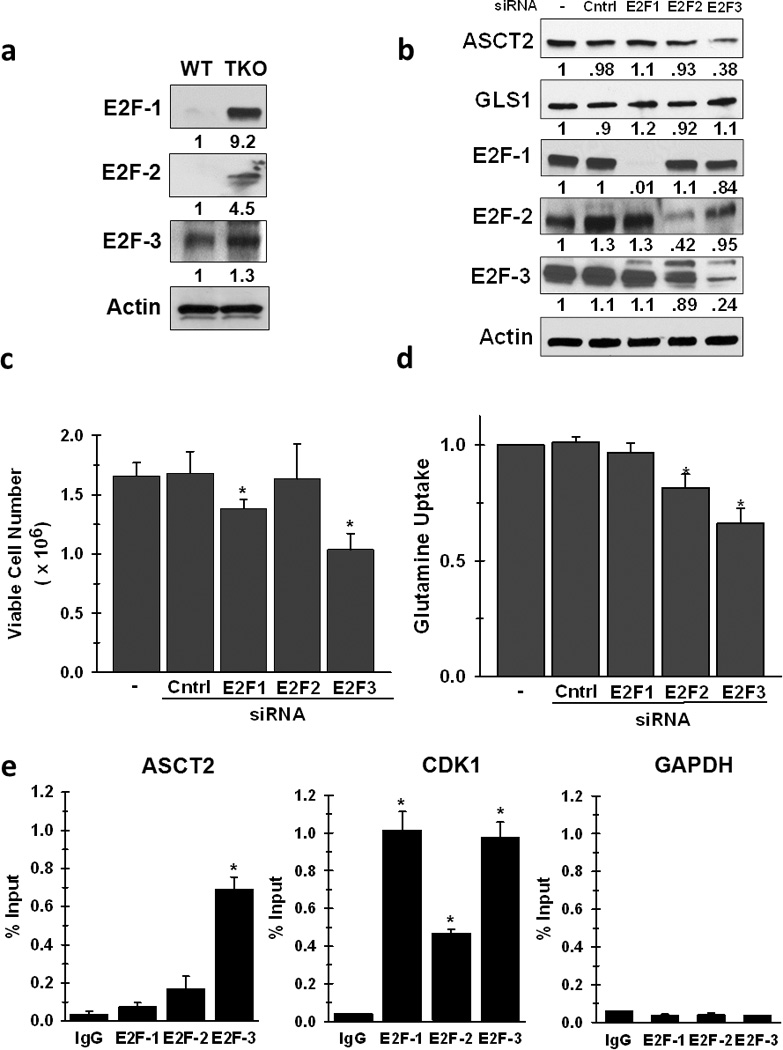
a. Protein expression of E2F-1, -2, and -3 in WT and TKO cells. Shown are representative images from three separate experiments, and densitometry analysis of relative protein expression with WT levels set to 1. * p < 0.01. b. ASCT2 and GLS1 protein expression was examined in TKO cells transiently transfected with siRNA specific for E2F-1, -2, or -3. Shown are representative images from three separate experiments, and densitometry analysis of relative expression of each protein was compared to the no siRNA controls, which was set to 1. c. Viable cell number was determined by hemocytometer enumeration after trypan blue staining 72 hours after siRNA transfection. Data are represented for each scenario as relative viable cell number (mean ± s.d.) compared to no siRNA control, which was set to 1. * p < 0.05. d. Glutamine uptake was determined by 14C-glutamine labeling in TKO cells 72 hours post-transfection with E2F-1, -2, and -3 siRNA. Data are represented as relative glutamine uptake (mean ± s.d.) compared to no siRNA control, which was set to 1. * p < 0.01. e. Association of E2F-1, -2, or -3 with the promoters of the mouse ASCT2, cdc2and GAPDH genes was assayed by chromatin immunoprecipitation analysis in TKO cells. Input or eluted chromatin was subjected to real-time PCR analysis using promoter-specific primers. Data are represented as the % input of the immunoprecipitated chromatin for each gene from three separate chromatin preparations. * p < 0.005.
Figure 7. Rb Regulates Glutamine Metabolism at Different Nodes Within Glutaminolysis.
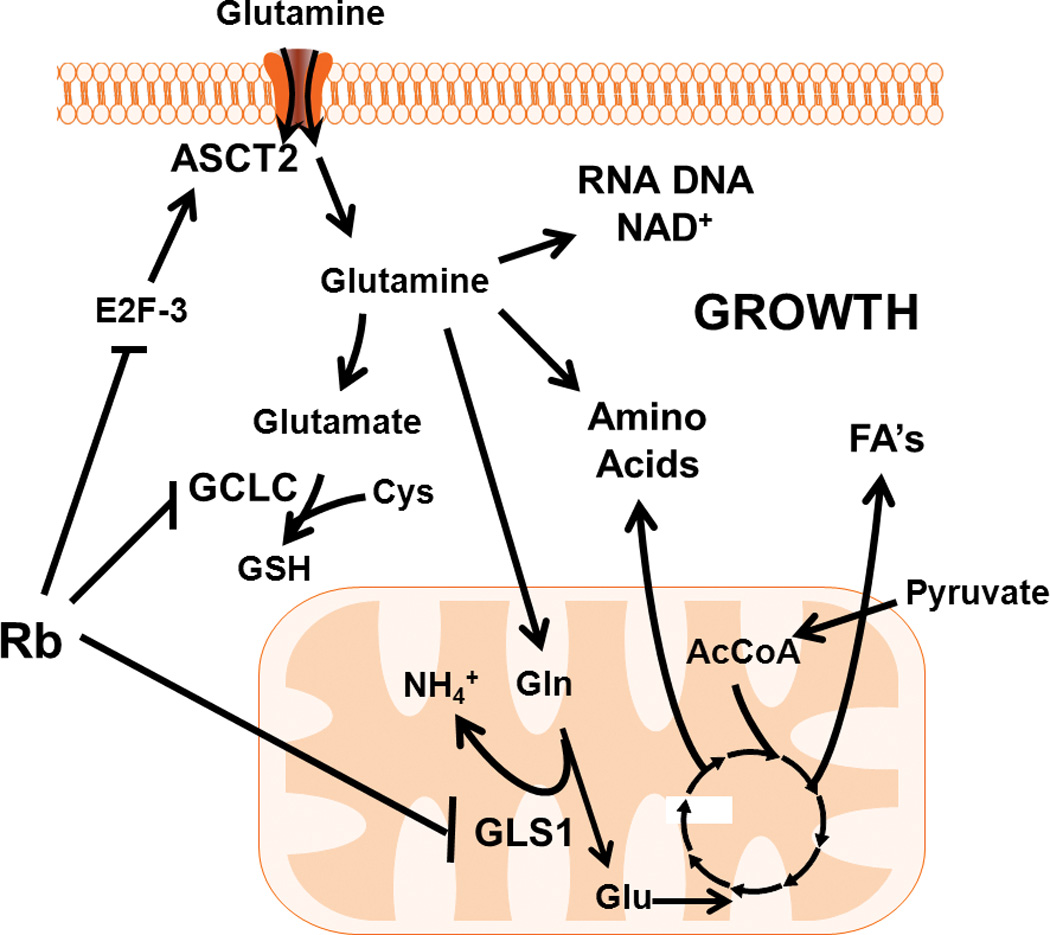
The Rb pathway controls glutamine consumption in part through E2F-3 mediated regulation of the glutamine transporter, ASCT2. Intracellularly, glutamine has several downstream biochemical fates, including glutathione, as anaplerotic carbon for the TCA cycle, and as nucleotides and amino acids. The Rb cascade can modulate glutathione synthesis by regulating the expression of gamma-glutamylcysteine ligase (GCLC), which is the rate-limiting step in glutathione production. For TCA anaplerosis, glutamine is converted to glutamate in the mitochondria by glutaminase 1 (GLS1), another Rb-family regulated enzyme, and can enter the TCA cycle as α-ketoglutarate. Glutamine derived carbon, within the TCA, can be further metabolized to oxaloacetate for conversion to additional amino acids, or combined with acetyl-CoA (AcCoA) from pyruvate for citrate synthesis, which is a precursor for fatty acid (FA) production. Ultimately, through regulation of both glucose and glutamine metabolism, the Rb pathway may act to efficiently modulate cellular growth not only by controlling cell cycle progression, but also through facilitating the production of the energy and macromolecules required for cell division. ASCT2: sodium-dependent neutral amino acid transporter type 2; Cys: cysteine; NH4+: ammonium; GSH: reduced glutathione; Gln: glutamine; Glu: glutamate; TCA: tricarboxylate acid cycle
Discussion
In the current study, we have found that genomic deletion of all three Rb family members causes a shift towards enhanced glutamine metabolism in part via the glutamine transporter ASCT2 and GLS1 that in turn leads to dependence on glutamine for cell proliferation and survival. Importantly, reconstitution of Rb-1 in the Rb-dysfunctional cells decreased glutamine uptake and suppressed ASCT2 expression. We also have found that Rb TKO MEFs, and not the WT MEFs, metabolize glutamine to increase mitochondrial function, ATP production and glutathione synthesis. Furthermore, the elevated ATP and ROS production observed in the Rb TKO cells was not due to greater mitochondrial mass since real-time PCR analysis revealed a reduction in the ratio of mitochondrial to nuclear DNA (Fig 2e) indicating that the mitochondria in Rb TKO MEFs are more metabolically active. This is in contrast to previous reports in normal differentiated adipocytes, where genomic loss of Rb-1 resulted in increased energy expenditure due to a significant increase in mitochondrial copy number (32). This discrepancy is unclear, yet may be due to multiple factors including cell type as well as extent of Rb pathway dysfunction. Taken together, our analysis on the effects of Rb loss and re-expression combined with our examination of direct E2F promoter occupancy support a direct role for the Rb pathway in regulating cell glutaminolysis.
Our analysis of E2F function on glutamine metabolism suggests that the E2F family members may regulate several independent but required cellular processes. E2F-3 suppression reduced both cell number and glutamine uptake and was functionally linked to the promoter of ASCT2. In addition, E2F-2 partially decreased glutamine uptake, yet suppression of E2F-2 expression yielded no effect on ASCT2 or GLS1 levels. These data indicate that there may be other Rb pathway regulated enzymes, beyond ASCT2, SLC38A5, and GLS1/2, that may contribute to the overall alteration in glutaminolysis driven by Rb dysfunction. E2F-1 knock-down significantly decreased cell proliferation in the Rb TKO cells yet had little effect on glutamine uptake. The effect on cell growth of Rb TKO cells mediated by E2F-1 siRNA could be due to its extensive involvement in regulating cell cycle mediators. However, a recent report has revealed that E2F-1 can enhance mitochondrial function through direct transcriptional regulation of multiple enzymes involved in oxidative phosphorylation (33). This alteration may, in turn, promote mitochondrial metabolism of available glutamine-derived carbon that is generated through the activities of E2F-3 and/or E2F-2 regulated protein targets. Furthermore, the Rb/E2F-1 cascade also has been observed to regulate the conversion of pyruvate to acetyl-CoA by directly modulating the expression of PDK4, which controls the activity of pyruvate dehydrogenase (34). These observations suggest that loss of Rb function may suppress the flux of glucose-derived carbons into the TCA cycle which, in turn, may increase reliance on alternative sources of anaplerotic carbon. Indeed, our data complement these results by extending the regulatory function of the Rb cascade to glutamine uptake and metabolism and by providing support for E2F members involved in suppressing this metabolic activity. Based on these new results, we believe that the ability of the Rb pathway to tightly regulate cellular growth not only stems from its well-known role in modulating cell cycle checkpoints, but also from its direct metabolic control of ATP and macromolecule production.
The Rb WT and Rb TKO cells are isogenic apart from genomic deletion of all three Rb family members (22). The Rb family contains three distinct members, and are known to exhibit overlapping functions (35). This compensatory activity was evident with respect to glutamine uptake wherein Rb-1−/− MEFs exhibited a 4-fold increase in glutamine uptake compared to WT MEFs, whereas a 6-fold increase was observed in the Rb TKO MEFs (Fig. 1a). While the Rb TKO MEFs were used to examine the complete loss of Rb function on glutamine metabolism, these data indicate that Rbl1 and/or Rbl2 also may play a role in regulating glutamine metabolism. Traditionally, the Rbl1 and Rbl2 members do not associate with the activator E2Fs, but interact primarily with E2F-4 or -5 (24). This, in turn, suggests that these E2Fs may also function to tether these Rb proteins to target genes involved in mediating glutaminolysis. However, Rbl1 has recently been observed to specifically control E2F-3 activity in regulating target gene expression (36). Since the independent regulatory mechanisms by Rb-1, Rbl1, and Rbl2 on glutamine metabolism remain unclear, the elucidation of the distinct contributions of each Rb family member and their respective E2F partners in mediating glutamine metabolism is a focus of on-going studies. That genomic deletion of all three Rb family members caused an established cancer-related metabolic shift to glutamine dependency strongly supports the conclusion that the Rb pathway has a previously under-recognized role in the regulation of cellular metabolism. Whether the metabolic effects observed in this study are due solely to direct Rb/E2F control on metabolic target genes or through additional downstream mediators whose expression is also altered by loss of Rb remains to be determined.
Genetic deletion or mutation of Rb is present in 100% of retinoblastomas, 90% of small cell lung carcinomas as well as in cancers of the bladder (70%), prostate (60%), liver (40%), and breast (30%) (25). Additionally, dysfunctional Rb extends to other cancer types, such as pancreatic (70%), colorectal (60%), and melanoma (60%), wherein the Rb family is inactivated by hyperphosphorylation by deregulated CDKs. Cumulatively, these frequencies suggest that therapies targeting the glutaminolytic pathway may be effective against a broad spectrum of tumor types. Indeed, inhibitors of GLS1, such as BPTES, are being utilized in pre-clinical settings, and have been shown to be effective in tumor xenograft models (10, 15, 37). Using in silico drug discovery methodologies (38, 39), we intend to identify novel small molecule antagonists of these enzymes for testing as anti-cancer agents. Additionally, the functional status of the Rb/E2F pathway in specific tumors may serve as a key biomarker that may predict responsiveness to these agents. We conclude that the ability of the Rb family to suppress glutamine metabolism contributes to its tumor suppressor activity and that these studies may identify novel targets for the development of anti-neoplastic therapies.
Materials and Methods
Cell Culture
Retinoblastoma protein family triple knock-out mouse embryonic fibroblasts (TKO MEFs) or Rb-1 null MEFs were generated as previously described (22). MEFs were cultured in DMEM (Gibco) supplemented with 10% FBS (Hyclone), 25mM glucose, 4mM glutamine and 50µg/mL gentamicin sulfate. Cells were maintained at 5% CO2 at 37°C.
Glutamine Uptake
WT, Rb-1−/−or TKO MEFs were washed and supplemented with 1 µM [U-14C]-glutamine for 5 minutes. Medium was aspirated, and cells were washed three times with glutamine deficient media. Sample c.p.m. was acquired on a liquid scintillation counter.
Real-time PCR
RNA was obtained from three separate preparations of WT and TKO MEFs using the RNeasy kit (Qiagen). Gene expression was determined using specific Taqman Gene Expression Assays (ABI) on a Step One Plus RT-PCR system.
Protein Expression
Western analysis was done on protein lysates from WT and TKO MEFs using antibodies against ASCT2 (Cell Signaling), GLS1 (Abnova), GCLC (ThermoScientific), E2F-1 (Cell Signaling), E2F-2 (Santa Cruz), E2F-3 (Santa Cruz), and β-actin (Pierce). Densitometry analysis was performed using Quantity One software.
Ammonia Excretion
Medium ammonia levels were determined using the Ammonia Assay Kit (Biovision). Secretion rate is presented as nmol / min / mg protein from triplicate samples from two independent experiments.
Stable Isotope Resolved Metabolomics Analysis by NMR
WT and TKO cells were seeded at 1 × 106 cells in 15 centimeter dishes. MEFs were then labeled for two cell doublings (WT: 60 hours, TKO: 48 hours) with DMEM supplemented with 25mM glucose, 10% dialyzed FBS, and 4mM [13C5,15N2]-glutamine. Metabolites were extracted twice with 10% TCA, and pooled extracts were then flash-frozen in LN2 and lyophilized. In order to resolve 13C-glutamine incorporation into glutamine (Q), glutamate (E), and GSH, a separate protocol for cell metabolite extraction under neutral pH was also performed as previously described (40). In both protocols, NMR spectra were recorded as described previously (38).
Oxygen Consumption and ATP Levels
A Seahorse Bioscience XF24 Extracellular Flux Analyzer was used to measure oxygen consumption in intact cells as described previously (41–43). The assay media consisted of DMEM supplemented with 25mM glucose and 4mM glutamine. In other studies, oxygen consumption was measured in WT and TKO MEFs cultured before and after addition of exogenous 4mM glutamine. ATP concentrations were determined using the ATP Determination Kit (Invitrogen).
Mitochondrial DNA Quantitative PCR
The ratio of mitochondrial to nuclear DNA was determined by real-time PCR using two sets of primers specific for either the mitochondrial or nuclear genome as previously described (32, 44). Real-time PCR was performed on serial dilutions of four separate cell DNA preparations in triplicate measurements.
Glutathione Levels
Total, reduced and oxidized glutathione levels were determined in cell lysates by using the Glutathione Assay Kit (Biovision), the GSH-GLO and GSSG/GSG-GLO Assay Kits (Promega). Data are presented as normalized total glutathione and the ratio of GSH to total glutathione between WT and TKO MEFs, or relative GSH levels in the TKO MEFs.
Reactive Oxygen Species
ROS production in WT and TKO MEFs was determined by DCF-DA (Invitrogen) fluorescence by flow cytometry or by a SpectraMax Fluorometer (Molecular Probes). Mitochondrial ROS was measured by MitoSOX fluorescence by flow cytometry.
Cell Proliferation and Viability
WT and TKO MEFs were plated in 12-well plates, and 16 hours post-seeding, media was replaced with either complete DMEM or DMEM without glutamine. Cell number and viability was determined by trypan blue exclusion and enumeration using a hemocytometer at 3, 6, 24, & 48 hours after media change. For alpha-ketoglutarate add-back experiments, 7 mM dimethyl-alpha-ketoglutarate was added concurrent with media change.
Cell Transfections
Cells were transfected with a plasmid encoding mouse Rb-1 (Origene: MG227287) using JetPrime reagent following manufacturer’s protocol (Polyplus). siRNA transfections were performed as previously described (39) and the following siRNAs were used: E2F-1: (Ambion: #s201266); E2F-2: (Ambion: #s109846); E2F-3: (Ambion: #s65238). Cell number, protein expression, and glutamine uptake were analyzed either 48 or 72 hours post-transfection as described above.
Chromatin Immunoprecipitation
ChIP assays were performed according to manufacturer’s protocol with minor modifications (Cell Signaling). ChIP was performed with antibodies raised against IgG (Cell Signaling), E2F-1 (Cell Signaling), E2F-2 (Santa-Cruz), and E2F-3 (Santa Cruz). Eluted chromatin was subjected to real-time PCR quantification for the mouse ASCT2, cdc2, and GAPDH promoters using specific primers (listed in supplementary material).
Statistics
Statistical significance for glutamine uptake, mRNA & protein expression, ammonia secretion, glutathione levels, ROS production, viable cell number, and ATP production between WT and TKO MEFs was determined by a two-sample, nonparametric, two-tailed t-test using Graph Pad Prism. p < 0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Jason Chesney, Sucheta Telang, and John Eaton for their insightful discussions, and thank Tyler Jacks for the kind gift of the TKO MEFs. NMR experiments were carried out at the James Graham Brown Cancer Center NMR facility, supported in part by the Brown Foundation and NCCRR grant 1P20 RR18733. This work was supported by a Center of Biomedical Research Excellence in Molecular Targets (3P20RR018733-09) grant from the National Center for Research Resources and by RR024489 (BGH).
Footnotes
Conflict of Interest
The authors state that there are no conflicts of interests with respect to these studies.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Dang CV, Lewis BC, Dolde C, Dang G, Shim H. Oncogenes in tumor metabolism, tumorigenesis, and apoptosis. J Bioenerg Biomembr. 1997 Aug;29(4):345–354. doi: 10.1023/a:1022446730452. [DOI] [PubMed] [Google Scholar]
- 3.Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med. 2008 Jun;49(Suppl 2):24S–42S. doi: 10.2967/jnumed.107.047258. [DOI] [PubMed] [Google Scholar]
- 4.Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A, Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007 Mar;274(6):1393–1418. doi: 10.1111/j.1742-4658.2007.05686.x. [DOI] [PubMed] [Google Scholar]
- 5.Warburg O, Negelein E. On the metabolism of cancer cells. Biochemische Zeitschrift. 1924;152:319–344. [Google Scholar]
- 6.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007 Dec 4;104(49):19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. 2007 Jul 2;178(1):93–105. doi: 10.1083/jcb.200703099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009 Apr 9;458(7239):762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008 Dec 2;105(48):18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012 Jan 4;15(1):110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011 Jan 19;481(7381):380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011 Jan 19;481(7381):385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaglio D, Soldati C, Vanoni M, Alberghina L, Chiaradonna F. Glutamine deprivation induces abortive s-phase rescued by deoxyribonucleotides in k-ras transformed fibroblasts. PLoS One. 2009;4(3):e4715. doi: 10.1371/journal.pone.0004715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lora J, Alonso FJ, Segura JA, Lobo C, Marquez J, Mates JM. Antisense glutaminase inhibition decreases glutathione antioxidant capacity and increases apoptosis in Ehrlich ascitic tumour cells. Eur J Biochem. 2004 Nov;271(21):4298–4306. doi: 10.1111/j.1432-1033.2004.04370.x. [DOI] [PubMed] [Google Scholar]
- 15.Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010 Nov 15;70(22):8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009 Nov 1;15(21):6479–6483. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006 Aug;16(4):253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 18.Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006 Jul 14;126(1):107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 19.Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010 Apr 20;107(16):7455–7460. doi: 10.1073/pnas.1001006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science. 2006 Jun 16;312(5780):1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 21.Dannenberg JH, van Rossum A, Schuijff L, te Riele H. Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev. 2000 Dec 1;14(23):3051–3064. doi: 10.1101/gad.847700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, et al. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 2000 Dec 1;14(23):3037–3050. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi C, Sasaki N, Kitajima S. Twists in views on RB functions in cellular signaling, metabolism and stem cells. Cancer Sci. 2012 Mar 26; doi: 10.1111/j.1349-7006.2012.02284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002 Dec;2(12):910–917. doi: 10.1038/nrc950. [DOI] [PubMed] [Google Scholar]
- 25.Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008 Sep;8(9):714–724. doi: 10.1038/nrc2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin Cancer Biol. 2005 Aug;15(4):254–266. doi: 10.1016/j.semcancer.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 27.Liu M, Liu H, Dudley SC., Jr Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res. 2010 Oct 15;107(8):967–974. doi: 10.1161/CIRCRESAHA.110.220673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clem BF, Clem AL, Yalcin A, Goswami U, Arumugam S, Telang S, et al. A novel small molecule antagonist of choline kinase-alpha that simultaneously suppresses MAPK and PI3K/AKT signaling. Oncogene. 2011 Jul 28;30(30):3370–3380. doi: 10.1038/onc.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007 Nov;27(21):7381–7393. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wells J, Boyd KE, Fry CJ, Bartley SM, Farnham PJ. Target gene specificity of E2F and pocket protein family members in living cells. Mol Cell Biol. 2000 Aug;20(16):5797–5807. doi: 10.1128/mcb.20.16.5797-5807.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Costantino ME, Montoya-Durango D, Higashi Y, Darling DS, Dean DC. The zinc finger transcription factor ZFHX1A is linked to cell proliferation by Rb-E2F1. Biochem J. 2007 Nov 15;408(1):79–85. doi: 10.1042/BJ20070344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dali-Youcef N, Mataki C, Coste A, Messaddeq N, Giroud S, Blanc S, et al. Adipose tissue-specific inactivation of the retinoblastoma protein protects against diabesity because of increased energy expenditure. Proc Natl Acad Sci U S A. 2007 Jun 19;104(25):10703–10708. doi: 10.1073/pnas.0611568104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blanchet E, Annicotte JS, Lagarrigue S, Aguilar V, Clape C, Chavey C, et al. E2F transcription factor-1 regulates oxidative metabolism. Nat Cell Biol. 2011 Sep;13(9):1146–1152. doi: 10.1038/ncb2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsieh MC, Das D, Sambandam N, Zhang MQ, Nahle Z. Regulation of the PDK4 isozyme by the Rb-E2F1 complex. J Biol Chem. 2008 Oct 10;283(41):27410–27417. doi: 10.1074/jbc.M802418200. [DOI] [PubMed] [Google Scholar]
- 35.Dannenberg JH, Schuijff L, Dekker M, van der Valk M, te Riele H. Tissue-specific tumor suppressor activity of retinoblastoma gene homologs p107 and p130. Genes Dev. 2004 Dec 1;18(23):2952–2962. doi: 10.1101/gad.322004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McClellan KA, Vanderluit JL, Julian LM, Andrusiak MG, Dugal-Tessier D, Park DS, et al. The p107/E2F pathway regulates fibroblast growth factor 2 responsiveness in neural precursor cells. Mol Cell Biol. 2009 Sep;29(17):4701–4713. doi: 10.1128/MCB.01767-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010 Sep 14;18(3):207–219. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther. 2008 Jan;7(1):110–120. doi: 10.1158/1535-7163.MCT-07-0482. [DOI] [PubMed] [Google Scholar]
- 39.Clem BF, Clem AL, Yalcin A, Goswami U, Arumugam S, Telang S, et al. A novel small molecule antagonist of choline kinase-alpha that simultaneously suppresses MAPK and PI3K/AKT signaling. Oncogene. 2011 Mar 21; doi: 10.1038/onc.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fan TW, Lane AN, Higashi RM, Yan J. Stable isotope resolved metabolomics of lung cancer in a SCID mouse model. Metabolomics. 2011 Jun 1;7(2):257–269. doi: 10.1007/s11306-010-0249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dranka BP, Benavides GA, Diers AR, Giordano S, Zelickson BR, Reily C, et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med. 2011 Nov 1;51(9):1621–1635. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hill BG, Dranka BP, Zou L, Chatham JC, Darley-Usmar VM. Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochem J. 2009 Nov 15;424(1):99–107. doi: 10.1042/BJ20090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perez J, Hill BG, Benavides GA, Dranka BP, Darley-Usmar VM. Role of cellular bioenergetics in smooth muscle cell proliferation induced by platelet-derived growth factor. Biochem J. 2010 Jun 1;428(2):255–267. doi: 10.1042/BJ20100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ciavarra G, Zacksenhaus E. Rescue of myogenic defects in Rb-deficient cells by inhibition of autophagy or by hypoxia-induced glycolytic shift. J Cell Biol. 2010 Oct 18;191(2):291–301. doi: 10.1083/jcb.201005067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.