

Abstract
Neutral lipid triglycerides, a main reserve for fat and energy, are stored in organelles called lipid droplets. The storage and release of triglycerides are actively regulated by several proteins specific to the droplet surface, one of which in insects is PLIN1. PLIN1 plays a key role in the activation of triglyceride hydrolysis upon phosphorylation. However, the structure of PLIN1 and its relation to functions remain elusive due to its insolubility and crystallization difficulty. Here we report the first solid-state NMR study on the Drosophila melanogaster PLIN1 in combination with molecular dynamics simulation to show the structural basis for its lipid droplet attachment. NMR spin diffusion experiments were consistent with the predicted membrane attachment motif of PLIN1. The data indicated that PLIN1 has close contact with the terminal methyl groups of the phospholipid acyl chains. Structure models for the membrane attachment motif were generated based on hydrophobicity analysis and NMR membrane insertion depth information. Simulated NMR spectra from a trans-model agreed with experimental spectra. In this model, lipids from the bottom leaflet were very close to the surface in the region enclosed by membrane attachment motif. This may imply that in real lipid droplet, triglyceride molecules might be brought close to the surface by the same mechanism, ready to leave the droplet in the event of lipolysis. Juxtaposition of triglyceride lipase structure to the trans-model suggested a possible interaction of a conserved segment with the lipase by electrostatic interactions, opening the lipase lid to expose the catalytic center.
Keywords: lipid storage droplet protein, triglyceride lipolysis, proton spin diffusion, magic-angle spinning, solid-state NMR, MD simulation
1. Introduction
Animals store most of excess energy in the form of neutral lipid triglycerides for later use as metabolic fuel. The hydrophobicity of triglycerides allows them to be densely packed into lipid droplets, providing an energy density 10 times that of hydrated proteins and carbohydrates [1]. The lipid droplets are composed of a triglyceride core surrounded by a monolayer of phospholipids and a variety of proteins [2]. Utilization of the stored triglycerides requires enzymatic breakdown (lipolysis) by lipases, while the surface layer of the droplet controls the accessibility of lipases to the stored triglycerides. Among the proteins surrounding the lipid droplet surface, proteins in the PAT family (named after three earliest members) have raised great interest in recent years. The PAT family consists of the mammalian Perilipin, ADRP, TIP47, S3-12, and OXPAT, as well as insect lipid storage droplet protein 1 (Lsd1) and 2 (Lsd2) [3]. Perilipin, ADRP, and Lsd1 constitutively attach to the lipid droplets, and they maintain fat storage and regulation of lipolysis. TIP47, S3-12, OXPAT, and arguably Lsd2 bind reversibly to the droplets; hypothetically they are responsible for the packaging of newly synthesized triglycerides into lipid droplets [3]. A new nomenclature has been recently proposed for the PAT-family of proteins [4]. Accordingly, from now on we will refer Lsd1 and Lsd2 as PLIN1 and PLIN2, respectively. Mammals and insects share significant conservation in the molecular mechanism of lipid droplet metabolism, highlighting the tremendous potential of using genetic technical advantages of insects to discover novel features of lipid homeostasis [3]. Studies of fruit fly models have established a correlation between triglyceride accumulation and the level of PLIN2 expression [5]. PLIN1 is found exclusively associated with lipid droplets [6]. It dynamically interacts with lipid droplet to control access of lipase to triglycerides thus regulates the lipids homeostasis. In contrast to mouse perilipin, which protects triglycerides from hydrolysis [7, 8], depletion of PLIN1 leads to adult-onset obesity [9] while overexpression of PLIN1 induces lipid droplet to shrink and aggregate [10]. PLIN1 serves as a lypolytic switch, which upon protein kinase A (PKA) mediated phosphorylation, promotes the activation of triglyceride lipolysis [11].
The association of these proteins on the surface of the lipid droplets is critical to their ability to properly regulate both storage and release of the triglycerides in the droplets. Despite the pressing need to understand the interaction between these proteins and the lipid droplets, progress has been hampered by the scarcity of three-dimensional structure information for these proteins. The first and so far the only structure determined was for the C-terminal TIP47 (residues 191-437) at 2.8 Å resolution using X-ray crystallography [12]. The structure consists of an α/β domain and a four-helix bundle that resembles the receptor-binding domain of apolipoprotein E. The deep hydrophobic cleft between the α/β domain and the four-helix bundle is consistent with binding to hydrophobic proteins and small molecules, rather than to the extended phospholipid membrane. This C-terminus construct was selected from one dozen truncations for soluble protein expression. Regrettably, it does not have the N-terminal 11-mer helical repeats that are probably responsible for reversible binding to lipid membranes.
Meanwhile, in vitro systems have recently become available for structural and functional studies. Recombinant PLIN1 has been purified and reconstituted in lipid droplet-like particles [13, 14]. Using an in vitro system, it was shown that phosphorylation of PLIN1 enhances the triglyceride lipase activity, demonstrating the direct connection between PLIN1 phosphorylation and the activation of lipolysis [13]. Hypothetically, PLIN1 phosphorylation causes changes on the droplet surface, making the internal triglycerides more accessible to the lipase [11]. The ability to reconstitute PLIN1 in lipoprotein particles opens the possibility to design structural studies to advance our understanding of the mechanisms of lipolysis regulation. Nevertheless, these lipoprotein particles are too large (~20 nm diameter) for solution NMR studies, and they are very difficult if not impossible to form diffraction quality single crystals for crystallography studies. Fortunately, several other structural techniques could be applied to this type of samples. For example, topologies of lipoprotein complexes have been determined using solid-state NMR [15, 16], fluorescence spectroscopy [15–18] and electron paramagnetic resonance (EPR) [19]. Among these, solid-state NMR is particularly suitable to study these lipoprotein complexes because three-dimensional structure details could be obtained [22–29]. Here we report both NMR experimental data and structural models that could be useful to advance our understanding of protein targeting to lipid droplet and the function of PLIN1.
2. Material and methods
2.1. Protein expression and purification
Isotopically enriched (13C, 15N) ISOGRO, 15NH4Cl and uniformly labeled 13C-glucose, were purchased from Sigma-Aldrich (St. Louis, MO). Lipid 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol (DMPG) was purchased from Avanti Polar Lipids (Alabaster, AL). Benzonase was purchased from EMD Millipore (Billerica, MA). The over-expression of PLIN1 (CG10374, NP_732904.2) as a fusion protein with thioredoxin-[His]6-Stag was carried out as previously reported [13] with slight modifications to incorporate stable isotopes for NMR studies. Transformed E. coli Rosetta cells with the recombinant plasmid (pET32-CG10374) were grown in 200 mL Luria broth medium at 37 °C until optical density 0.8 at 600 nm. The bacteria pellet was collected by centrifugation, and cultured in 1 liter M9 minimal medium containing reagents enriched with NMR-active stable isotopes (13C-glucose and 15NH4Cl). The medium was supplemented with properly labeled algae extracts (ISOGRO) to boost protein yield. When optical density reached 0.8, protein expression was induced by addition of 1 mM IPTG. After 6 hours, cells were harvested by centrifugation. Thioredoxin-Lsd1 fusion protein was purified essentially as previously described [12]. The final protein pellet was resuspended in 20 mM Tris, pH 8.0, 6 M Urea, 150 mM NaCl, 10 mM dithiothreitol and a solution of protein stock (1.7 mg/mL) was stored in the freezer.
2.2. Reconstitution of thioredoxin–PLIN1 in lipoprotein particles and thrombin cleavage
Thioredoxin-PLIN1/DMPG complexes were prepared as previously described with a final lipid to protein ratio of 70:1 [12]. After exhaustive dialysis, thioredoxin-PLIN1/DMPG complexes were brought to 60% (w/v) sucrose and subjected to ultracentrifugation in a sucrose density gradient (30 to 60 % (w/v)). The distinct white band floating at a density of 1.17g/ml was collected as a single fraction. Complexes were sedimented by ultracentrifugation in an aqueous buffer and resuspended in a buffer containing 5 mM Na2HPO4, 0.15 M NaCl, 0.1% octylglucoside at pH 7.4 and incubated with thrombin (1 unit/mg of protein) for 15 h at 4 °C to cleave the thioredoxin-[His]6 tag. After centrifugation, the pellet containing PLIN1/DMPG complex was washed with 5 mM phosphate buffer (pH 7.5) and excess of water was removed in the speed vac for 1 h. Based on proton NMR signal intensities, the sample contained about 35% wt of water. These complexes were previously reported to have apparent diameter of 20 nm [13], and they are likely small unilamellar vesicles (SUVs) [20]. The head groups of anionic lipids, such as DMPG, facilitate interaction with positively charged protein sidechain groups and they are important for membrane attachment of the protein. Neutral lipid DMPC was also tested, but it resulted in dramatic protein loss by sticking to the dialysis membranes and the centrifuge tubes. DMPG provided a cost effective way to obtain isotopically enriched PLIN1 lipoprotein complexes.
2.3 NMR spectroscopy
All Solid-state NMR experiments were carried out on a 600 MHz Varian INOVA spectrometer and a triple resonance magic-angle spinning (MAS) probe with a 1.6 mm spin module. All spectra were acquired with a MAS rate at 13.3 kHz. For 13C 1D and 13C-13C 2D experiments, the proton 90° pulse was 2.2 μs, cross polarization (CP) contact time 0.7 ms, locking fields of 73 kHz on 1H and 80 kHz on 13C channels, 100 kHz two pulse phase modulation (TPPM) decoupling [21], and dipolar-assisted rotational resonance (DARR) recoupling [22]. For proton spin diffusion experiment, the proton-detected NHH pulse sequence (Fig. S1 in Supplementary Data) was modified from the CHH sequence [23] with MISSISSIPPI solvent suppression [24] and an additional T2 filter (300 μs) [25] to suppress signals from the less mobile protein molecule (Fig. S2 in Supplementary Data). The CP locking fields were 73 and 60 kHz on 1H and 15N channels, respectively. Contact time for the first CP (1H to 15N) was 1 ms, and 0.6 ms for the second CP (15N to 1H). Other experimental details can be found in figure captions.
2.4. MD simulation and spectral simulation
All MD simulations were performed on up to 600 processors on a linux cluster supercomputer using software GROMACS 4.5.5 and GROMOS96 54A7 force field combined with DMPG lipid interaction parameters with simple point charge-extended (SPCE) water model [36, 37]. The non-bonded van der Waals interactions were estimated using Lennard-Jones potential with cutoff value of 1.2 nm and the bonds were constrained by linear constraint solver (LINCS) algorithm [26]. Electrostatic forces and energies were calculated using Particle-Mesh Ewald (PME) summation algorithm with cutoff value of 1.2 nm too [27].
Two equilibration phases, constant volume (NVT) and constant pressure (NPT) ensembles, were subsequently carried out, each with 1 fs time steps. In the first phase, the system was coupled to a strong temperature bath using V-rescale coupling [28] with temperature coupling constant of τT = 0.1 ps to maintain system temperature at 300 K. In the second phase, Parrinello-Rahman pressure coupling [29] with coupling constant τP = 5.0 ps to maintain the pressure semi-isotropically at 1 bar and a weak Nose-Hoover temperature coupling with a coupling constant τT = 0.5 ps [30–32] was used to ensure a true NPT ensemble. A total of 15 ns equilibration was followed by a 50 ns production run of molecular dynamics (MD) in 2 fs step size, during which temperature and pressure were maintained using weak coupling methods (Nose-Hoover with τT = 0.5 ps and Parrinello-Rahman with τP = 2 ps).
For a given structure model, backbone and β-carbon chemical shifts were predicted by shiftX [33]. Based on these chemical shifts, 13C-13C 2D spectra were simulated by program peaks2ucsf in the Sparky package (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco) with assistance of a custom computer script.
3. Results and discussion
3.1 Spectra of 13C,15N-PLIN1
Initially, 13C 1D spectrum on 13C, 15N-uniformly labeled PLIN1 was performed to assess resolution and sensitivity, providing feedback for sample preparation process. The spectrum was acquired at several temperatures to assess cross polarization dynamics (Fig. 1). Overall signal intensity increased by 30% when the temperature was lowered from −15 to −37 °C, due to the fact that cross polarization from proton to 13C is more efficient when the dipolar coupling is strong in rigid molecules. The lipid methylene intensity increased by 50% instead, indicating that the mobility of acyl chains is reduced at lower temperatures than the protein, which presumably binds to the surface of the lipid membrane. From these 1D spectra, signals can be identified for carbonyl, aromatic ring, Cα, and side chain carbons.
Figure 1.

13C cross polarization spectra of 13C,15N-PLIN1 lipoprotein complexes at different temperatures. Spectra were acquired on a 600 MHz instrument with the sample spinning at 13.3 kHz. For each spectrum 128 scans were accumulated. The data were apodized with 40 Hz line broadening. Calibrated sample temperature values are labeled in the figure.
Several 13C-13C 2D correlation spectra were obtained to examine the chemical shift dispersion among amino acid types, and to analyze the secondary structures based on chemical shifts (the resonance position in a spectrum, in ppm unit) [34]. The 13C–13C 2D spectrum (Fig. 2), acquired with 10 ms DARR mixing [22], shows peaks in all the expected locations. For example, the serine, threoine, alanine, and isoleucine correlations have been labeled in the Figure. Correlations observed in isolated regions of the spectrum (e.g., Ala Cα-Cβ) provide a means to performing analysis of secondary structure. Further analysis within the unique chemical shift ranges of various residue types showed that the Cα, Cβ and CO (carbonyl carbon) signals were observed with chemical shifts indicative of their secondary structure. For example, 47% of the Ala Cα-Cβ correlations intensities (Fig. 2 inset) are found in the area consistent with Cα and Cβ chemical shifts characteristic of a-helical structures; 31% of the intensities are found in the area charateristic of turn or random coil structures; 22% in the area characteristic of b-sheet. These numbers are consistent with the analysis based on circular dichroism spectrum: 34% a-helix, 16% b-sheets, 50% turns or random coil [13]. Fig. 5B shows the 13C–13C 2D spectrum with 50 ms DARR mixing to create longer-range correlations. Among the many new peaks, the isoleucine Cδ1-Cγ2 and the inter-residue Cα-Cα correlations are also observed. Moreover, the individual outlying peaks allow estimation of linewidth to be about 0.5 ppm.
Figure 2.
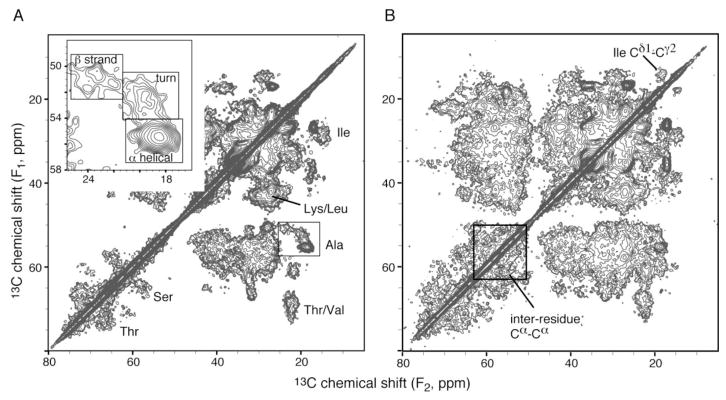
13C-13C 2D correlation NMR spectra of the 13C,15N-PLIN1 sample with 10 ms (A) and 50 ms (B) DARR mixing. The inset is the expansion of the alanine Cα-Cβ region, with three boxes marking the characteristic chemical shift ranges for the three types of secondary structure. Data were acquired on a 600 MHz instrument with the sample spinning at 13.3 kHz. Each spectrum took 42 hours, with 96 scans per row. Sample temperature was −37 °C.
Figure 5.

The trans-model of PLIN1 membrane-anchoring motif. (A) Starting model constructed based on hydrophobicity of predicted helices (hydrophobic H6 and H8, amphipathic H7 and H9) [13] and NMR membrane insertion information. (B) Top view of the model after 50 ns MD simulation in DMPG lipid bilayer. Alpha helices are shown in purple, coils in gray, turns in cyan, and 282EPENQARP289 motif in green. Lipids of the top leaflet are shown in orange, with the phosphorus shown in yellow sphere. (C) Side view of the model showing proximity between helices 6 and 8 with the lipid methyl groups. The lipids from top layer are shown in orange lines while those from bottom layer in blue. (D) Juxtaposition of the triglyceride lipase structure (PDB 4TGL) to the PLIN1 model. PLIN1 is shown in black, with the 282EPENQARP289 motif shown in green, alpha carbon of E282 and E284 in red ball, alpha carbon of R288 in blue ball. TGL is shown in gray, with the opened lid (residues 82 to 96) in purple, the exposed catalytic center (S144, D203, H257) in CPK molecular models, alpha carbon of R86 in blue ball, and alpha carbon of D91 in red ball.
3.2. Membrane insertion by spin diffusion
The attachment of PLIN1 on the surface of the lipid droplets is critical to its function in the activation of triglyceride hydrolysis. The four helices predicted in the central region have been hypothesized to be droplet targeting motifs based on their hydrophobicity (hydrophobic H6 and H8, amphipathic H7 and H9) [13]. To verify this hypothesis and to probe the depth of membrane insertion, NHH spin diffusion NMR experiment was performed at 45 °C, higher than the gel to liquid-crystalline phase transition temperature (23 °C) of DMPG (Fig. 3A). This experimental temperature was chosen to give relatively sharp lipid methyl and methylene signals in proton spectra (Fig. 3B). The proton resonances are assigned based on the published lipid NMR data [42, 43]. Particularly, water resonates ca. 4.7 ppm, acyl chain methylenes at ca. 1.3 ppm, and the terminal methyl groups of the acyl chains at ca. 0.8 ppm. In NHH spin diffusion experiments (Fig. S1 in Supplementary Data), proton polarization was first transferred to 15N by cross polarization and all remaining proton signals were then wiped off. The polarization was transferred back to protein proton and propagated to water and lipids via spin-diffusion (tm). The initial spectrum (tm = 0 ms, Fig. 3C) shows weak residue amide signals not completely removed by 300 μs T2 filter (also see Fig. S2 in Supplementary Data); longer T2 delays hurt experimental sensitivity. Nevertheless, the aliphatic region (0–4 ppm) was free of interfering protein signals. The non-zero water signal likely originated from chemical exchange with lysine and arginine side chain amine protons, which could occur during 15N to proton cross-polarization transfer. With increasing diffusion time (4 and 36 ms in Fig. 3C), water and lipid signals grew stronger.
Figure 3.
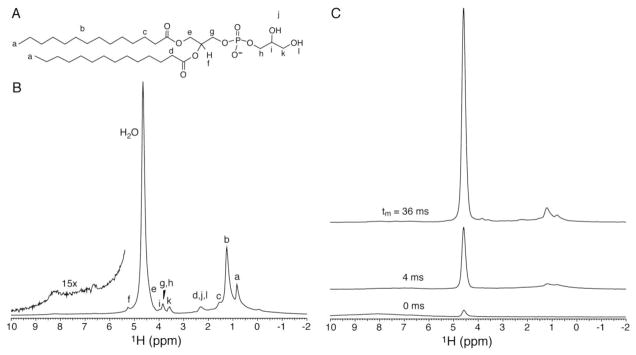
(A) DMPG molecule with proton designations. (B) Direct proton excitation spectrum on uniform 13C,15N-PLIN1 reconstituted into DMPG vesicles at 45°C. The amide region is magnified in the inset. (C) NHH spin diffusion spectra with diffusion time as indicated in the figure; 600 scans were acquired for each spectrum.
The dependence of peak intensities on serial diffusion times, namely, spin diffusion buildup curves, are shown in Fig. 4. The buildup rate of how fast a buildup curve reaches its plateau relates to the distance between the protein and water or lipid groups. The curve of the terminal CH3 groups of the acyl chains in Fig. 4 exhibits a much faster buildup rate than water, indicating that a region of PLIN1 has close contact with the lipid acyl chains. This region of PLIN1 is buried in the middle of the bilayer in the native-like lipoprotein complexes; in the native lipid droplets, it very likely reaches the interface between the lipid monolayer and the triglyceride core. This deeply buried region should be critical to droplet attachment for PLIN1. Moreover, when PLIN1 is activated by phosphorylation, this region may be involved in opening the phospholipid monolayer to grant lipase access to the stored triglycerides.
Figure 4.

Proton 1D spin diffusion data buildup curves as a function of mixing time. The CH2 and CH3 curves are also rescaled for easy comparison of buildup rates. The fast growing CH3 curve (down triangle) indicates a close contact between protein and the phospholipid acyl chain terminals, meaning that this part is deeply buried inside the lipid bilayer. While the CH2 group (up triangle) shows a clear biphasic behavior indicating that the there are two types of interactions with CH2 groups from the protein, possibly the one buried inside the bilayer and the one in the cytosol part. The experiment was performed at 45 °C and data points were corrected for T1 relaxation.
The CH2 buildup curve clearly shows a biphasic behavior, which can be decomposed into two exponential growth components (Fig. S3 in Supplementary Data). The fast component has a buildup rate similar to the CH3 curve, very likely arising from a region of PLIN1 in close contact with the acyl chain. The slow component may be attributed to long-range diffusion from cytosolic regions of PLIN1. The buildup rate of water is also relatively fast, indicating close contact of water with the protein. The rate is slower than the rates of CH2 and CH3 in close contact with the protein, due to a smaller dipolar interaction resulted from fast motion of water molecules.
The proton spin diffusion data on water, CH2 and CH3 protons can provide a semi-quantitative evaluation of the protein regions located in different environments. The relative saturation intensities are roughly 85%, 8.5%, and 6.5% for water, CH2, and CH3 respectively, according to Fig. 3 and taking into account that half of the CH2 intensity arises from close contact (Fig. S3). The sum of the latter two, 15%, represents percentage of the PLIN1 molecule anchored in the lipid membrane; this value is in very good agreement with the percentage (13%) of highly hydrophobic regions (residues 249 to 275 and 290 to 318) with regard to total number of amino acids (431) in the protein [13].
3.3. Model building
So far, there is no structure or structure model for any PAT proteins bound to membrane. The conserved PAT domain of the PLIN1 is highly soluble and unlikely to participate in lipid interaction. No transmembrane domain was found in PLIN1 using HMMTOP server [35]. Bioinformatics analysis indicated that four predicted helices might participate in membrane targeting: hydrophobic helices H6 (249-261) and H8 (290-300) as well as amphipathic helices with high hydrophobic moment H7 (265-275) and H9 (301-318) [13]. The spin diffusion NMR experiments above corroborated this hypothesis. It is therefore interesting to build structural models for the membrane-binding domain, contained in the segment stretching from residue 249 to 318. This segment also includes a highly conserved hydrophilic motif 282EPENQARP289 that could act as a modulator of lipolysis [6].
Two models of the most probable membrane-binding motif (residues 249 to 318) containing predicted helices 6 to 9 [13] were manually constructed based on hydrophobicity distribution: trans-model in Fig. 5A and cis-model in Fig. S4A in the Supplementary Data. The amphipathic helices H7 and H9 most likely lie in the membrane-cytosol interface. The loop between H7 and H8 is so placed as to expose the hydrophilic conserved 282EPENQARP289, which could be functionally important. This requires the hydrophobic helices H6 and H8 to be placed parallel to the membrane surface. Had these two helices been aligned perpendicular to the membrane surface, this hydrophilic stretch would have been pulled into the membrane. First, the Cα trace was designed with assistance of Visual Molecular Dynamics (VMD) [36] and then the all-atom molecule structure was generated by Structural Alphabet based protein Backbone Builder from alpha Carbon trace (SABBAC) 1.3 [37]. The two starting models (shown in Fig. 5A and Fig. S4) were energy minimized in vacuum using GROMACS 4.5.5 [38] and then embedded in a bilayer containing 512 DMPG molecules, with the amphipathic helices right above the lipid phosphate groups. The atomic level coordinates and interaction parameters for well-equilibrated DMPG lipid bilayer [39] were downloaded from Lipidbook [40]. These models were put in periodic triclinic boxes, solvated with adequate water and counterions were added to obtain electrically neutral system. These systems were then taken through steepest descent energy minimization and found to converge to physically realistic minimum energy value with maximum force less than 100 kJ/mol/nm. Then a total of 15 ns equilibration was performed, followed by a 50 ns production run of molecular dynamics (see details in Material and Methods).
The final structure of the trans-model is shown in Fig. 5B and 5C. All helices were able to maintain the helical structures, but helices 7 and 8 slightly unwound. In the final structure, H6 and H8 both have close contacts with the terminal methyl groups of the acyl chains from both leaflets, agreeing with the spin diffusion data (Fig. 5C). The acyl chains of these lipids from the top leaflet wrapped around the helices to accommodate the perturbation caused by the protein. Such perturbation also caused the two leaflets to come close locally, allowing the terminal methyl groups from the bottom leaflet contact H6 and H8. Interestingly, lipids from the bottom leaflet were getting very close to the surface in the region enclosed by the helices. This may imply that in real lipid droplet, triglyceride molecules might be brought close to the surface by the same mechanism, ready to leave the droplet in the event of lipolysis.
In Fig. 5D, the crystal structure of a fungus triglyceride lipase [41] is juxtaposed to the PLIN1 model. In this structure, the lid (82SSSIRNWIADLTFVP96, with residues 83 to 84 and 91 to 95 as hinges and charged residues underlined) of the lipase is propped open by diethyl p-nitrophenyl phosphate to expose a patch of hydrophobic area of 800 Å2 and the catalytic center residues S144, D203, H257. It is interesting that the footage of the lipase matches with PLIN1 trans-model, and that the lipase lid has a similar orientation to the 282EPENQARP289 (charged residues underlined) motif of PLIN1. More importantly, two possible electrostatic interactions, one between the two glutamates (282E and 284E) on PLIN1 and 86R on the lipase and the other between 288R on PLIN1 and 91D on lipase, may be responsible to open the lipase lid in place of diethyl p-nitrophenyl phosphate. There is no structure of any Drosophila triglyceride lipase, however, sequence alignments show that several Drosophila fat body triglyceride lipases share conserved amino acids with the lid segment of the fungus lipase (Fig. 6). The charged residues 86R and 91D of the fungus lipase have their counterparts in the Drosophila lipases (Fig. 6). Specifically, RLRNFTND of two isoforms of CG8552, which have been considered to be activated by PLIN1 in insects [3, 13], could facilitate electrostatic interaction for three pairs of amino acids with 282EPENQARP289 of PLIN1.
Figure 6.

Sequence alignments of fungus Rhizomucor miehei triglyceride lipase chain A (PDB ID: 4TGL) [41] with Drosophila melanogaster triglyceride lipases: two isoforms of CG8552 (NP_001188714.1 and NP_001188715.1) [42], CG11055 (NP_611463.1) [42], and CG8823 (NP_477331.1) [43]. The alignments were performed using program SIM [44]. 86R and 91D of the fungus lipase and corresponding charged residues in other proteins are underlined.
The interaction between the two proteins may also cause reorganization of the PLIN1 structure and further perturbation to the local phospholipid molecules. Energy released from the electrostatic interactions could convert to mechanical energy, causing the lipase to push against H7 and H9 of PLIN1 (Fig. 5D). This could results in an increased area for the region enclosed by the helices, creating a passage for the stored triglyceride molecules, which were already very close to the surface, to diffuse toward the catalytic center. The interaction of PLIN1 and triglyceride lipase is probably modulated by phosphorylation and Ca2+. Phosphorylation of PLIN1 promotes lipase activity in hydrolyzing triglycerides stored inside the lipid droplet [11, 13] and Ca2+ is also an activator of lipolysis [45]. These facts suggest that phosphorylation and binding of Ca2+ could promote conformational changes affecting certain protein regions. These changes would affect the interaction of the protein with lipid, affecting the accessibility of the lipases to the triglyceride molecules, or the interaction of PLIN1 with lipases and/or other proteins required in the activation process. Thus, future studies of the structures of PLIN1 with and without Ca2+ may identify structural changes that would explain the role of certain protein regions in the function of PLIN1.
The starting and final structures of the cis-model are shown in Fig. S4. Helix 8 is completely unwound and helix 9 also significantly changed its structure. Neither the trans-model nor cis-model penetrates into the bottom layer of DMPG bilayer, in agreement with the proposed function of attaching the protein to the phospholipids monolayer covering the lipid droplet.
3.4. Model verification by NMR data
Backbone and β-carbon chemical shifts were predicted from structures by program shiftX [33]. Based on these chemical shifts, 13C-13C 2D spectra were simulated and compared with experimental spectra (Fig. 6 and Fig. S5). The simulated spectrum of trans-model agrees very well with experiments. The Cβ-CO peaks of T262 and T291 (lower left corner of Fig. 7A) do not have corresponding peaks in the experimental spectrum acquired with 10 ms DARR mixing, which is chosen for establishing short-range (1- and 2-bond) resonance correlations. Matching peaks are found for the experimental spectrum acquired with 50 ms DARR for long-range correlations (Fig. S5B). These two threonine residues possibly undergo unfavorable dynamics, resulting in weaker dipolar coupling between Cβ and CO. On the contrary, more unmatched peaks are found for the cis-model (Fig. 7B and Fig. S5). The T291 and T 262 Cβ-CO peak (lower left corner of Fig. 7B) do not have matching peak even for the spectrum acquired with 50 ms DARR mixing (Fig. S5B). A280, T300, and S303 are also mismatched. Therefore, the cis-model does not agree with NMR data.
Figure 7.
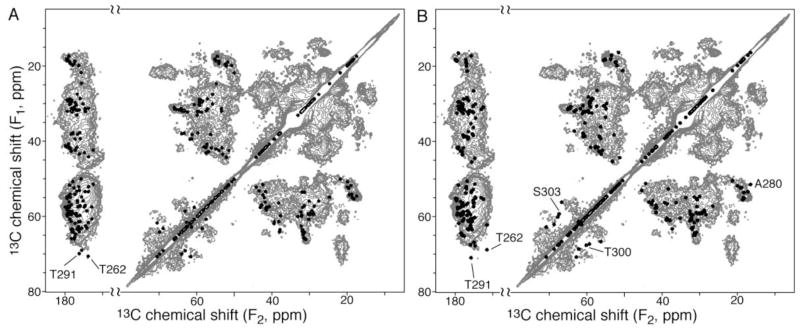
Verification of structure models by 13C-13C 2D data. The experimental spectrum (gray) was obtained with 10 ms DARR mixing [22]. Simulated spectra (black) for the trans-model (A) and cis-model (B) only consist of Cα-Cβ, Cα-CO, and Cβ-CO correlations, without other side chain carbons.
Although the trans-model agreed well with NMR data, the real structure may deviate from it. First, other regions of the protein might significantly affect the actual structure of the four helices. Second, it is uncertain whether PLIN1 is oligomerized in the active form, and oligomerization has not been taken into consideration in the current model.
4. Conclusion
NMR spin diffusion experiments were consistent with the predicted membrane attachment motif of PLIN1, and they indicated that some regions of PLIN1 have a deep contact with the phospholipid acyl chains near the bilayer center. For a native lipid storage droplet that is covered by a lipid monolayer, PLIN1 could penetrate to the interface of the monolayer and the triglyceride core. Two structure models for the membrane attachment motif were generated based on hydrophobicity analysis and NMR membrane insertion depth information, followed by optimization in lipid environment. Both models consist of four membrane interacting elements that are roughly parallel to the membrane surface. Two amphipathic elements stay on the membrane surface, and two hydrophobic elements are buried deeper. Simulated NMR spectra for the trans-model agreed with experimental spectra. Juxtaposition of the triglyceride lipase structure to the PLIN1 trans-model suggests a possible interaction of the conserved sequence (EPENQARP), which is on a long loop between lipid binding elements, with the lipase. The long loop could bind to the lipase lid domain by electrostatic interactions and open the lid to expose the catalytic center. Interaction with the lipase could also cause reorganization of the membrane attachment elements of PLIN1, leading to an increased area for the region enclosed by the membrane attachment motif. A passage may be created by this process for the stored triglyceride molecules, which are already very close to the surface due to perturbation by PLN1 attachment, to diffuse toward the catalytic center. Therefore, this structural model could help design future experiments to elucidate the role of PLIN1 in lipolysis.
Supplementary Material
Highlights.
First NMR study on lipoprotein complexes of lipid storage droplet protein 1
Membrane anchoring elements and deep insertion confirmed by spin diffusion NMR
Basis for membrane attachment shown by models built with MD simulation
Electrostatic interactions with lipase proposed to participate in activation of hydrolysis
Acknowledgments
The authors greatly appreciate financial support from National Institutes of Health (GM097713 and GM64677). The computing for this project was performed at the OSU High Performance Computing Center (OSUHPCC). This work was supported in part through instrumentation funded by the National Science Foundation through grant OCI–1126330. We thank Dr. Margaret Eastman (Oklahoma Statewide Shared NMR Facility) for technical assistance.
Abbreviations
- Lsd1
lipid storage droplet protein 1
- PAT family
Perilipin
- ADRP
TIP 47 family
- MD
molecular dynamics
- DMPG
1,2-dimyristoyl-sn-glycero-3-phosphoglycerol
- MAS
magic-angle spinning
- CP
cross polarization
- DARR
dipolar-assisted rotational resonance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wolins NE, Brasaemle DL, Bickel PE. A proposed model of fat packaging by exchangeable lipid droplet proteins. FEBS Lett. 2006;580:5484–5491. doi: 10.1016/j.febslet.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 2.Tauchi-Sato K, Ozeki S, Houjou T, Taguchi R, Fujimoto T. The Surface of lipid droplets Is a phospholipid monolayer with a unique fatty acid composition. J Biol Chem. 2002;277:44507–44512. doi: 10.1074/jbc.M207712200. [DOI] [PubMed] [Google Scholar]
- 3.Bickel PE, Tansey JT, Welte MA. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim Biophys Acta Mol Cell Biol Lipids. 2009;1791:419–440. doi: 10.1016/j.bbalip.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kimmel AR, Brasaemle DL, McAndrews-Hill M, Sztalryd C, Londos C. Adoption of PERILIPIN as a unifying nomenclature for the mammalian PAT-family of intracellular lipid storage droplet proteins. J Lipid Res. 2010;51:468–471. doi: 10.1194/jlr.R000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gronke S, Beller M, Fellert S, Ramakrishnan H, Jackle H, Kuhnlein RP. Control of fat storage by a Drosophila PAT domain protein. Curr Biol. 2003;13:603–606. doi: 10.1016/s0960-9822(03)00175-1. [DOI] [PubMed] [Google Scholar]
- 6.Arrese EL, Mirza S, Rivera L, Howard AD, Chetty PS, Soulages JL. Expression of lipid storage droplet protein-1 may define the role of AKH as a lipid mobilizing hormone in Manduca sexta. Insect Biochem Mol Biol. 2008;38:993–1000. doi: 10.1016/j.ibmb.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez-Botas J, Anderson JB, Tessier D, Lapillonne A, Chang BHJ, Quast MJ, Gorenstein D, Chen KH, Chan L. Absence of perilipin results in leanness and reverses obesity in Lepr db db mice. Nat Genet. 2000;26:474–479. doi: 10.1038/82630. [DOI] [PubMed] [Google Scholar]
- 8.Tansey JT, Sztalryd C, Gruia-Gray J, Roush DL, Zee JV, Gavrilova O, Reitman ML, Deng CX, Li C, Kimmel AR, Londos C. Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6494–6499. doi: 10.1073/pnas.101042998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beller M, Bulankina AV, Hsiao HH, Urlaub H, Jackle H, Kuhnlein RP. PERILIPIN-Dependent Control of Lipid Droplet Structure and Fat Storage in Drosophila. Cell Metab. 2010;12:521–532. doi: 10.1016/j.cmet.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Marcinkiewicz A, Gauthier D, Garcia A, Brasaemle DL. The phosphorylation of serine 492 of perilipin A directs lipid droplet fragmentation and dispersion. Journal of Biological Chemistry. 2006;281:11901–11909. doi: 10.1074/jbc.M600171200. [DOI] [PubMed] [Google Scholar]
- 11.Patel RT, Soulages JL, Hariharasundaram B, Arrese EL. Activation of the lipid droplet controls the rate of lipolysis of triglycerides in the insect fat body. J Biol Chem. 2005;280:22624–22631. doi: 10.1074/jbc.M413128200. [DOI] [PubMed] [Google Scholar]
- 12.Hickenbottom SJ, Kimmel AR, Londos C, Hurley JH. Structure of a lipid droplet protein: the PAT family member TIP47. Structure. 2004;12:1199–1207. doi: 10.1016/j.str.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 13.Arrese EL, Rivera L, Hamada M, Mirza S, Hartson SD, Weintraub S, Soulages JL. Function and structure of lipid storage droplet protein 1 studied in lipoprotein complexes. Arch Biochem Biophys. 2008;473:42–47. doi: 10.1016/j.abb.2008.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arrese EL, Rivera L, Hamada M, Soulages JL. Purification and characterization of recombinant lipid storage protein-2 from Drosophila melanogaster. Prot Pept Lett. 2008;15:1027–1032. doi: 10.2174/092986608785849191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez MC, Toledo JD, Tricerri MA, Garda HA. The central type Y amphipathic alpha-helices of apolipoprotein AI are involved in the mobilization of intracellular cholesterol depots. Archives of biochemistry and biophysics. 2008;473:34–41. doi: 10.1016/j.abb.2008.02.021. [DOI] [PubMed] [Google Scholar]
- 16.Prieto ED, Garda HA. Membrane insertion topology of the central apolipoprotein A-I region. Fluorescence studies using single tryptophan mutants. Biochemistry. 2011;50:466–479. doi: 10.1021/bi1009634. [DOI] [PubMed] [Google Scholar]
- 17.Garda HA, Arrese EL, Soulages JL. Structure of apolipophorin-III in discoidal lipoproteins - Interhelical distances in the lipid-bound state and conformational change upon binding to lipid. J Biol Chem. 2002;277:19773–19782. doi: 10.1074/jbc.M110089200. [DOI] [PubMed] [Google Scholar]
- 18.Soulages JL, Arrese EL. Fluorescence spectroscopy of single tryptophan mutants of apolipophorin-III in discoidal lipoproteins of dimyristoylphosphatidylcholine. Biochemistry. 2000;39:10574–10580. doi: 10.1021/bi0007223. [DOI] [PubMed] [Google Scholar]
- 19.Lai AL, Moorthy AE, Li Y, Tamm LK. Fusion activity of HIV gp41 fusion domain is related to its secondary structure and depth of membrane insertion in a cholesterol-dependent fashion. Journal of molecular biology. 2012;418:3–15. doi: 10.1016/j.jmb.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cruciani O, Mannina L, Sobolev AP, Segre CC3A. An Improved NMR Study of Liposomes Using 1-Palmitoyl-2-oleoyl-sn-glycero-3-phospatidylcholine as Model. Molecules. 2006;11:334–344. doi: 10.3390/11050334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear decoupling in rotating solids. J Chem Phys. 1995;103:6951–6958. [Google Scholar]
- 22.Takegoshi K, Nakamura S, Terao T. 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett. 2001;344:631–637. [Google Scholar]
- 23.Luo W, Hong M. A 1D sensitivity-enhanced 1H spin diffusion experiment for determining membrane protein topology. Sol St Nucl Magn Reson. 2006;29:163–169. doi: 10.1016/j.ssnmr.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 24.Zhou DH, Rienstra CM. High-performance solvent suppression for proton-detected solid-state NMR. J Magn Reson. 2008;192:167–172. doi: 10.1016/j.jmr.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumashiro KK, Schmidt-Rohr K, Murphy OJ, Ouellette KL, Cramer WA, Thompson LK. A Novel Tool for Probing Membrane Protein Structure: Solid-State NMR with Proton Spin Diffusion and X-Nucleus Detection. J Am Chem Soc. 1998;120:5043–5051. [Google Scholar]
- 26.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–1472. [Google Scholar]
- 27.Darden T, York D, Pedersen L. Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 28.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101:014101–014107. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 29.Parrinello M, Rahman A. Polymorphic Transitions in Single-Crystals - a New Molecular-Dynamics Method. J Appl Phys. 1981;52:7182–7190. [Google Scholar]
- 30.Nose S. A Molecular-Dynamics Method for Simulations in the Canonical Ensemble. Mol Phys. 1984;52:255–268. [Google Scholar]
- 31.Nose S, Klein ML. Constant Pressure Molecular-Dynamics for Molecular-Systems. Mol Phys. 1983;50:1055–1076. [Google Scholar]
- 32.Hoover WG. Canonical Dynamics - Equilibrium Phase-Space Distributions. Phys Rev A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 33.Neal S, Nip AM, Zhang H, Wishart DS. Rapid and accurate calculation of protein 1H, 13C and 15N chemical shifts. J Biomol NMR. 2003;26:215–240. doi: 10.1023/a:1023812930288. [DOI] [PubMed] [Google Scholar]
- 34.Frericks H, Zhou D, Yap L, Gennis R, Rienstra C. Magic-angle spinning solid-state NMR of a 144 kDa membrane protein complex: E. coli cytochrome bo3 oxidase. J Biomol NMR. 2006;36:55–71. doi: 10.1007/s10858-006-9070-5. [DOI] [PubMed] [Google Scholar]
- 35.Tusnády GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2011;17:849–850. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
- 36.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 37.Maupetit J, Gautier R, Tuffery P. SABBAC: online Structural Alphabet-based protein BackBone reconstruction from Alpha-Carbon trace. Nucleic Acids Res. 2006;34:W147–151. doi: 10.1093/nar/gkl289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pronk S, Páll Sr, Schulz R, Larsson P, Bjelkmar Pr, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piggot TJ, Holdbrook DA, Khalid S. Electroporation of the E. coli and S. Aureus Membranes: Molecular Dynamics Simulations of Complex Bacterial Membranes. J Phys Chem B. 2011;115:13381–13388. doi: 10.1021/jp207013v. [DOI] [PubMed] [Google Scholar]
- 40.Domanski J, Stansfeld P, Sansom MP, Beckstein O. Lipidbook: A Public Repository for Force-Field Parameters Used in Membrane Simulations. J Membrane Biol. 2010;236:255–258. doi: 10.1007/s00232-010-9296-8. [DOI] [PubMed] [Google Scholar]
- 41.Derewenda U, Brzozowski AM, Lawson DM, Derewenda ZS. Catalysis at the interface: the anatomy of a conformational change in a triglyceride lipase. Biochemistry. 1992;31:1532–1541. doi: 10.1021/bi00120a034. [DOI] [PubMed] [Google Scholar]
- 42.Hoskins RA, Carlson JW, Kennedy C, Acevedo D, Evans-Holm M, Frise E, Wan KH, Park S, Mendez-Lago M, Rossi F, Villasante A, Dimitri P, Karpen GH, Celniker SE. Sequence Finishing and Mapping of Drosophila melanogaster Heterochromatin. Science. 2007;316:1625–1628. doi: 10.1126/science.1139816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zinke I, Kirchner C, Chao LC, Tetzlaff MT, Pankratz MJ. Suppression of food intake and growth by amino acids in Drosophila: the role of pumpless, a fat body expressed gene with homology to vertebrate glycine cleavage system. Development. 1999;126:5275–5284. doi: 10.1242/dev.126.23.5275. [DOI] [PubMed] [Google Scholar]
- 44.Huang X, Miller W. A time-efficient, linear-space local similarity algorithm. Adv Appl Math. 1991;12:337–357. [Google Scholar]
- 45.Arrese EL, Flowers MT, Gazard JL, Wells MA. Calcium and cAMP are second messengers in the adipokinetic hormone-induced lipolysis of triacylglycerols in Manduca sexta fat body. J Lipid Res. 1999;40:556–564. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.