

Abstract
Because many cancers harbor mutations that confer resistance to apoptosis, there is a need for therapeutic agents that can trigger alternative forms of cell death. Methuosis is a novel form of nonapoptotic cell death characterized by accumulation of vacuoles derived from macropinosomes and endosomes. Previous studies identified an indole-based chalcone, 3-(5-methoxy-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (MOMIPP), that induces methuosis in human cancer cells. Herein, we describe the synthesis of related 2-indolyl substituted pyridinylpropenones and their effects on U251 glioblastoma cells. Increasing the size of the 2-indolyl substituent substantially reduces growth inhibitory activity and cytotoxicity but does not prevent cell vacuolization. Computational models suggest that the results are not due to steric-driven conformational effects. The unexpected uncoupling of vacuolization and cell death implies that the relationship between endosomal perturbations and methuotic cell death is more complex than previously realized. The new series of compounds will be useful in further defining the molecular and cellular mechanisms underlying methuosis.
Keywords: Methuosis, vacuolization, glioblastoma multiforme, indole-based pyridinylpropenones, chalcones
With the exception of newer ‘targeted’ agents, the majority of drugs developed for cancer chemotherapy operate by triggering caspase-dependent apoptotic cell death via DNA damage,1 disruptions of the cytoskeleton,2 or induction of endoplasmic reticulum (ER) stress.3 However, many cancers harbor mutations in genes that are essential to promote an efficient apoptotic response (e.g., PTEN, p53, pRB), thus limiting their susceptibility to these approaches.4 Cancer cells that survive after treatments with apoptosis-inducing drugs often give rise to recurrent drug-resistant tumors.5,6 These challenges have spurred interest in discovering other mechanisms that can induce cell death. A number of distinct nonapoptotic cell death pathways have now been identified,7,8 and a recent review describes a broad spectrum of small molecules known to induce these pathways in cancer cells.9
Working predominantly with human glioblastoma cells, our group has defined a unique form of cell death termed methuosis.10−12 A hallmark of cells undergoing methuosis is extensive cytoplasmic vacuolization. The vacuoles arise from the merger of fluid-filled macropinosomes with clathrin-independent endosomal compartments under conditions where endosomal recycling and/or lysosomal trafficking are defective. Consequently, large vacuoles accumulate until they fill much of the cytoplasmic space. The vacuolated cells eventually lose membrane integrity, detach from the substratum, and die without the typical features of apoptosis. Methuosis is considered to be caspase-independent since it can occur in the presence of caspase inhibitors.10,12
We recently described a series of small, chalcone-like molecules that can induce methuosis in glioblastoma and other tumor cells.12,13 The most potent of these indole-based compounds was 3-(5-methoxy-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (MOMIPP, 1, Figure 1). MOMIPP induces methuosis in cultured glioblastoma cells at low micromolar concentrations.13 The indolyl- and pyridinyl-moieties demonstrate a high degree of structural specificity for induction of methuosis, as small modifications led to compounds that were inactive.13 For example, switching the position of the substituent from the para- to either the meta- or the ortho-positions so as to produce isomeric versions of 2 (the des-methyl predecessor of MOMIPP), abolished the induction of vacuolization and cell death. In general, previous structural modifications that prevent vacuolization also appear to minimize the loss of cell viability, suggesting that vacuolization is a key factor in the cell death mechanism. The foregoing findings, coupled with the reversibility of the effects of MOMIPP upon removal of the drug from the test medium, suggest that MOMIPP exerts its biological activity by interacting with discrete protein targets in a reversible manner.
Figure 1.

Structures of lead compound 1 (MOMIPP), its des-methyl predecessor (2), and its 3-pyridinyl-analogue (3, meta-MOMIPP). The latter has now been prepared to confirm its use as an inactive control during biological testing assays and studies directed toward the identification of relevant protein targets.
Interestingly, our previous structure–activity relationship (SAR) studies revealed that when a simple methyl-group is added to the 2-position of the indole ring, there is a significant increase in methuosis-inducing activity compared to the parent scaffold, which bears a hydrogen atom at this location (2).13 In the present report, we applied this same manipulation to our prior meta-pyridine analogue of 2 to confirm that the latter isomer still attenuates potency and that the combination resulting in 3 can thus provide for a very weakly active control that is otherwise identical in structure to lead compound 1. In addition, we continued to modify the 2-position with hydrocarbons having increased length and branching in order to further explore the effects upon vacuolization and viability within U251 glioblastoma cells.
Lead compound 1 and its novel isomer 3 were prepared from commercially available 5-methoxy-2-methylindole in a manner similar to our previously published methods, namely, formylation followed by condensations with 4-acetylpyridine and 3-acetylpyridine, respectively.13 Compound 2 was prepared from commercially available 5-methoxyindole and 4-acetylpyridine in a manner identical to our previously published method.13 Alternatively, the series of probes having larger alkyl groups in the 2-position required synthesizing the indole-system for each of the desired substituents. Thus, the syntheses of probes 18–21 (Scheme 1) began with 4-methoxy-2-methylaniline to which the addition of a Boc-group via di-tert-butyldicarbonate in refluxing THF was used to protect the amine (4). Acylation14 of the 2-methyl-group was accomplished using sec-butyllithium as a base. The resulting dilithio species enabled regioselective acylation with various synthesized Weinreb amides155–8 to produce intermediates 9. Derivatives of 9 were washed with 1 N HCl to neutralize base and then used directly in the next step. A more rigorous purification was not necessary as spontaneous cyclization was observed to occur slowly under neutral and ambient conditions. Removal of the N-Boc protecting group accompanied by controlled cyclization was performed in one-pot fashion using excess TFA to provide the key 2-substituted 5-methoxyindoles1410–13. Formylation reactions16 were performed using Vilsmeier–Haack acylation conditions yielding aldehydes 14–17, which were conveniently isolated as precipitates from basic solutions after treatment with 1 N NaOH. Claisen–Schmidt condensation17 of the aldehydes with 4-acetylpyridine yielded the series of novel 2-substituted indole-based pyridinylpropenones 18–21.
Scheme 1. Synthesis of 2-Substituted Indole-Based Pyridinylpropenones.

Reagents and conditions: (a) THF, sec-butyllithium, −40 to −50 °C; (b) 5–8, −50 to −10 °C; (c) TFA/DCM; (d) POCl3, DMF, 0 °C; (e) 4-acetylpyridine, piperidine, MeOH, reflux.
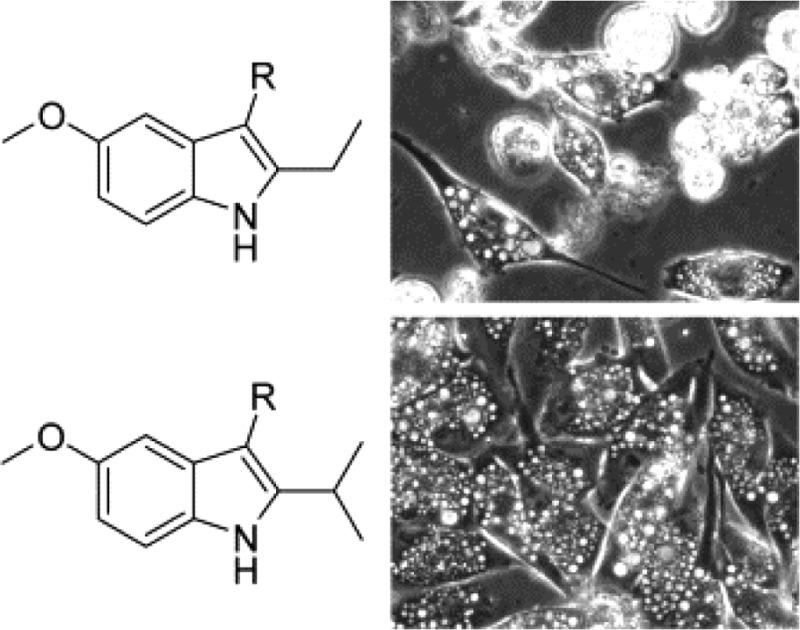
Known compounds 1 and 2, isomer 3, and probes 18–21, as well as a DMSO vehicle control, were tested in U251 glioblastoma cells using a sulforhodamine B (SRB) assay to assess their growth inhibition (GI) properties after two-day exposures of each test agent across a concentration range from 0 to 20 μM. Fifty-percent inhibition (GI50) values were derived from the resulting dose–response curves (Supporting Information) and are recorded in Table 1. The GI50 for the ethyl substitution, 18, was not statistically different from lead compound 1 (MOMIPP). In contrast, each of the other substitutions on the indole ring had a negative impact on growth inhibitory activity, with GI50 values increasing to >15 μM. The difference between compounds 1 and 18 compared to the other 2-substitued indole compounds was even more striking when evaluated by morphological and cell viability criteria. The typical progression of methuosis involves the initial formation of vacuoles, which can be detected as early as 2–3 h after addition of MOMIPP. Over the next 24–48 h, the progressive accumulation and enlargement of vacuoles is followed by cell rounding and detachment, with the majority of the detached cells exhibiting signs of disrupted membrane integrity. In our previous SAR studies comparing MOMIPP and a directed library of analogues,12,13 the compounds that most effectively triggered vacuolization also caused cell death, while those that failed to induce vacuoles were not cytotoxic. This led to the concept that accumulation of vacuoles is an important contributing factor in the methuosis death program. To evaluate the relationship between vacuolization and cell death using the current series of compounds, cells were examined by phase-contrast microscopy after one and two days of treatment (Figure 2). At 10 μM, the ethyl derivative (18) produced morphological effects indistinguishable from those observed with the same concentration of MOMIPP (1). After one day, the cells were sparse and heavily vacuolated, and by day two, many of the vacuolated cells had detached and lost membrane integrity. In contrast, cells treated with 3, the 3-pyridinyl isomer of MOMIPP, did not accumulate vacuoles and resembled the vehicle control cells in their ability to proliferate to high density by day two. Cells treated with compounds containing the H (2), n-Pr (19), i-Pr (20), and i-Bu (21) substituents at the 2-position of the indole ring were extensively vacuolated, but surprisingly, they remained attached and continued to increase in density. Trypan blue dye-exclusion assays clearly demonstrated that the progressive rounding and detachment of the vacuolated cells treated with 1 and 18 reflected a substantial loss of cell viability (Table 1 and Supporting Information). However, viability was only minimally affected in the vacuolated cells treated with 2, 19, 20, and 21 (Table 1).
Table 1. Cell Growth, Viability and Computational Chemistry Results.
| cmpd | substa | GI50b | % viablec | stscd | ststd | scscd | scstd |
|---|---|---|---|---|---|---|---|
| DMSOe | NAf | NA | 96.7 ± 0.6 | NA | NA | NA | NA |
| 2 | H | 18.6 | 91.2 ± 0.7 | 1.1 | 2.0 | 0.0 | 0.5 |
| 1 | Me | 2.1g | 38.1 ± 3.7 | 0.0 | 0.7 | 0.5 | 1.1 |
| 18 | Et | 3.2g | 34.6 ± 1.1 | 0.0 | 0.7 | 0.4 | 1.0 |
| 19 | n-Pr | 15.3 | 83.9 ± 2.3 | 0.0 | 0.6 | 0.1 | 1.0 |
| 20 | i-Pr | >20 | 78.8 ± 1.1 | 0.0 | 1.3 | 1.1 | 1.9 |
| 21 | i-Bu | 16.6 | 87.4 ± 1.5 | 0.0 | 0.5 | 0.7 | 1.5 |
| 3h | Me | 16.9 | 96.2 ± 1.3 | NA | NA | NA | NA |
Substituents at the indole’s 2-position.
Concentration of test agent in μM causing 50% inhibition of cell growth compared to DMSO control.
Percent of viable cells (mean ± SD), based on Trypan blue dye exclusion assays performed on three separate cultures treated with each compound at 10 μM for 2 days.
Energies in kcal/mol for each of the four conformations studied by computational chemistry. The nomenclature for these conformations is described in detail within the text and in Figure 3.
Vehicle control for delivery of test agents via DMSO.
Not applicable.
Dose–response curves for the Me and Et derivatives were repeated six more times. The means (±SD) for all seven determinations were as follows: Me, 2.5 ± 0.6; Et, 2.8 ± 0.5. The difference was not significant at p < 0.2 (Student’s t test).
This compound has a 3-pyridyl arrangement, while all others have the 4-pyridyl arrangement shown to be important for activity.
Figure 2.

Effects of MOMIPP (1) and related 2-indolyl-substituted pyridinylpropenones on morphology of U251 glioblastoma cells. Cells were observed by phase contrast microscopy on days one and two after the addition of the indicated compounds at 10 μM. Vacuoles appear as clusters of phase-lucent puncta within the cytoplasm of the cells. Rounded cells that are poorly focused in the cultures treated with 1 and 18 have detached from the surface of the dish.
The initial increase in cytotoxicity observed when substituting from H to either Me or Et followed by the abrupt falloff thereafter when substituting to n-Pr, i-Pr, and i-Bu is intriguing. Explanations for such an SAR trend are likely associated with switching the H to a more electron donating and lipophilic alkyl group. Possible consequences from substitution include (i) a subtle but potentially broad-ranging enhancement in the electronic features of the central pharmacophore such as a favorable impact upon the structurally embedded double-vinylogous-amide; (ii) an enhanced interaction with a hydrophobic pocket within this distinct locale of the target protein; or (iii) some combination of these factors. However, for all of these possibilities, it is also clear that their benefit must be achieved by encompassing only a compact region of space before the presence of additional bulk becomes deleterious to potency. This requirement can potentially arise from a limitation derived from either (iv) the existence of a tight steric boundary within this distinct pocket of the target or (v) an influence upon the molecule itself in terms of sterically perturbing a conformational arrangement that is preferred for overall target interaction. While assessment of possibilities (i)–(iv) will require further studies to identify the protein target(s) that mediate methuosis, factor (v) is amenable to computational studies, which, in turn, can also help to inform the design of future probes. Our computational assessment is summarized below. A more elaborate description of the computational studies can be found in the Supporting Information.
Table 1 lists the energies from molecular-mechanics calculations for the various conformations that can be adopted by compounds 1 and 2 as well as probes 18–21. An implicit solvent model was used so as to simulate the low polarity environment within a protein’s ligand-binding pocket. Since NMR data indicate that the double bond located α,β to the ketone possesses trans-stereochemistry (discernible by higher coupling constants) in all compounds, only this arrangement was considered. To address the single bonds, these compounds can be treated as double rotors wherein there is rotation between the indole ring and the connecting chain, and again about the C–C bond in the C=C–C=O moiety. The conformational possibilities that can be derived from these arrangements and their appropriate nomenclature are depicted in Figure 3. The first two letters, “st” or “sc” (s-trans and s-cis, respectively), refer to the relative position of the formal C2–C3 double bond of the indole and the C=C bond of the connecting chain. The second two letters refer to the relative conformational arrangement across the C=C–C=O system. From this molecular construct, the pyridine ring can be expected to adopt its most favorable position when optimizing the corresponding stsc, stst, scsc, and scst conformations. For the case of the 2-propyl substituent, a gauche conformation with respect to the C2 atom of the indole ring was accepted at the onset of the geometry optimization.
Figure 3.

Structural depictions of conformations considered during the computational studies. See text for descriptions of their associated nomenclature.
Only for the case of the smallest 2-substituent, namely, compound 2 having an H atom, is the most favorable conformation scsc. For lead compound 1 and all of the other probes 18–21, the stsc conformer is the most stable. This conformation (Figure 3) maintains an s-trans relationship between the C2–C3 bond of pyrrole and the C=C bond of the connecting chain, and an s-cis conformation across the C=C–C=O moiety. Apparently, the increasing size of the 2-substituent influences the connecting chain to turn farther away from the added bulk. Also notable is that the indole-CCCO substructure moiety is generally not coplanar. Torsion angles about the rotational bonds deviate by up to 30° from a fully coplanar system corresponding to 0° for sc and 180° for st, respectively. The C=C–C=O moiety is s-cis in all the sxsc (x = t or c) preferred structures, and the pyridine ring is generally out of the O=C–C4 (pyridine) plane by about 30°. While it is tempting to speculate that the noted conversion from an scsc to an stsc conformational preference when going from H to Me or E , may represent yet another possible explanation for at least the initial portion of the SAR trend discussed above, the actual energy differences are rather small. Thus, the more appropriate conclusion to draw from a purview of Table 1 is that the modeling studies predict small, up to at most 2 kcal/mol, relative conformer energies across all of these arrangements. When taken within the context of simultaneously interacting with the ligand-binding region on a protein surface, such small energy differences likely would not prevent rotations in order to adopt any conformation (even if not of the lowest relative ligand energy) that could most favorably bind to the protein. This suggests that the second part of the SAR trend, wherein there is a falloff in cytotoxicity with increasing size of the 2-substituent, is likely not due to some conformational effect and, instead, may result from a steric boundary that resides in this specific locale of the ligand-binding pocket.
In conclusion, the studies reported herein provide several new insights regarding the activity of the methuosis-inducing indolyl-substituted pyridinylpropenones. The results confirm that switching the pyridine N from the 4-position within lead compound 1 to the 3-position so as to provide 3 eliminates the ability of the compound to induce vacuolization and substantially reduces potency in growth inhibition and cytotoxicity assays. This is important because it implies that the compounds that elicit vacuolization and methuotic cell death are acting through one or more distinct protein targets that have very specific structural requirements for interaction with their ligands. In this regard, the SAR associated with growth inhibition and cytotoxicity suggests that when MOMIPP is bound to its protein target(s), there may be a pocket near the 2-position of the indole ring that can tolerate small groups capable of favorably influencing the electronics of the parent ligand or directly enhancing interactions with the target protein. Our computational studies suggest that while the addition of steric bulk to the 2-position can affect the preferred conformations adopted by the connecting chain, these effects are small and likely to be relevant only when the compounds are dissolved in nonpolar solvents. Thus, when considered within the context of the energies typically encountered when ligands associate with protein targets, all conformational possibilities are likely to be available to analogues having various levels of steric bulk in the 2-position, at least up to that of an i-Bu group. Taken together, the overall SAR suggests that further exploration of the impact of this key region on biological activity should be undertaken with reasonably small substituents that have a range of electronic, lipophilic, and hydrogen bonding properties.
A major novel finding from these studies is the demonstration that increasing the size of aliphatic substituents at the 2-position of the indolyl moiety does not reduce vacuolization but substantially reduces cytotoxicity. The surprising dissociation of vacuolization and cell death revealed by this series of compounds has not been observed in previous SAR studies. Several possible explanations can be envisioned to account for this observation. One is that the vacuoles induced by the n-Pr, i-Pr, and i-Bu compounds are functionally different from those induced by the Me or Et substituted compounds, so that their accumulation has a less severe impact on intracellular vesicular trafficking and cellular metabolism. Alternatively, the cytotoxic compounds, MOMIPP (1) and 18 may have unique pleiotrophic effects on specific targets beyond those that lead to endosomal vacuolization. In this scenario, endosomal vacuolization remains an essential contributing factor to methuosis but must be combined with other cellular insults in order to cause eventual metabolic collapse and cell death. The current series of analogues should prove to be very useful in future studies aimed at dissecting the underlying mechanisms of methuosis and the relationship between endosomal vacuolization and cell death.
Acknowledgments
We thank the NIH for financial support (R01Ca115549) and Dr. Yong Wah Kim for maintenance of the NMR facility.
Glossary
Abbreviations
- MOMIPP
3-(5-methoxy-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one
- SAR
structure–activity relationship
- DMF
dimethyl formamide
- POCl3
phosphorus oxychloride
- MeOH
methanol
- DMSO
dimethyl sulfoxide
- SRB
sulforhodamine B
- THF
tetrahydrofuran
- ER
endoplasmic reticulum
- TFA
trifluoroacetic acid
- NA
not applicable
Supporting Information Available
Experimental protocols, computational methods, supporting biological figures, and characterization of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by the NIH (R01CA115495) and by the Harold and Helen McMaster Endowment for Biochemistry and Molecular Biology.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hurley L. H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [DOI] [PubMed] [Google Scholar]
- Dumontet C.; Jordan M. A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat. Rev. Drug Discovery 2010, 9, 790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonthal A. H. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem. Pharmacol. 2013, 85, 653–666. [DOI] [PubMed] [Google Scholar]
- Delbridge A. R.; Valente L. J.; Strasser A. The role of the apoptotic machinery in tumor suppression. Cold Spring Harbor Perspect. Biol. 2012, 4, a008789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M.; Fojo T.; Bates S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [DOI] [PubMed] [Google Scholar]
- Abdullah L. N.; Chow E. K. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2013, 2, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L.; Vitale I.; Abrams J. M.; Alnemri E. S.; Baehrecke E. H.; Blagosklonny M. V.; Dawson T. M.; Dawson V. L.; El-Deiry W. S.; Fulda S.; Gottlieb E.; Green D. R.; Hengartner M. O.; Kepp O.; Knight R. A.; Kumar S.; Lipton S. A.; Lu X.; Madeo F.; Malorni W.; Mehlen P.; Nunez G.; Peter M. E.; Piacentini M.; Rubinsztein D. C.; Shi Y.; Simon H. U.; Vandenabeele P.; White E.; Yuan J.; Zhivotovsky B.; Melino G.; Kroemer G. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzaler P.; Watson C. J. Killing a cancer: what are the alternatives?. Nat. Rev. Cancer 2012, 12, 411–424. [DOI] [PubMed] [Google Scholar]
- Kornienko A.; Mathieu V.; Rastogi S. K.; Lefranc F.; Kiss R. Therapeutic agents triggering nonapoptotic cancer cell death. J. Med. Chem. 2013, 56, 4823–4839. [DOI] [PubMed] [Google Scholar]
- Overmeyer J. H.; Kaul A.; Johnson E. E.; Maltese W. A. Active Ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol. Cancer Res. 2008, 6, 965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhanot H.; Young A. M.; Overmeyer J. H.; Maltese W. A. Induction of nonapoptotic cell death by activated Ras requires inverse regulation of Rac1 and Arf6. Mol. Cancer Res. 2010, 8, 1358–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overmeyer J. H.; Young A. M.; Bhanot H.; Maltese W. A. A chalcone-related small molecule that induces methuosis, a novel form of non-apoptotic cell death, in glioblastoma cells. Mol. Cancer 2011, 10, 69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. W.; Overmeyer J. H.; Young A. M.; Erhardt P. W.; Maltese W. A. Synthesis and evaluation of indole-based chalcones as inducers of methuosis, a novel type of nonapoptotic cell death. J. Med. Chem. 2012, 55, 1940–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark R. D.; Muchowski J. M.; Fisher L. E.; Flippin L. A.; Repke D. B.; Souchet M. Preparation of indoles and oxindoles from N-(tert-butoxycarbonyl)-2-alkylanilines. Synthesis 1991, 10, 871–878. [Google Scholar]
- Nahm S.; Weinreb S. M. N-Methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett. 1981, 22, 3815–3818. [Google Scholar]
- Gillard J. W.; Belanger P. Metabolic synthesis of arylacetic acid antiinflammatory drugs from arylhexenoic acids 0.2. Indomethacin. J. Med. Chem. 1987, 30, 2051–2058. [DOI] [PubMed] [Google Scholar]
- Van Order R. B.; Lindwall H. G. Indole-3-aldehyde and certain of its condensation products. J. Org. Chem. 1945, 10, 128–133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.