

Abstract
For the development of new therapies, proof-of-concept studies in large animal models that share clinical features with their human counterparts represent a pivotal step. For inherited retinal dystrophies primarily involving photoreceptor cells, the efficacy of gene therapy has been demonstrated in canine models of stationary cone dystrophies and progressive rod–cone dystrophies but not in large models of progressive cone–rod dystrophies, another important cause of blindness. To address the last issue, we evaluated gene therapy in the retinitis pigmentosa GTPase regulator interacting protein 1 (RPGRIP1)-deficient dog, a model exhibiting a severe cone–rod dystrophy similar to that seen in humans. Subretinal injection of AAV5 (n = 5) or AAV8 (n = 2) encoding the canine Rpgrip1 improved photoreceptor survival in transduced areas of treated retinas. Cone function was significantly and stably rescued in all treated eyes (18–72% of those recorded in normal eyes) up to 24 months postinjection. Rod function was also preserved (22–29% of baseline function) in four of the five treated dogs up to 24 months postinjection. No detectable rod function remained in untreated contralateral eyes. More importantly, treatment preserved bright- and dim-light vision. Efficacy of gene therapy in this large animal model of cone–rod dystrophy provides great promise for human treatment.
Introduction
Inherited retinal dystrophies form a phenotypically and genetically heterogeneous group of blinding diseases characterized by the progressive degeneration of photoreceptor cells.1 They are commonly classified according to the rate of photoreceptor cell loss, stationary or progressive disorders, and the subtype of photoreceptors functionally affected first: cone dystrophy (dysfunction of cones only),2 cone–rod dystrophy (dysfunction of cones then rods),2,3 and rod–cone dystrophy (dysfunction of rods then cones).1 When both rod and cone responses are severely impaired within the first years of life (rod–cone or cone–rod dystrophies), the term Leber congenital amaurosis is usually used.
The pathological involvement of photoreceptors in inherited retinal degeneration (IRD) may be the result of mutations in genes/loci mainly expressed in photoreceptors themselves (cones and/or rods) or in the retinal pigment epithelium cells that are essential for the maintenance of the function, structural integrity, and survival of photoreceptors.4 Primary genetic mutations in cones generally affect only cone function/survival (cone dystrophy).2 By contrast, rod-specific mutations primarily affect rods but can often result in secondary loss of cone-mediated function and vision (rod–cone dystrophy).1 When the causal gene is expressed in both subtypes of photoreceptors, cone function loss can slightly precede rod damage (cone–rod dystrophy) or inversely (rod–cone dystrophy) depending on the implicated gene, causal mutations, and/or genetic modifiers.4 Retinal pigment epithelium–initiated dystrophies, which also hinder the function of both types of photoreceptors, can be associated with cone–rod or rod–cone phenotypes.4
IRDs are currently incurable, but for those acquired by recessive (50–60%) or X-linked (5–15%) inheritance,1 gene addition therapy holds great promise. Indeed, over the past decade, gene transfer has resulted in significant morphological and/or functional improvements in a dozen different rodent models of IRD including models of cone, cone–rod and rod–cone dystrophies.5,6,7
Large models (dogs, cats, or pigs) are more amenable than rodents to vision testing, and their longevity enables long-term follow-up. More importantly, the retinal distribution, density, and proportion of rods and cones in these larger animals more closely match those of primates.8
Recently, gene therapy targeting photoreceptors was successfully used in two canine models of stationary cone dystrophy caused by a defect in the cone-specific Cngb3 gene (Cngb3−/− and Cngb3m/m dogs),9 in the Rcd1 canine model of progressive rod–cone dystrophy caused by a defect in the rod-specific Pde6β gene10, and in two canine models of progressive rod–cone dystrophy linked to a defect in the Rpgr gene, expressed in both rods and cones (XLPRA1 and XLPRA2 dogs).11 These results strongly support the translation of gene therapy for stationary cone and progressive rod–cone dystrophies into the clinic. The efficacy of photoreceptor gene therapy in a large model of cone–rod dystrophy remains, however, to be demonstrated.
A closed research colony of miniature longhaired dachshund (MLHD) provides a highly relevant model for validating potential gene therapies for cone–rod dystrophies. Indeed, these dogs display a severe early onset cone–rod dystrophy (Cord1) that allows fairly rapid assessment of the effects of gene therapy on both retinal function and vision. Their cone function is totally absent at 2 months of age, and their rod function rapidly declines from 2 to 12 months of age.12,13 In addition, their retinal degeneration has been linked to a locus containing the GTPase regulator interacting protein 1 (Rpgrip1) gene,14 a known cause of recessive Leber congenital amaurosis (4–6% of all cases)15,16,17,18,19 and cone–rod dystrophy (1% of all cases)20 in humans. In mice, the Rpgrip1 gene encodes multiple protein isoforms; among those, RPGRIP1α1 is specifically expressed in the connecting cilium and outer segments of photoreceptor cells.21,22,23 The RPGRIP1α1 protein isoform anchors RPGR21,24 and is thought to control microtubule organization in the connecting cilium and the molecular trafficking between the inner and outer segments of photoreceptors.25 RPGRIP1 also appears to be required for the development and maintenance of photoreceptor outer segments.26
Gene therapy for RPGRIP1 deficiencies has previously been evaluated in the Rpgrip1 knockout mouse, providing encouraging results.27,28 In the initial study, delivery of the murine Rpgrip1 cDNA into photoreceptors of Rpgrip1−/− mice at postnatal day 20 (P20) using a rAAV2/2 restored the localization of both RPGRIP1α1 and RPGR in the connecting cilium and led to a significant slowing of photoreceptor function loss at 5 months postinjection (mpi) (loss of 6% of electroretinographic (ERG) b-wave amplitude per month in treated eyes compared with 22% per month in untreated contralateral eyes).27 The second study used a rAAV2/8 and a human RPGRIP1 cDNA, leading to a preservation of one-third of initial photoreceptor function in treated eyes at 5 mpi. At this age, ERG responses cannot be further recorded in untreated eyes.28
Herein, we evaluate the efficacy of rAAV-mediated Rpgrip1 gene transfer in the MLHD-Cord1 dog. We found that gene addition therapy can restore the functional deficit of cone photoreceptors and prevent the retinal degeneration and vision loss in this large animal model of cone–rod dystrophy.
Results
An AAV2/5 vector carrying the eGFP under the control of the human RK promoter transduces both rods and cones in the canine retina
As RPGRIP1 deficiencies affect both cone and rod functions,15,16,17,18,19,20 effective gene therapy in the MLHD-Cord1 dog would require efficient transgene expression in both subtypes of photoreceptors. In this study, we chose to use rAAV2/5 or rAAV2/8 vectors, as they have been shown to be excellent vectors for transducing photoreceptors in mice, dogs,29 pigs,30 and nonhuman primates.31,32 Concerning the promoter, several studies have demonstrated that the human RK promoter mediates efficient and specific transgene expression in both cones and rods, following subretinal injection, in the murine28,33,34,35,36,37,38 and the nonhuman primate retina,32 suggesting that this promoter may be ideally used in a clinical setting. This was reinforced with its recent successful use in a gene replacement therapy study in the XLPRA2 canine model of rod–cone dystrophy linked to a defect in both rods and cones.11 However, these findings are inconsistent with previous results showing that in the dog, the human RK promoter drives rAAV-mediated expression exclusively in rods, except when high titer was used (3 × 1012 genome copies).39
To determine whether the human RK promoter can drive expression in both cones and rods in the dog and is suitable for our gene replacement study, we subretinally delivered a rAAV2/5 vector expressing enhanced green fluorescent protein (eGFP) under the control of the RK promoter (1 × 1011 genome copies, Figure 1a) into two dogs, NA2 and NA3 (Table 1). As reported earlier,29 the area transduced, determined by live indirect ophthalmoscopy, perfectly matched with the vector subretinal bleb (Figure 1b,c). eGFP fluorescence peaked at 6 weeks postinjection (data not shown) and remained stable for at least 9 mpi, the time when the animals were sacrificed for histological assessment (Figure 1c). Histological sections were performed in the vector-exposed area. Epifluorescence microscopy showed eGFP expression limited to photoreceptor cells (cell bodies, inner/outer segments) (Figure 1d). Notably, a robust transduction of cones was detected (Figure 1f–h). Cones were identified based on two criteria: their well-defined shape and their labeling with L/M opsin (Figure 1e–h), a known marker of L/M cone outer segments, and at high exposure, of L/M cone inner segments (Figure 1h). Recombinant AAV2/5-RK-eGFP transduced on average 49 ± 21% of L/M cones (number of double-labeled eGFP/opsin cones per field at ×20 magnification, n = 2 eyes). These results confirmed that the RK promoter could drive efficient and specific rAAV-mediated transgene expression into both cones (at least L/M) and rods in the canine retina, further supporting its application for gene transfer in MLHD-Cord1 dog.
Figure 1.

Green fluorescent protein (GFP) expression in rod and cone photoreceptors after subretinal injection of AAV2/5-RK-eGFP in the normal canine retina. (a) Schematic structure of recombinant AAV2/5-RK-eGFP vector. It encodes the eGFP reporter gene under the control of the human RK promoter (−112 to +87 bp region of the proximal promoter). (b) Retinal detachment created by the subretinal injection of AAV2/5-RK-eGFP vector 20 minutes after injection in the right eye of dog NA2. (c) Live fluorescent eGFP signal at 9 mpi in the right eye of dog NA2. (d) Image of native eGFP fluorescence (green) in the injected area of the right eye of dog NA2 after nuclei counterstaining with DAPI (blue). (e) Images of native eGFP (green) and (f) L/M opsin immunolabeling (red) in the injected area of the right eye of dog NA2. (g–h) Overlay images were taken at high exposure to 568 nm to allow the staining of cone inner segments (ISs). Arrows indicate eGFP-positive cones. Scale bar = 30 µm. BGHpA, bovine growth hormone polyadenylation signal; DAPI, 4′,6-diamidino-2-phenylindole; eGFP, enhanced GFP; GCL, ganglion cell layer; INL, inner nuclear layer; ITR, inverted terminal repeat; ONL, outer nuclear layer; OS, outer segment.
Table 1. List of dogs included in the study.
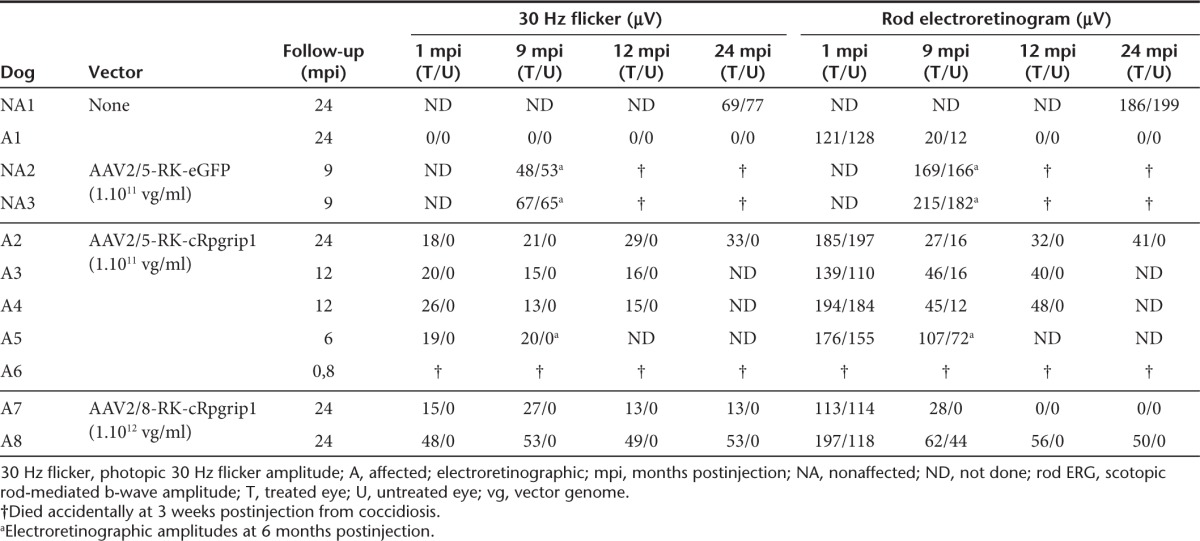
Production of therapeutic rAAV vectors and subretinal delivery in the MLHD-Cord1 dog
Given the efficacy and specificity of the AAV-RK vector system, we subsequently produced rAAV2/5 and rAAV2/8 vectors encoding canine Rpgrip1 (cRpgrip1) under the control of the same promoter (Figure 2a). We first isolated and subcloned the cRpgrip1 cDNA. Seven overlapping fragments covering the entire Rpgrip1 coding sequence were amplified using polymerase chain reaction (PCR) and RNA-ligase–mediated RACE (RLM-RACE) PCR (Supplementary Figure S1a) using primers designed against human, bovine and rodent RPGRIP1 consensus sequences and putative canine sequences (Supplementary Table S1). The cRpgrip1 open reading frame spans 3630 bp, contains 24 coding exons, and encodes a predicted protein of 1209 amino acid (Supplementary Figure S1b and Table S2), as recently published.40 Then, a total of seven MLHD-Cord1 dogs (A2–A8) were injected subretinally with these therapeutic vectors at 1 month of age (see below). Dogs A2–A6 received AAV2/5-RK-cRpgrip1, whereas dogs A7 and A8 received AAV2/8-RK-cRpgrip1 (Table 1). All injections were performed unilaterally on the nasal superior quadrant of the retina, except for dog A3 that was treated in temporal.
Figure 2.

Reverse transcriptase–polymerase chain reaction (PCR) amplification of transgene (wild-type) and native (mutant) Rpgrip1 transcripts from A6 retinas at 3 weeks postinjection. (a) Schematic structure of recombinant AAV2/5-RK-cRpgrip1 and AAV2/8-RK-cRrpgrip1 vectors. Both encode the canine Rpgrip1 cDNA (cRpgrip1) under the control of the human RK promoter (−112 to +87 bp region of the proximal promoter). (b) PCR amplification using 184F and 317R primers encompasses a fraction of exons 1–3 of the canine Rpgrip1 gene. MLHD-Cord1 dogs have been shown previously to carry an insertion of 44 bp in exon 2 of the cRpgrip1 gene (polyA tract insertion of 29 nucleotides flanked by 15-bp duplication). Thus, PCR amplification results in a 198-bp product when mutant Ins44 cDNAs are used as templates and in a 154-bp product when wild-type (vector-derived) cDNAs templates are used. Arrows indicate the positions of the 184F and 317R primers. The black box indicates the position of the Ins44 mutation in the cRpgrip1 gene. (c) Agarose gel electrophoresis of cRpgrip1 and β-actin RT-PCR. Lanes 1 and 2 show products from Rpgrip1+/+ and Rpgrip1+/− untreated control retinas, respectively. Lanes 3 and 4 show products from untreated and AAV2/5-RK-cRpgrip1-treated retinas of dog A6 at 3 weeks postinjection, respectively. Lane 5 shows the product from the SSV9-RK-cRpgrip1 vector plasmid. +/+, Rpgrip1+/+ retina; bp, base pairs; cDNA Rpgrip1; SSV9-RK-cRpgrip1 vector plasmid; Ins44, 44bp insertion mutation in the cRpgrip1 exon 2; ladder, 1 kb molecular size ladder; NA1, nonaffected Rpgrip1+/− retina; T, treated retina; U, untreated retina.
During the progression of this study, dog A6 died accidentally at 3 weeks postinjection. Its retinas were collected to analyze the expression of cRpgrip1 by reverse transcriptase–PCR. Previous studies have suggested that a polyA tract insertion of 29 nucleotides flanked by a 15-bp duplication (Ins44), in the coding exon 2 of the cRpgrip1 gene, contributes to retinal degeneration in the MLHD-Cord1 dog.14 We, therefore, developed an allele-specific reverse transcriptase–PCR to ensure discrimination between wild-type (transgene) and Ins44 mutant (endogeneous) Rpgrip1 transcripts in treated retinas (Figure 2b). The 154-bp amplification product derived from transgene RNA templates was detected in A6 treated retina (Figure 2c). On the contrary, untreated Rpgrip1−/− A6 eye yielded only the mutant RNA (198 bp) (Figure 2c). This result confirmed that vector-encoded cRpgrip1 was expressed in transduced photoreceptors.
Gene therapy reduces the progressive photoreceptor degeneration in MLHD-Cord1 retinas
In the MLHD-Cord1 dog, photoreceptor degeneration becomes evident by histology at ~2 months of age and is almost complete at 28 months of age.12,13 It is associated with a dramatic reduction of the retinal vasculature between 5 months and 2 years of age.13 To determine whether rAAV-mediated cRpgrip1 gene addition can prevent, delay, slow, or stop this morphological damage, three complementary analyzes were performed in all treated dogs at different times after injection. First, funduscopy and time-domain optical coherence tomography (TD-OCT) were performed from 3 to 24 mpi to monitor the kinetic of retinal degeneration in treated and untreated eyes (Figure 3 and Supplementary Figure S2). Second, spectral-domain OCT (SD-OCT) was performed at the latest time point to determine the extent of retinal preservation in treated eyes (Figure 4a and Supplementary Figure S3a). Finally, the eyes of the dog A2 were examined by histology and immunohistochemistry at 24 mpi to monitor the effect of gene transfer on the morphology of each photoreceptor cell type (Figure 4b,c and Supplementary Figure S3b).
Figure 3.

In vivo assessment of retinal morphology in dogs A2 and A8 at 3, 9, 12, and 24 mpi. (a) Fundus photographs and time-domain optical coherence tomography (OCT) images obtained for the right and left eyes of control nonaffected, untreated dog NA1 at 24 months of age. (b) Fundus photographs and retinal cross-sectional images obtained for the treated and untreated eyes of dog A2 treated with AAV2/5-RK-cRpgrip1 at 3, 9, 12, and 24 months postinjection (mpi). (c) Fundus photographs and retinal cross-sectional images obtained for the treated and untreated eyes of dog A8 treated with AAV2/8-RK-cRpgrip1 at 3, 9, 12, and 24 mpi. White-dotted circles on fundus photographs schematically represent areas of the treated retinas exposed to rAAV vectors. OCT scans were acquired on a horizontal line shown on the fundus images (white lines). The localization and the size of the white lines represent the localization and the size of the OCT scans. Retinal thicknesses at the same location (13 mm above the optic nerve head) were measured using calibrated calipers and are indicated on the OCT scans. m, months of age; T, treated eye; U, untreated eye.
Figure 4.
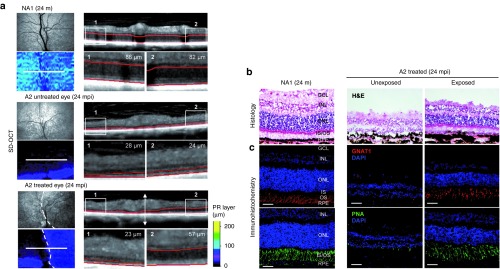
In vivo and postmortem assessment of photoreceptor (PR) layer thickness in dog A2 at 24 months postinjection (mpi). (a) Topographies of PR layer and spectral-domain optical coherence tomography (SD-OCT) scans (obtained 13 mm above the optic nerve head) from the right eye of dog NA1 (top) and both untreated (middle) and treated (bottom) eyes of dog A2 at 24 mpi. White-dotted circles on fundus photographs schematically represent the area of the treated A2 retina exposed to AAV2/5-RK-cRpgrip1 vector. OCT scans were acquired on a horizontal line shown on the fundus images (white lines). PR layer (outer nuclear layer (ONL) + outer segments (OSs) and inner segments (ISs)) was defined semi-automatically (red). PR layer thickness was measured in two normalized (1) temporal and (2) nasal locations. (b,c) Microscopic images of retinal sections from the treated eye of dog A2 at 24 mpi and the right eye of age-matched control dog NA1. Serial retinal cryosections encompassing the vector-exposed and the vector-unexposed areas of the treated retina were (b) stained with hematoxylin and eosin (H&E) (top) or (c) immunolabeled against GNAT1 (red, middle) or peanut agglutinin-FITC (green, bottom). Primary GNAT1 antibody was detected with Alexa 546–conjugated goat antirabbit IgG. Scale bar = 30 µm. GCL, ganglion cell layer; INL, inner nuclear layer; m, months of age; RPE, retinal pigment epithelium.
Kinetics of retinal degeneration in MLHD-Cord1 untreated and treated eyes. The first analysis is illustrated by representative funduscopy and TD-OCT data obtained from dogs NA1 (nonaffected control; Figure 3a), A2 (rAAV2/5-treated; Figure 3b), and A8 (rAAV2/8-treated; Figure 3c) at 3, 9, 12, and 24 mpi.
In agreement with previous data,13 an obvious reduction of retinal vasculature was observed from 3 to 24 mpi in untreated MLHD-Cord1 eyes (Figure 3b,c, bottom) compared with normal eyes (Figure 3a). A reduction of retinal vasculature was also observed in both rAAV2/5- and rAAV2/8-treated eyes from 3 to 24 mpi (Figure 3b,c, top). However, this progressive attenuation of retinal vasculature in treated eyes seemed reduced compared with untreated eyes. Indeed, at 24 mpi, both rAAV2/5- and rAAV2/8-treated eyes displayed a partially preserved retinal vasculature (Figure 3b,c, top right), whereas only a few central vessels remained visible in the untreated eyes (Figure 3b,c, bottom right). This time course of preservation of retinal vasculature correlated with those of central retinal thickness. Indeed, both treated and untreated eyes showed a progressive reduction of the central retinal thickness from 3 to 24 mpi compared with normal eyes (Figure 3a and Supplementary Figure S2). However, at all the four time points explored, treated eyes had a thicker central retina than uninjected eyes, and the difference of thickness between both eyes remained relatively constant over time in all treated dogs (Supplementary Figure S2). For example, at both 3 and 24 mpi, the treated retina of dogs A2 (Figure 3b and Supplementary Figure S2) and A8 (Figure 3c and Supplementary Figure S2) were 4 and 10% thicker than their contralateral untreated retina, respectively. These results suggest an early therapeutic effect of gene transfer on retinal morphology. Despite the fact that this result was observed in the five treated dogs, we cannot exclude that they all randomly received the treatment in their better eye (it is not possible to perform a TD-OCT examination at the time of injection).
Topography of photoreceptor preservation in MLHD-Cord1 treated eyes. SD-OCT allows outer nuclear layer thickness assessment in vivo. This examination was performed at the latest time point for dogs A2, A3, A4, A5, A7, A8, and an age-matched control dog (NA1) to determine the extent of the therapeutic effects of gene therapy on retinal degeneration (Figure 4a and Supplementary Figure S3a).
For example, for the A2 treated eye, SD-OCT scans demonstrated a stark contrast between vector-exposed and vector-unexposed regions (Figure 4a). Vector-exposed regions (A2) had a much thicker outer nuclear layer (ONL) along with nearly normal appearing inner/outer segments (57 µm; Figure 4a), accounting for 69% of the photoreceptor layer thickness of the control retina (NA1) in a similar region (82 µm; Figure 4a). By contrast, vector-unexposed regions had a homogeneously thin ONL and no inner/outer segments (23 µm; Figure 4a), which represented only 27% of the thickness observed in the normal retina (86 µm; Figure 4a). A similar pattern of localized photoreceptor thickness in the region exposed to the rAAV vector was observed across all retinas examined. It was more pronounced at late time points (Supplementary Figure S3a).
Morphology of long-term preserved photoreceptors in MLHD-Cord1 treated eyes. To further determine which subtypes of photoreceptors were maintained in transduced area of MLHD-Cord1 treated eye, dog A2 was sacrificed at 24 mpi, and its retinas were analyzed by histology. Retina cryosections containing the vector-exposed region in the temporal half of the section and vector-unexposed regions in the nasal half were stained with eosin/hematoxylin (Figure 4b) or labeled against rod- (GNAT1) and cone-specific markers (peanut lectin agglutinin (PNA), L/M opsin, and S opsin) (Figure 4c and Supplementary Figure S3b). None of the four tested antibodies raised against the murine or human Rpgrip1 antigens showed specific staining on canine retinal flat mounts, cryo- or paraffin-embedded sections (data not shown).
Histological data (Figure 4b,c and Supplementary Figure S3b) confirmed our in vivo results (Figure 4a). The ONL in the vector-unexposed part of the treated retina was reduced to 2–3 rows of photoreceptor nuclei, indicating a near complete loss of photoreceptors. In this region, no staining of cone- or rod- outer segments can be detected (Figure 4c, left and Supplementary Figure S3b). By contrast, transduced regions showed a well-preserved ONL with 11–13 rows of photoreceptor nuclei remaining (Figure 4b, middle), representing 52–62% of normal ONL thickness (21 rows; Figure 4b, right). Preserved photoreceptors displayed well-organized outer segments and were positive for all tested cone and rod markers similar to the normal retina (Figure 4c and Supplementary Figure S3b). Thus, in the treated regions of MLHD-Cord1 retina, both subtypes of photoreceptor seem to be morphologically preserved over the long term.
Gene therapy restores cone function and partially preserves rod function in Rpgrip1−/− dogs
We used full-field flash electroretinogram to analyze rescue of the hallmark functional deficits of Rpgrip1 deficiency in treated dogs. ERGs were performed at different time points following vector delivery, starting at 1 mpi. Time course analysis was over 24 mpi for dogs A2, A7, and A8, 12 mpi for dogs A3 and A4, and 6 mpi for dog A5 (Table 1).
Both AAV2/5 and AAV2/8 treatments led to a significant restoration of cone-mediated ERG responses compared with untreated contralateral eyes as early as 1 mpi (Table 1, Figures 5b,c and 6a,c, and Supplementary Figures S4 and S5). Importantly, this restored cone function remained stable over time, up to 24 mpi, the longest period of exploration (Table 1, Figures 5 and 6a,c, and Supplementary Figure S5). Restored cone function varied from 18% (13 µV) to 72% (53 µV) of the normal cone function (NA1, 73 µV; Table 1 and Figure 6e,g). This difference did not seem to be related to rAAV serotypes but can be explained by small interanimal variations in the extent/localization of the vector subretinal bleb and/or alteration of cones at the time of treatment.
Figure 5.

Bilateral full-field electroretinographic traces from dogs NA1, A2, and A8 at 1, 12, and 24 months following subretinal injection. (a) Electroretinographic trace from control nonaffected, untreated dog NA1 at 24 months of age. (b) Electroretinographic traces from dog A2 treated with AAV2/5-RK-cRpgrip1 at 1, 12, and 24 months postinjection (mpi). (c) Electroretinographic traces from dog A8 treated with AAV2/8-RK-cRpgrip1 at 1, 12 and 24 mpi. The top two recordings are low- and high-intensity dark-adapted responses, whereas the bottom two recordings show light-adapted responses (responses to single flash and 30 Hz flicker stimuli, respectively). m, months of age; T, treated eye; U, untreated eye.
Figure 6.

Kinetics of cone function recovery and rod function preservation in treated dogs. (a,b) Amplitudes of (a) 30 Hz flicker and (b) rod-b wave responses for dog A2 treated with AAV2/5-RK-cRpgrip1 from 1 to 24 months postinjection (mpi). (c,d) Amplitudes of (c) 30 Hz flicker and (b) rod-b wave responses for dog A8 treated with AAV2/8-RK-cRpgrip1 from 1 to 24 mpi. (e,g) Percentages of normal NA1 cone function (73 µV) recovered in MLHD-Cord1 treated dogs at (e) 1 and (g) 24 mpi. (f,h) Percentages of normal NA1 rod function (192 µV) preserved in MLHD-Cord1 treated dogs at (f) 1 and (h) 24 mpi. Treated eyes are shown in dark and untreated eyes in white. ERG, electroretinogram. MLHD, miniature longhaired dachshund.
At 1 mpi, no significant difference on rod b-wave amplitudes were observed between treated (113–197 µV) and untreated eyes (110–197 µV) of MLHD-Cord1 dogs (Table 1, Figures 5 and 6b,d, and Supplementary Figures S4 and S5). These rod-mediated responses were all highly similar to that observed in the age-matched Rpgrip1+/− control dog (NA1, 192 µV) (Table 1 and Figure 6f). In the untreated eyes of MLHD-Cord1 dogs, rod ERG responses initially present at 1 mpi decreased dramatically during the following months and were totally lost at 12 mpi (Table 1, Figures 5 and 6b,d, and Supplementary Figure S4). A similar decrease in rod b-wave amplitude was observed in the treated eye of dog A7 (Table 1 and Supplementary Figure S4). Interestingly, in dogs A2, A3, A4, and A8, although the rod ERG amplitude decreased during the first months following vector administration, it stabilized in the treated eyes from 9 mpi suggesting a partial and stable preservation of rod function (Table 1, Figure 6b,d, and Supplementary Figure S4). At 24 mpi, preserved rod responses of the rAAV2/5-treated eye of dog A2 represent 22% (41 µV) of those recorded at 1 mpi (185 µV) and 21% of that recorded in an age-matched Rpgrip1+/− dog (192 µV) (Table 1 and Figure 6h). Likewise, rod responses of the rAAV2/8-treated eye of dog A8 were 25% (50 µV) of its baseline response (197 µV) and 26% of that recorded in the control dog (192 µV; Table 1 and Figure 6h). At this age, no detectable response remained in untreated eyes.
Gene therapy preserves vision of MLHD-Cord1 dogs in both dim and bright light
To determine whether cone function rescue and/or rod function preservation protected visual function, we used an obstacle avoidance course to evaluate visually guided behavior in dogs A2, A3, A4, A7, and A8 at different times after treatment. Dim- and bright-light conditions were used to evaluate rod- and cone-dominated vision, respectively (Table 2).
Table 2. Evolution of bright- and dim-light vision in MLHD-Cord1 treated dogs.
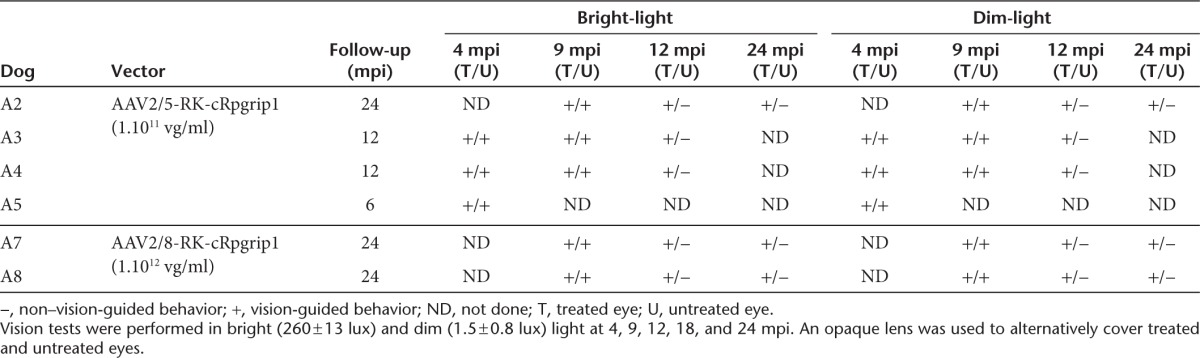
At 4 and 9 mpi, under both light conditions, none of the treated dogs showed behavioral signs of blindness regardless of which eye was covered (data not shown). However, at 12, 18, and 24 mpi, all dogs tested consistently avoided obstacles when their untreated eye was covered with an opaque lens, whereas they showed many difficulties in navigating around the panels when their treated eye was occluded (Table 2). These results were confirmed by a significant variation in transit time. For example, in bright light, dog A7 completed the obstacle course in 8 seconds with its untreated eye covered, whereas it took 27 seconds with its treated eye covered (Supplementary Video S1, part I). Similar results were obtained in dim-light condition, with dog A7 completing the obstacle course in 8 seconds with the untreated eye covered and in 33 seconds with the treated eye covered (Supplementary Video S1, part II). Together, these results strongly demonstrate that both rAAV2/5- and rAAV2/8-mediated cRpgrip1 expression preserved both dim- and bright-light vision in the MLDH-Cord1 dog.
Discussion
In this study, we have demonstrated the efficacy of gene replacement therapy in a large animal model of cone–rod dystrophy, the MLHD-Cord1 dog.12,13 Using rAAV2/5 and rAAV2/8 vectors expressing the cRpgrip1 cDNA under the control of the photoreceptor-specific human RK promoter, we have shown an increase of photoreceptor survival, a significant restoration of cone function, and a partial preservation of rod function for up to 24 mpi. Dim- and bright-light vision was also preserved in treated animals. Collectively, these results strongly indicate that gene therapy is a possible approach for treating recessive cone–rod dystrophies.
Animal models of retinal dystrophies that share clinical features with their human counterparts play a crucial role for the development and validation of new therapies. In this study, we confirmed that our MLHD-Cord1 dogs manifest an early onset and severe progressive cone–rod dystrophy, consistent with previous characterization of this research colony.12,13 All the dogs examined exhibited undetectable cone function from the earliest age measured (1 month of age). They had a nearly normal rod function at 1 month that rapidly declined thereafter. This dysfunction of photoreceptors was associated with a progressive loss of photoreceptor cells, as shown by the nearly complete loss of ONL nuclei at 24 months. This phenotype was highly conserved in all our Rpgrip1−/− dogs, despite small interindividual variations in the onset/speed of rod function and photoreceptor cell loss. This phenotype strongly correlates with the ophthalmologic abnormalities seen (i) in the Rpgrip1 knockout mice in which photoreceptor degeneration begins at P15 and is almost complete by 5 months of age25 and (ii) in patients with RPGRIP1 mutations that display a degeneration of both rod and cone photoreceptors and experience a severe loss of color vision and central acuity early in life.15,16,17,18,19,20 Of note, this MLHD-Cord1 phenotype differs from that of other Rpgrip1−/− dogs in the pet population41 or in a crossbred research colony42 that have the same genetic mutation but display a variable cone function and no change of rod function over time. Several hypotheses have been formulated to explain this genotype/phenotype discrepancy, including that the Cord1 phenotype involves a second gene that controls the penetrance or expression of cRpgrip141,42 or that cRpgrip1 itself is the modifier gene.43
In this article, we showed that AAV2/5-RK-cRpgrip1 or AAV2/8-RK-cRpgrip1 gene transfer restored 18–72% of normal cone function in treated MLHD-Cord1 eyes, as early as 1 mpi. Notably, this rescued cone function remained stable for at least 24 mpi (the longest time point explored), which supports a long-lasting and stable restoration of cone function after a single vector injection. This study demonstrates that gene therapy can restore cone function in a large animal model of RPGRIP1 deficiency. All previous reports of Rpgrip1 gene therapy have been focusing on the Rpgrip1 knockout mouse, which exhibits nearly normal cone function before photoreceptor degeneration.25 In this Rpgrip1−/− mice, subretinal injection of AAV2/8-RK-hRPGRIP1 at P14 led to the preservation of fourfold higher cone ERG responses compared with untreated eyes at 5 months of age, accounting for ~33% of wild-type levels. There was no decline in cone function in treated eyes from 2 to 5 months of age, whereas untreated eyes experienced a monthly decline of 25% of the cone amplitude.28 In our MLHD-Cord1 dogs, the cone function recovery (mean: 34 ± 14% of nonaffected dog) was within the range of that previously obtained after rAAV-mediated gene therapy in murine models of cone–rod dystrophy with nonfunctional cones (primary genetic defect in both rods and cones), including the GC1KO36,37,44 (from 20 to 65% of wild-type function) and GC1/GC2 knockout mice (from 29 to 44% of wild-type function).38
In addition to this sustained cone function recovery, we evaluated the effect of cRpgrip1 gene replacement on rod photoreceptor function. Indeed, MLHD-Cord1 dogs have been shown also to exhibit an extinction of rod function over time.12,13 Interestingly, all along the first 6–9 months after treatment, this progressive loss of rod function remained unchanged in all treated Rpgrip1−/− dogs (n = 5). This indicates that subretinal delivery of AAV2/5-RK-cRpgrip1 or AAV2/8-RK-cRpgrip1 at 1 month of age, in over 35% of the total retinal surface, was insufficient to entirely stop or obviously delay the initiation of rod dysfunction in this model. Nevertheless, continuous monitoring of rod function up to 24 mpi revealed that in four out of the five treated dogs, cRpgrip1 gene transfer also had a significantly positive effect on rod function. Indeed, from 9 mpi, four dogs (rAAV2/5, n = 2 and rAAV2/8, n = 2) showed a better preservation of b-wave amplitude in treated eyes compared with their untreated eyes. At 12 mpi, rod-mediated responses of the untreated eye were almost flat, whereas the treated eye of dog A2, A3, A4, and A8 maintained a substantial ERG response (21, 29, 25, and 26% of normal eyes, respectively). Interestingly, in the two dogs followed up to 24 mpi, A2 (rAAV2/5) and A8 (rAAV2/8), this preserved rod function remained stable for the 12 following months, suggesting that rod function loss was halted in these treated eyes after the initial decrease. This long-term preserved rod function was 22 and 25% of baseline for A2 and A8 treated eye, respectively. It may represent a fraction of preserved rods with reduced but stabilized function and/or a smaller fraction of totally potent rods that remain healthy over time. It is interesting to note that 3 murine models of progressive cone–rod dystrophy with initial normal rod function, the Rpgrip1−/−,27 the Aipl1h/h,45 and the Bbs4-null mice,46 displayed a similar kinetic of rod function preservation after gene therapy, i.e., an initial decrease of rod function followed by a stabilization.
Although the molecular events causing or leading to photoreceptor death in MLHD-Cord1 dogs are not completely understood, all our in vivo and postmortem observations indicated that localized AAV2/5-RK-cRpgrip1 or AAV2/8-RK-cRpgrip1 injection at 1 month of age resulted in localized photoreceptor preservation up to 24 mpi. At this age, photoreceptor loss was nearly complete in vector-unexposed regions. This result is similar to the confined photoreceptor preservation previously observed in the XLPRA1 and XLPRA2 dog models that display a progressive rod–cone dystrophy linked to a genetic defect in both rods and cones11 and in the Rcd1 dog, a model of progressive rod–cone dystrophy that carries a genetic defect in rods only.10
Interestingly, our analysis also revealed that despite evidence of better photoreceptor preservation in the vector-exposed area of MLHD-Cord1 treated eyes, advancing retinal thinning was still observed in all treated eyes from 3 to 24 mpi. A similar progressing retinal dystrophy has previously been observed in several canine gene therapy models, but remarkably, not all. In both XLPRA211 and Rpe65−/−47 dogs treated after the onset of apparent retinal degeneration, a gradual loss of photoreceptor nuclei was demonstrated in the vector-exposed area (from 3 to 24 mpi in the XLPRA2 dog treated at 5 weeks of age (as clearly shown by SD-OCT on Supplementary Figure S1 of ref. 11) and at 1.3 years postinjection in the Rpe65−/− dog treated between 4.9 and 6.6 years of age). By contrast, in both Rcd110 and Rpe65−/−47,48 dogs treated before the onset of the disease, retinal degeneration was stably prevented in the vector-exposed area (up to 30 mpi for the Rcd1 dog treated at P20 (Petit, L, unpublished data, 2013) and up to 11 years postinjection for the Rpe65−/− dog treated before 2.4 years of age).47,48 The alteration stage of some photoreceptors at the age of the treatment/transgene expression may be responsible for this different outcome in dog models of IRD,47 as previously suggested in murine models of IRD.38,49,50 Interestingly, it was recently demonstrated in Rpe65−/− dogs and RPE65-deficient patients treated at late stages of the disease that, advancing photoreceptor degeneration is not associated with a gradual loss of restored photoreceptor function,47 strongly suggesting that it mainly reflects the gradual loss of functionally silent (nonrescued) photoreceptors. Long-term results from two of our treated dogs with persistent rescued cone and preserved rod function from 12 to 24 mpi suggest that it is also the case in the MLHD-Cord1 dog.
The question then is whether these preclinical data on MLHD-Cord1 dogs are predictive for future treatment of RPGRIP1-deficient patients? Human RPGRIP1 deficiencies are associated with Leber congenital amaurosis15,16,17,18,19 and cone–rod dystrophy,20 depending on the causative mutations and/or genetic modifiers. In all cases, RPGRIP1-deficient patients display an early onset and severe photoreceptor dysfunction and vision loss. However, the onset and progression of photoreceptor cell loss has been relatively poorly assessed. Importantly, a study reported that one patient with RPGRIP1 deficiencies retains central retinal architecture and a substantial ONL for long periods after the total loss of visual function.17 In our study, both rAAV2/5- and rAAV2/8-mediated gene transfer had therapeutic effects in the MLHD-Cord1 dog. Similar cone and rod function improvements were observed despite rAAV2/8 (1012 vg/ml) was injected at a 10-fold higher titer than rAAV2/5 (1011 vg/ml), as previously observed and discussed for the Pde6β−/− treated dog.10 This observation reinforces the potential of AAV2/5 associated with the human RK promoter for targeting photoreceptors in humans, as recently demonstrated in the nonhuman primate.32 The positive effects on the major pathologic hallmarks that are found in the MLHD-Cord1 dog suggest that patients with RPGRIP1 or other cone–rod dystrophies might benefit from gene addition therapy.
Materials and Methods
Amplification of the canine Rpgrip1 cDNA
Poly(A) mRNA isolation and cDNA synthesis. Poly(A) messenger RNAs (mRNAs) were extracted and purified from snap-frozen canine retinas using the Oligotex direct mRNA kit (Qiagen, Courtaboeuf, France) according to the manufacturer's instructions. cDNAs were synthesized using random hexamer primers and reverse transcriptase (Transcriptor High Fidelity, Roche Diagnostics, Meylan, France) according to the supplier's protocol.
PCR and RLM-RACE PCR. Human, bovine and rodent RPGRIP1 cDNA sequences were aligned with the canine genomic sequence. Sequences of homology were used for primer design (Table S1) to amplify seven overlapping fragments covering the entire predicted cRpgrip1 coding sequence (Rpgrip1-1 to Rpgrip1-7) (Supplementary Figure S1). Both 3′ and 5′ ends of cRpgrip1 transcript were obtained with RLM-RACE PCR (Ambion, Applied Biosystems, Courtaboeuf, France) according to the manufacturer's instructions. The 5′ end of the cRpgrip1 cDNA was obtained in two steps using Rpgrip1-1F (5′ RACE adaptor primer)/Rpgrip1-1R (sequence specific primer in exon 6) as outer primers and 1nestedF (5′ RACE adaptor primer)/1nestedR (sequence specific primer in exon 4) as inner primers. The 3′ end of the cRpgrip1 cDNA was obtained in two steps using Rpgrip1-7F (sequence specific primer in exon 19)/Rpgrip1-7R (3′ RACE adaptor primer) as outer primers and 7nestedF (sequence specific primer in exon 19–20/7nestedR (3′ RACE adaptor primer) as inner primers.
PCR amplification was performed using KOD Hot Start Polymerase (Novagen, Merck Eurolab, Fontenay sous Bois, France) under the following conditions: initiation at 95°C for 2 minutes, followed by 35 cycles of denaturation, annealing, and extension (depending on the amplicon; Supplementary Table S1). PCR products were then analyzed on a 1.5% agarose gel, extracted (NucleoSpin Extract II kit, Macherey Nagel, Hoerdt, France), cloned into the pS972 vector (Promega, Charbonnières, France), and sequenced with standard SP6 and T7 forward and reverse primers.
Rpgrip1-1 to Rpgrip1-7 PCR fragments were assembled using endogenous restriction sites present in the cRpgrip1 cDNA. The cloned cRpgrip1 cDNA was verified by sequencing and was identical to the recently published canine Rpgrip1 reference sequence (GenBank HM021768).40
Vector plasmids construction and production of rAAV vectors. Recombinant AAV2/5-RK-eGFP, AAV2/5-RK-cRpgrip1, and AAV2/8-RK-cRpgrip1 vectors were produced by triple transfection of 293 cells according to previously reported methods using the SSV9-RK-eGFP or SSV9-RK-cRpgrip1 vector plasmids.10 These constructs expressed either eGFP or cRpgrip1 cDNA directly under the control of the short human RK promoter (−112 to +87 bp)33 and the bovine growth hormone polyadenylation signal (BGHpA), flanked by two AAV2 inverted terminal repeat sequences.
For the SSV9-RK-eGFP vector plasmid construction, the 588-bp cytomegalovirus (CMV) promoter of a parental SSV9-CMV-eGFP plasmid was replaced with the 238-bp human RK promoter (kindly provided by Shahrokh Khani, State University of New-York, Buffalo, NY). For the SSV9-RK-cRpgrip1 plasmid construction, the 1340-bp eGFP-WRPE sequence of the SSV9-RK-eGFP plasmid was replaced with the 3861-bp cRpgrip1 cDNA. The resulting SSV9-RK-eGFP and SSV9-RK-cRpgrip1 constructs were verified by sequencing.
Viral vector titers were determined by dot blot and by quantitative real-time PCR and are expressed as vector genomes per millilitre (vg/ml). The final vector titers of AAV2/5-RK-eGFP, AAV2/5-RK-cRpgrip1, and AAV2/8-RK-cRpgrip1 were 1 × 1011, 1 × 1011, and 1 × 1012 vg/ml, respectively.
Animals. A total of eight affected Rpgrip1−/− and 3 Rpgrip1+/− MLHD dogs were used in this study (Table 1). The first four Rpgrip1−/− individuals of the colony were kindly provided by Cathryn Mellersh (Animal Health Trust, Lanwades Park, Kentford, Newmarket, Suffolk, UK).14 Animals were maintained at the Boisbonne Center (ONIRIS, Nantes-Atlantic College of Veterinary Medicine, Food Science and Engineering, Nantes, France) under a 12/12-hour light/dark cycle. All procedures involving animals were performed in compliance with the Association for Research in Vision and Ophthalmology statement for the use of animals in ophthalmic and vision research.
Subretinal administration of rAAV vectors. Subretinal injection of AAV2/5-RK-eGFP vector was performed bilaterally on two nonaffected Rpgrip1+/− dogs at 7 months of age. Subretinal injections of AAV2/5-RK-cRpgrip1 and AAV2/8-RK-cRpgrip1 were performed unilaterally on seven affected Rpgrip1−/− dogs at 1 month of age (Table 1). All subretinal injections were performed under general anesthesia as previously published.10 All the vectors were injected at their maximal titer (AAV2/5-RK-eGFP, 1011 vg/ml; AAV2/5-RK-cRpgrip1, 1011 vg/ml; and AAV2/8-RK-cRpgrip1, 1012 vg/ml). The volume injected varied from 90 to 140 µl. At the time of injection, the location and extent of the subretinal blebs were recorded on fundus photographs or schematic fundus drafts when it was not possible to obtain clear fundus photography at the time of injection.
In vivo GFP fluorescence imaging and GFP expression analysis. GFP fluorescence in dogs NA2 and NA3 was monitored at weekly intervals by fluorescence retinal imaging using a Canon UVI retinal camera connected to a digital imaging system (Lhedioph Win Software, Lheritier SA, Saint-Ouen-l'Aumône, France).
At 9 mpi, dogs were euthanized by intravenous injection of pentobarbital sodium (Vétoquinol, Lure, France). Eyecups were fixed for 48 hours in Bouin's solution (Laurylab, Saint Fons, France), embedded in paraffin, and 7-µm sections were prepared. Before immunostaining, sections were deparaffinized in toluene and rehydrated with phosphate-buffered saline solution. This method allows good preservation of cone photoreceptor shape but slightly reduces eGFP fluorescence. Nonspecific antigen binding was blocked by incubating sections in blocking solution (20% normal goat serum (Invitrogen), 0.1% Triton X-100 in phosphate-buffered saline) for 45 minutes at room temperature. Sections were then incubated overnight at 4°C with rabbit anti-LM opsin (1:200; Millipore, Molsheim, France) in blocking solution. After three washes in 0.1% Triton X-100 in phosphate-buffered saline solution, slides were incubated with the Alexa fluor 546–conjugated goat antirabbit IgG (1:250; Invitrogen) for 2 hours at room temperature. Cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen), and sections were mounted in Prolong Gold antifade reagent (Invitrogen). Sections were observed with an epifluorescence microscope (Nikon, Champigny-sur-Marne, France). eGFP signals were observed directly under 488 nm. To facilitate the colocalization of eGFP and L/M opsin, the immunolabeled sections were highly exposed to 568 nm to observe Alexa fluor 546 signals in cone inner segments. Images were captured with a digital camera (Nikon) (30 slices analyzed by canine retina, n = 4).
Reverse transcriptase–PCR. Total RNA was isolated from individual snap-frozen retinas of Rpgrip1+/+, Rpgrip1+/−, and Rpgrip1−/− dogs using TRIzol Reagent (Invitrogen). Rnase-free TURBO Dnase I (Ambion) was used according to the manufacturer's instructions to remove contaminating DNA before generation of cDNA by reverse transcription. Nine hundred nanograms of total RNA were reverse-transcribed using oligo-dT primers and Transcriptor High Fidelity reserve transcriptase (Roche Diagnostics) in a total volume of 20 µl as per the manufacturer's instructions.
Integrity of cDNA was determined by amplification of a 660-bp sequence of the canine β-actin gene using β-actinF (5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′) and β-actinR (5′-CTAGAAGCATTTGCGGTGGACGATGGAGGG-3′) primers. For cRpgrip1 amplification, 184F (5′-CCAGTGTCGAAAGGTAAGAAT-3′) and 317R (5′-TCCTCGCCCTTTTGATCTCAT-3′) primers were designed to amplify a 154-bp or 198-bp product specific for the wild-type or mutant Ins44 cDNA, respectively (Figure 3). The amount of cDNA used in β-actin and cRpgrip1 PCR was 40 ng. Both PCRs were performed using KOD Hot Start DNA Polymerase (Novagen) and a Veriti thermocycler (Applied Biosystems). Conditions of amplification were as follows: an initial denaturation step at 95°C for 2 minutes, followed by 40 cycles at 95°C for 20 seconds, 58°C for 10 seconds, and 70°C for 7 seconds. Actin and cRpgrip1 PCR products were resolved on a 1.5 and 4% agarose gel electrophoresis, respectively.
Fundus photography and TD-OCT. Fundus photography and TD-OCT were performed bilaterally on all treated dogs every 2 months after treatment (except dog A6). Before clinical examinations, pupils were fully dilated by topical administration of 0.3% atropine (Alcon Cusi SA, Barcelona, Spain), tropicamide (Novartis, Annonay, France), and phenylephrine hypochloride (Novartis). Dogs were anesthetized by intravenous injection of xylazine (BayerHealth Care, Shawnee Mission, UK) and ketamine (Rhone Merieux, Lyon, France).
Fundus photographs were taken with a Canon UVI retinal camera connected to a digital imaging system (Lhedioph). TD-OCT was performed using a 3-mm horizontal line scan of the retina (Stratus 3000; Carl Zeiss S.A.S., Le Pecq, France). Mann–Whitney paired t-test was used to statistically analyze the results from treated and untreated eyes. A P value <0.05 was considered significant (confidence intervals = 95%). Statistical analyzes were performed using GraphPad Prism for Macintosh (GraphPad, Software, San Diego, CA).
Spectral-domain OCT. SD-OCT was performed bilaterally on dogs A2, A7, and A8 at 24 mpi. Pupils were dilated as described above, and dogs were anesthetized following premedication by intravenous injection of thiopental sodium (Specia Laboratories, Paris, France) followed by isoflurane gas inhalation.
SD-OCT was performed with 30° horizontal scans placed across the vector bleb temporal boundary (Spectralis Heidelberg Engineering, Heidelberg, Germany). Scans covered retinal regions of 13 × 8 mm (13 line scans of 1 × 8 mm, 23 averaging). Postacquisition processing of OCT data was performed with a commercial program following manufacturer's instructions (Heidelberg). The photoreceptor layer (ONL + outer and inner segments) was defined semi-automatically, and its thickness was automatically calculated.
Postmortem analysis of photoreceptor preservation in MLHD-Cord1–treated eyes. Dog A2 and age-matched untreated dog NA1 were sacrificed 24 mpi as described above. Eyes were fixed for 2 hours in 4% paraformaldehyde in phosphate-buffered saline solution before removal of the anterior chamber and the lens. Eyecups were embedded in optimal cutting temperature compound (OCT Cryomount, Microm Microtech, Francheville, France) and snap frozen in a dry isopentane bath. Cryosections of 12–15 μm thickness were prepared and stored until use.
For morphological examination, sections were stained with hematoxylin–eosin before imaging by transmitter light microscopy (Nikon). For immunohistochemical studies, sections were immunolabeled as previously described with the following antibodies: PNA-FITC (Vector Laboratories, Bulingame, CA, USA; 1:250), rabbit anti-L/M opsin (Millipore; 1:250), rabbit anti-S opsin (Millipore; 1:250), and rabbit anti-GNAT1 (Santa-Cruz Technology, Heidelberg, Germany; 1:250). Primary antibodies were revealed with the secondary antibody Alexa 546–conjugated goat antirabbit IgG (Invitrogen) at 1:250. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole, and sections were mounted in Mowiol antifade reagent (Calbiochem, San Diego, CA). Tissues were viewed on an epifluorescence microscope (Nikon), and images were captured with a digital camera (Nikon).
Electroretinography. Retinal function of AAV2/5-RK-cRpgrip1- or AAV2/8-RK-cRpgrip1-treated Rpgrip1−/− dogs and age-matched untreated Rpgrip1−/− and Rpgrip1+/− controls were evaluated using bilateral full-field flash electroretinogram. Initial ERG measurements were recorded at 1 mpi and every subsequent 2 months, up to 4 (dog A5), 12 (dogs A3 and A4), or 24 mpi (dogs A2, A7, and A8).
Both pupils of animals were topically dilated as described above. Dogs were dark adapted for 20 minutes and anesthetized following premedication by intravenous injection of thiopental sodium (Specia Laboratories) followed by isoflurane gas inhalation. Hydroxypropulmethylcellulose (Laboratoires Théa, Clermont-Ferrand, France) was applied to each eye to prevent corneal dehydration during recordings.
A computer-based system (Neuropack MEB 9102K, Nikon KohdenO, Tokyo, Japan) and contact lens electrodes (ERG-jet; Microcomponents SA, Grenchen, Switzerland) were used. ERGs were recorded in a standardized fashion according to the International Society for Clinical Electrophysiology of Vision recommendations using a protocol described previously.29 Mann–Whitney paired t-test was used to statistically analyze the results from treated and untreated eyes. A P value <0.05 was considered significant (confidence intervals = 95%). Statistical analyzes were performed using GraphPad Prism for Macintosh (GraphPad Software).
Behavioral studies. Ambulation of treated Rpgrip1−/− dogs through an obstacle avoidance course in bright- (260 ± 13 lux) and dim-light (1.5 ± 0.8 lux) conditions was evaluated at 4, 9, 12, 18, and 24 mpi as previously described.10 An opaque lens was used to alternatively cover the treated or the untreated eye, and obstacle panel combinations were randomly determined for each test to prevent the dogs memorizing the positions of the obstacle panels. All dim-light films were cleared before mounting with commercial Final Cut Pro 7 software (Apple, Cork, Ireland) using a standardized protocol. For each dog, the transit time and the number of collisions from three trials on the same day were used for data analysis. Vision-guided behavior (referred as + in Table 2) was determined for a mean transit time ≤10 seconds and a mean number of collisions ≤1. Non–vision-guided behavior (referred as – in Table 2) was determined for a mean transit time ≥20 seconds and/or a mean number of collisions ≥3.
SUPPLEMENTARY MATERIAL Figure S1. Canine Rpgrip1 amplification strategy. Figure S2. Gene therapy did not halt retinal degeneration in treated MLHD-Cord1 eyes. Figure S3. Gene therapy prevented long-term photoreceptor degeneration in all treated MLHD-Cord1 eyes. Figure S4. Bilateral full-field electroretinographic traces from dogs A7, A3, A4, and A5. Figure S5. Statistical analysis of ERG responses in treated and untreated MLHD-Cord1 eyes. Table S1. Primers used for the amplification of cRpgrip1 cDNA. Table S2. Characterization of the exon–intron junctions of the cRpgrip1 gene. Video S1. Assessment of bright- and dim-light vision of dog A7 at 12 months postinjection.
Acknowledgments
We thank Cathryn Mellersh (Animal Health Trust, Newmarket, Suffolk, UK) for the gift of the first MLHD-Cord1 dogs and Shahrokh Khani (State University of New-York, Buffalo, NY, USA) for the gift of the human RK promoter. We thank the Vector Core (http://www.vectors.univ-nantes.fr) for production of the rAAV vectors, the staff of the Boisbonne Center and Thibaut Larcher (INRA/ONIRIS, Nantes, France) for animal care, and Mireille Ledevin (INRA/ONIRIS, Nantes, France) for technical assistance. We also thank Knut Stieger (Justut-Liebig-University Giessen, Germany) for critical reading of the manuscript. This work was supported by unrestricted grants from the Association Française contre les Myopathies, the INSERM, the Fondation pour la Thérapie Génique en Pays de la Loire, and the Agence Nationale pour la Recherche.
Supplementary Material
Assessment of bright- and dim-light vision of dog A7 at 12 months postinjection.
References
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- Michaelides M, Hardcastle AJ, Hunt DM, Moore AT. Progressive cone and cone-rod dystrophies: phenotypes and underlying molecular genetic basis. Surv Ophthalmol. 2006;51:232–258. doi: 10.1016/j.survophthal.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Hamel CP. Cone rod dystrophies. Orphanet J Rare Dis. 2007;2:7. doi: 10.1186/1750-1172-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander AI, Black A, Bennett J, Cremers FP. Lighting a candle in the dark: advances in genetics and gene therapy of recessive retinal dystrophies. J Clin Invest. 2010;120:3042–3053. doi: 10.1172/JCI42258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Bainbridge JW, Ali RR. Gene supplementation therapy for recessive forms of inherited retinal dystrophies. Gene Ther. 2012;19:154–161. doi: 10.1038/gt.2011.161. [DOI] [PubMed] [Google Scholar]
- Colella P, Auricchio A. Gene therapy of inherited retinopathies: a long and successful road from viral vectors to patients. Hum Gene Ther. 2012;23:796–807. doi: 10.1089/hum.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye SE, Boye SL, Lewin AS, Hauswirth WW. A comprehensive review of retinal gene therapy. Mol Ther. 2013;21:509–519. doi: 10.1038/mt.2012.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieger K, Lhériteau E, Lhéariteau E, Moullier P, Rolling F. AAV-mediated gene therapy for retinal disorders in large animal models. ILAR J. 2009;50:206–224. doi: 10.1093/ilar.50.2.206. [DOI] [PubMed] [Google Scholar]
- Komáromy AM, Alexander JJ, Rowlan JS, Garcia MM, Chiodo VA, Kaya A, et al. Gene therapy rescues cone function in congenital achromatopsia. Hum Mol Genet. 2010;19:2581–2593. doi: 10.1093/hmg/ddq136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit L, Lhériteau E, Weber M, Le Meur G, Deschamps JY, Provost N, et al. Restoration of vision in the pde6β-deficient dog, a large animal model of rod-cone dystrophy. Mol Ther. 2012;20:2019–2030. doi: 10.1038/mt.2012.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran WA, Cideciyan AV, Lewin AS, Iwabe S, Khanna H, Sumaroka A, et al. Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc Natl Acad Sci USA. 2012;109:2132–2137. doi: 10.1073/pnas.1118847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turney C, Chong NH, Alexander RA, Hogg CR, Fleming L, Flack D, et al. Pathological and electrophysiological features of a canine cone-rod dystrophy in the miniature longhaired dachshund. Invest Ophthalmol Vis Sci. 2007;48:4240–4249. doi: 10.1167/iovs.04-0737. [DOI] [PubMed] [Google Scholar]
- Lhériteau E, Libeau L, Stieger K, Deschamps JY, Mendes-Madeira A, Provost N, et al. The RPGRIP1-deficient dog, a promising canine model for gene therapy. Mol Vis. 2009;15:349–361. [PMC free article] [PubMed] [Google Scholar]
- Mellersh CS, Boursnell ME, Pettitt L, Ryder EJ, Holmes NG, Grafham D, et al. Canine RPGRIP1 mutation establishes cone-rod dystrophy in miniature longhaired dachshunds as a homologue of human Leber congenital amaurosis. Genomics. 2006;88:293–301. doi: 10.1016/j.ygeno.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Dryja TP, Adams SM, Grimsby JL, McGee TL, Hong DH, Li T, et al. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am J Hum Genet. 2001;68:1295–1298. doi: 10.1086/320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber S, Perrault I, Hanein S, Barbet F, Ducroq D, Ghazi I, et al. Complete exon-intron structure of the RPGR-interacting protein (RPGRIP1) gene allows the identification of mutations underlying Leber congenital amaurosis. Eur J Hum Genet. 2001;9:561–571. doi: 10.1038/sj.ejhg.5200689. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Aleman TS, Sumaroka A, Schwartz SB, Roman AJ, et al. Leber congenital amaurosis caused by an RPGRIP1 mutation shows treatment potential. Ophthalmology. 2007;114:895–898. doi: 10.1016/j.ophtha.2006.10.028. [DOI] [PubMed] [Google Scholar]
- Fakhratova M.2013Identification of a novel LCA6 mutation in an Emirati family. Ophthalmic Genet(doi:10.3109/13816810.2012.755552). [DOI] [PubMed]
- Khan AO, Abu-Safieh L, Eisenberger T, Bolz HJ, Alkuraya FS. The RPGRIP1-related retinal phenotype in children. Br J Ophthalmol. 2013;97:760–764. doi: 10.1136/bjophthalmol-2012-303050. [DOI] [PubMed] [Google Scholar]
- Hameed A, Abid A, Aziz A, Ismail M, Mehdi SQ, Khaliq S. Evidence of RPGRIP1 gene mutations associated with recessive cone-rod dystrophy. J Med Genet. 2003;40:616–619. doi: 10.1136/jmg.40.8.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepman R, Bernoud-Hubac N, Schick DE, Maugeri A, Berger W, Ropers HH, et al. The retinitis pigmentosa GTPase regulator (RPGR) interacts with novel transport-like proteins in the outer segments of rod photoreceptors. Hum Mol Genet. 2000;9:2095–2105. doi: 10.1093/hmg/9.14.2095. [DOI] [PubMed] [Google Scholar]
- Hong DH, Yue G, Adamian M, Li T. Retinitis pigmentosa GTPase regulator (RPGRr)-interacting protein is stably associated with the photoreceptor ciliary axoneme and anchors RPGR to the connecting cilium. J Biol Chem. 2001;276:12091–12099. doi: 10.1074/jbc.M009351200. [DOI] [PubMed] [Google Scholar]
- Mavlyutov TA, Zhao H, Ferreira PA. Species-specific subcellular localization of RPGR and RPGRIP isoforms: implications for the phenotypic variability of congenital retinopathies among species. Hum Mol Genet. 2002;11:1899–1907. doi: 10.1093/hmg/11.16.1899. [DOI] [PubMed] [Google Scholar]
- Boylan JP, Wright AF. Identification of a novel protein interacting with RPGR. Hum Mol Genet. 2000;9:2085–2093. doi: 10.1093/hmg/9.14.2085. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Hong DH, Pawlyk B, Yue G, Adamian M, Grynberg M, et al. The retinitis pigmentosa GTPase regulator (RPGR)- interacting protein: subserving RPGR function and participating in disk morphogenesis. Proc Natl Acad Sci USA. 2003;100:3965–3970. doi: 10.1073/pnas.0637349100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won J, Gifford E, Smith RS, Yi H, Ferreira PA, Hicks WL, et al. RPGRIP1 is essential for normal rod photoreceptor outer segment elaboration and morphogenesis. Hum Mol Genet. 2009;18:4329–4339. doi: 10.1093/hmg/ddp385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlyk BS, Smith AJ, Buch PK, Adamian M, Hong DH, Sandberg MA, et al. Gene replacement therapy rescues photoreceptor degeneration in a murine model of Leber congenital amaurosis lacking RPGRIP. Invest Ophthalmol Vis Sci. 2005;46:3039–3045. doi: 10.1167/iovs.05-0371. [DOI] [PubMed] [Google Scholar]
- Pawlyk BS, Bulgakov OV, Liu X, Xu X, Adamian M, Sun X, et al. Replacement gene therapy with a human RPGRIP1 sequence slows photoreceptor degeneration in a murine model of Leber congenital amaurosis. Hum Gene Ther. 2010;21:993–1004. doi: 10.1089/hum.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Meur G, Weber M, Péréon Y, Mendes-Madeira A, Nivard D, Deschamps JY, et al. Postsurgical assessment and long-term safety of recombinant adeno-associated virus-mediated gene transfer into the retinas of dogs and primates. Arch Ophthalmol. 2005;123:500–506. doi: 10.1001/archopht.123.4.500. [DOI] [PubMed] [Google Scholar]
- Mussolino C, della Corte M, Rossi S, Viola F, Di Vicino U, Marrocco E, et al. AAV-mediated photoreceptor transduction of the pig cone-enriched retina. Gene Ther. 2011;18:637–645. doi: 10.1038/gt.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe LH, Bell P, Maguire AM, Cearley CN, Xiao R, Calcedo R, et al. Dosage thresholds for AAV2 and AAV8 photoreceptor gene therapy in monkey. Sci Transl Med. 2011;3:88ra54. doi: 10.1126/scitranslmed.3002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye SE, Alexander JJ, Boye SL, Witherspoon CD, Sandefer KJ, Conlon TJ, et al. The human rhodopsin kinase promoter in an AAV5 vector confers rod- and cone-specific expression in the primate retina. Hum Gene Ther. 2012;23:1101–1115. doi: 10.1089/hum.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khani SC, Pawlyk BS, Bulgakov OV, Kasperek E, Young JE, Adamian M, et al. AAV-mediated expression targeting of rod and cone photoreceptors with a human rhodopsin kinase promoter. Invest Ophthalmol Vis Sci. 2007;48:3954–3961. doi: 10.1167/iovs.07-0257. [DOI] [PubMed] [Google Scholar]
- Sun X, Pawlyk B, Xu X, Liu X, Bulgakov OV, Adamian M, et al. Gene therapy with a promoter targeting both rods and cones rescues retinal degeneration caused by AIPL1 mutations. Gene Ther. 2010;17:117–131. doi: 10.1038/gt.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku CA, Chiodo VA, Boye SL, Goldberg AF, Li T, Hauswirth WW, et al. Gene therapy using self-complementary Y733F capsid mutant AAV2/8 restores vision in a model of early onset Leber congenital amaurosis. Hum Mol Genet. 2011;20:4569–4581. doi: 10.1093/hmg/ddr391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye SE, Boye SL, Pang J, Ryals R, Everhart D, Umino Y, et al. Functional and behavioral restoration of vision by gene therapy in the guanylate cyclase-1 (GC1) knockout mouse. PLoS ONE. 2010;5:e11306. doi: 10.1371/journal.pone.0011306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihelec M, Pearson RA, Robbie SJ, Buch PK, Azam SA, Bainbridge JW, et al. Long-term preservation of cones and improvement in visual function following gene therapy in a mouse model of leber congenital amaurosis caused by guanylate cyclase-1 deficiency. Hum Gene Ther. 2011;22:1179–1190. doi: 10.1089/hum.2011.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye SL, Peshenko IV, Huang WC, Min SH, McDoom I, Kay CN, et al. AAV-mediated gene therapy in the guanylate cyclase (RetGC1/RetGC2) double knockout mouse model of Leber congenital amaurosis. Hum Gene Ther. 2013;24:189–202. doi: 10.1089/hum.2012.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran WA, Boye SL, Boye SE, Chiodo VA, Lewin AS, Hauswirth WW, et al. rAAV2/5 gene-targeting to rods:dose-dependent efficiency and complications associated with different promoters. Gene Ther. 2010;17:1162–1174. doi: 10.1038/gt.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova T, Zangerl B, Goldstein O, Acland GM, Aguirre GD. Structural organization and expression pattern of the canine RPGRIP1 isoforms in retinal tissue. Invest Ophthalmol Vis Sci. 2011;52:2989–2998. doi: 10.1167/iovs.10-6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyadera K, Kato K, Boursnell M, Mellersh CS, Sargan DR. Genome-wide association study in RPGRIP1(-/-) dogs identifies a modifier locus that determines the onset of retinal degeneration. Mamm Genome. 2012;23:212–223. doi: 10.1007/s00335-011-9384-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova T, Iwabe S, Boesze-Battaglia K, Pearce-Kelling S, Chang-Min Y, McDaid K, et al. Exclusion of RPGRIP1 ins44 from primary causal association with early-onset cone-rod dystrophy in dogs. Invest Ophthalmol Vis Sci. 2012;53:5486–5501. doi: 10.1167/iovs.12-10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyadera K, Brierley I, Aguirre-Hernández J, Mellersh CS, Sargan DR. Multiple mechanisms contribute to leakiness of a frameshift mutation in canine cone-rod dystrophy. PLoS ONE. 2012;7:e51598. doi: 10.1371/journal.pone.0051598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye SL, Conlon T, Erger K, Ryals R, Neeley A, Cossette T, et al. Long-term preservation of cone photoreceptors and restoration of cone function by gene therapy in the guanylate cyclase-1 knockout (GC1KO) mouse. Invest Ophthalmol Vis Sci. 2011;52:7098–7108. doi: 10.1167/iovs.11-7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MH, Smith AJ, Pawlyk B, Xu X, Liu X, Bainbridge JB, et al. Gene therapy for retinitis pigmentosa and Leber congenital amaurosis caused by defects in AIPL1: effective rescue of mouse models of partial and complete Aipl1 deficiency using AAV2/2 and AAV2/8 vectors. Hum Mol Genet. 2009;18:2099–2114. doi: 10.1093/hmg/ddp133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons DL, Boye SL, Hauswirth WW, Wu SM. Gene therapy prevents photoreceptor death and preserves retinal function in a Bardet-Biedl syndrome mouse model. Proc Natl Acad Sci USA. 2011;108:6276–6281. doi: 10.1073/pnas.1019222108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Jacobson SG, Beltran WA, Sumaroka A, Swider M, Iwabe S, et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci USA. 2013;110:E517–E525. doi: 10.1073/pnas.1218933110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acland GM, Aguirre GD, Bennett J, Aleman TS, Cideciyan AV, Bennicelli J, et al. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther. 2005;12:1072–1082. doi: 10.1016/j.ymthe.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Sothilingam V, Garcia Garrido M, Tanimoto N, Becirovic E, Koch F, et al. Gene therapy restores vision and delays degeneration in the CNGB1(-/-) mouse model of retinitis pigmentosa. Hum Mol Genet. 2012;21:4486–4496. doi: 10.1093/hmg/dds290. [DOI] [PubMed] [Google Scholar]
- Pang JJ, Dai X, Boye SE, Barone I, Boye SL, Mao S, et al. Long-term retinal function and structure rescue using capsid mutant AAV8 vector in the rd10 mouse, a model of recessive retinitis pigmentosa. Mol Ther. 2011;19:234–242. doi: 10.1038/mt.2010.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Assessment of bright- and dim-light vision of dog A7 at 12 months postinjection.