

Abstract
Mitsugumin 53 (MG53) is a member of the membrane repair system in skeletal muscle. However, the role(s) of MG53 in the unique functions of skeletal muscle have not been addressed, although it is known that MG53 is expressed only in skeletal and cardiac muscle. In the present study, MG53-binding proteins were examined along with proteins that mediate skeletal muscle contraction and relaxation using the binding assays of various MG53 domains and quadrupole time-of-flight mass spectrometry. MG53 binds to sarcoplasmic reticulum Ca2+-ATPase 1a (SERCA1a) via its tripartite motif (TRIM) and PRY domains. The binding was confirmed in rabbit skeletal muscle and mouse primary skeletal myotubes by co-immunoprecipitation and immunocytochemistry. MG53 knockdown in mouse primary skeletal myotubes increased Ca2+-uptake through SERCA1a (more than 35%) at micromolar Ca2+ but not at nanomolar Ca2+, suggesting that MG53 attenuates SERCA1a activity possibly during skeletal muscle contraction or relaxation but not during the resting state of skeletal muscle. Therefore MG53 could be a new candidate for the diagnosis and treatment of patients with Brody syndrome, which is not related to the mutations in the gene coding for SERCA1a, but still accompanies exercise-induced muscle stiffness and delayed muscle relaxation due to a reduction in SERCA1a activity.
Keywords: Mitsugumin 53 (MG53), SR Ca2+-ATPase 1a (SERCA1a), Tripartite Motif (TRIM), PRY domain, Phospholamban, Sarcolipin
1. INTRODUCTION
The sarcoplasmic reticulum (SR) acts as an internal Ca2+-store and the Ca2+ in the SR is the major Ca2+ source for the contraction of skeletal muscle [1; 2]. During skeletal muscle contraction, ryanodine receptor 1 is responsible for the channel that releases Ca2+ from the SR to the myoplasm [1; 2]. To reduce myoplasmic Ca2+ level to the resting state (nano-molar range) during muscle relaxation and, at the same time, to re-fill the SR with Ca2+, SR Ca2+-ATPase (SERCA) uptakes Ca2+ from the myoplasm to the SR [3]. SERCA1a is the main isoform of adult skeletal muscle (more than 70% in all types of skeletal muscle fibers) [4; 5].
Mitsugumin 53 (MG53, also called TRIM72), a muscle-specific tripartite motif (TRIM) family protein, contains a TRIM domain at the N-terminus and a PRY domain followed by a SPRY domain at the C-terminus [6; 7; 8; 9; 10]. The TRIM domain consists of a Ring domain harboring ubiquitin E3 ligase activity, a B-box harboring a zinc-binding moiety and two coiled-coil domains [6; 7]. MG53 is a member of the membrane repair system [8; 9], and an injury of the sarcolemmal membrane in mouse skeletal muscle fibers leads to a resealing of the injured membrane by gathering the intracellular vesicles coated with MG53 [8]. During this process, the oligomerization of MG53 through the oxidation of the thiol group of cysteine at 242 and a leucine zipper motif between the two coiled-coil domains are needed for the intracellular vesicle trafficking to the injury site [11]. Dysferlin, polymerase I and transcript release factor, and non-muscle myosin type IIA interact with MG53 to facilitate the vesicle trafficking to the injury site [12; 13; 14]. The binding of caveolin 3 to MG53 moderates the robust vesicle trafficking to the injury site [9; 14].
MG53 knockout mice show progressive skeletal myopathy associated with defective membrane repair [8]. Subcutaneous injection of purified MG53 to a mouse model of Duchenne muscular dystrophy alleviates the muscle pathology by promoting membrane repair [15]. Muscle-specific overexpression of MG53 in a hamster model of muscular dystrophy also ameliorates the pathology by enhancing membrane repair [16]. Interestingly, defective membrane repair by altered subcellular localization of MG53 is thought to be the cause of human muscular dystrophy in association with mutations in caveolin 3 [14].
Until now, studies on MG53 have been focused on MG53-mediated membrane repair, as mentioned above. There have been only a few reports of other roles of MG53 in skeletal muscle. In an MG53 knockdown study of the C2C12 cell line, MG53 facilitates the differentiation of C2C12 cells to myotubes by enhancing vesicle trafficking and membrane fusion [9]; and, another study shows that MG53 attenuates the insulin-like growth factor (IGF)-induced differentiation of C2C12 [17]. Considering that MG53 is expressed only in skeletal and cardiac muscle [8], that MG53 is related to skeletal muscle diseases [8; 14; 15; 16], and that MG53 plays a certain role in muscle cell differentiation [9; 17], it is possible that MG53 also participates in unique functions of skeletal muscle such as muscle contraction and relaxation. Therefore, in the present study, we searched MG53-binding proteins among proteins mediating skeletal muscle contraction and relaxation using biochemical and cellular approaches. We found that MG53 directly binds to SERCA1a via its TRIM and PRY domains and attenuates SERCA1a activity in skeletal muscle.
2. MATERIALS AND METHODS
2.1. cDNA construction and protein expression of GST-fused MG53 proteins
Using the full-length mouse MG53 cDNA (GenBank accession number: NM_001079932) as a template, GST-fused full-length MG53 or MG53 domains were synthesized and expressed in E. coli (DH5α), as previously described [18].
2.2. Preparation of triad vesicles and binding assay of GST-fused MG53 proteins with triad proteins
The triad vesicles were prepared, as previously described [19; 20]. All surgical interventions and pre- and post-surgical animal care were provided, as previously described [21; 22]. Binding assays were performed as previously described [18] and presented in the Supplementary Material 2.
2.3. In-gel digestion, protein identification by quadrupole time-of-flight mass spectrometry (qTOF MS), and database searches
Protein bands obtained from the binding assay were subjected to in-gel digestion with trypsin, as previously described [20]. The digested peptide solution was subjected to qTOF MS and database searches for protein identification, as described in the Supplementary Material 2.
2.4. Cell culture, co-immunoprecipitation, immunoblot assay, and immunocytochemistry
Mouse primary skeletal myoblasts were derived from mouse skeletal muscle, then proliferated and differentiated to myotubes, as previously described [21; 22; 23]. The myotubes were solubilized and subjected to co-immunoprecipitation, as previously described [20] and presented in the Supplementary Material 2.
2.5. MG53 knockdown by small interference RNA (siRNA) and quantitative real-time PCR (qPCR)
Sequences of two different siRNAs for mouse MG53 (Supplementary Material 3) were selected using siRNA design software (Dharmacon RNAi Technologies, Lafayette, CO). Transfection of the siRNAs to mouse primary skeletal myotubes and qPCR were performed as previously described [21] and presented in the Supplementary Material 3.
2.6. Preparation of myotube homogenate and the oxalate-supported 45Ca2+-uptake experiment
The MG53 knockdown myotubes were homogenated, as described in the Supplementary Material 2. The myotube homogenate was subjected to the oxalate-supported 45Ca2+-uptake experiment, as previously described [24] and presented in the Supplementary Material 2.
2.7. Statistical analysis
The results are presented as the mean ± S.E. The significant differences were analyzed using a paired t-test (GraphPad InStat, v2.04, GraphPad Software, La Jolla, CA). The differences were considered to be significant at p < 0.05.
3. RESULTS AND DISCUSSION
3.1. MG53 binds to SERCA1a via its TRIM and PRY domains
To investigate the MG53-binding proteins among proteins mediating the contraction and relaxation of skeletal muscle, first, cDNAs for five GST-fused MG53 proteins were constructed (Fig. 1A and Supplementary Material 1): GST-TRIM, GST-PRY, GST-SPRY, GST-PRY-SPRY, and GST-MG53 (full-length). Each GST-fused MG53 protein was expressed in E. coli and the bacterial cell lysate was separated on a SDS-PAGE gel and stained with Coomassie Brilliant Blue (Fig. 1B). The GST-fused MG53 proteins were successfully expressed. For binding assays, affinity beads were prepared by immobilizing each GST-fused MG53 protein on GST beads and the affinity beads were incubated with the solubilized triad vesicle sample from ‘rabbit’ skeletal muscle. The triad vesicles are composed of junctional SRs and t-tubules that are enriched portions with triad proteins that mediate the contraction and relaxation of skeletal muscle [1; 2; 20]. The proteins that were bound to the affinity beads were separated at three different percentages of SDS-PAGE gels (7, 10 and 12% for a clear view of proteins with different molecular weights) and were stained with Coomassie Brilliant Blue in order to evaluate the proteins that were specifically bound to the GST-fused MG53 proteins (Fig. 1C). The bands for the proteins bound to GST itself were excluded from consideration. Nine bands appeared as proteins that were bound to the GST-fused MG53 proteins, and the GST-fused MG53 proteins showing the nine bands are summarized in Fig. 1D.
Figure 1. Binding assays of GST-fused MG53 proteins with triad proteins.
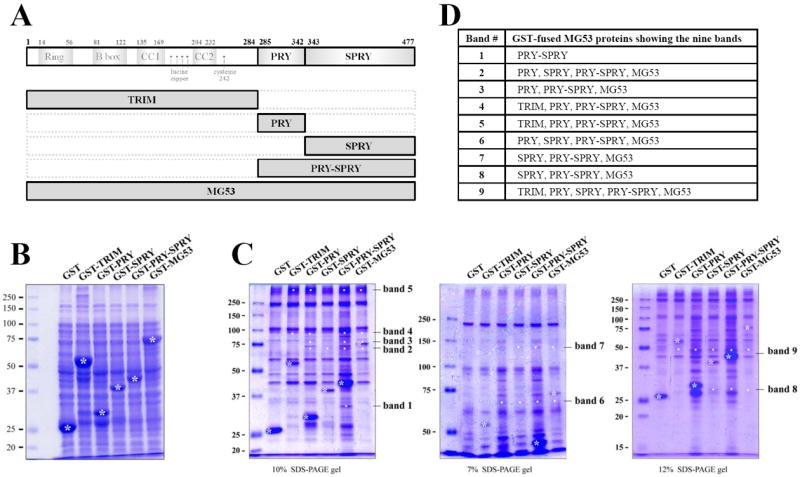
(A) Schematic diagrams of full-length mouse MG53 and domains. Numbers indicate the sequence of amino acids. (B) GST-fused MG53 proteins expressed in E.coli were separated on a SDS-PAGE gel (10%) and stained with Coomassie Brilliant Blue staining. GST-fused MG53 proteins are indicated by white asterisks. (C) The bound proteins obtained from the binding assays of GST-fused MG53 proteins with the triad proteins from rabbit skeletal muscle were separated on three different percentages of SDS-PAGE gels and stained with Coomassie Brilliant Blue. GST was used as a negative control. GST or GST-fused MG53 proteins are indicated by white asterisks. The specifically bound proteins to the GST-fused MG53 proteins are indicated by white dots. The newly appearing nine bands compared with the GST control are indicated on the right (bands 1 to 9). (D) The GST-fused MG53 proteins showing the nine bands are summarized.
The nine bands were subjected to in-gel digestion and to qTOF MS for protein identification. Supplementary Material 4 and Table 1 show the results of q-TOF MS and database searches. Band 1 was identified as a mouse MG53 fragment that would bind only to PRY-SPRY (Figs. 1C and 1D), suggesting that MG53 could homo-oligomerize through an inter-domain formed by PRY and SPRY domains but not by each PRY or SPRY domain. Bands 2, 3, 6, and 9 were identified as non-specifically bound proteins that originated from the E. coli lysate during the binding assay. Band 4 was identified as a protein complex composed of SERCA1a that originated from ‘rabbit’ skeletal muscle and two other non-specifically bound proteins that originated from either the E. coli lysate or from pasteurella. Band 5 was also identified as SERCA1a like band 4, suggesting that SERCA1a could be a MG53-binding protein. Considering that bands for SERCA1a would bind to TRIM, PRY, PRY-SPRY, and to a full-length MG53 but not to SPRY (Fig. 1D), the TRIM and PRY domains of MG53 were involved in binding to SERCA1a. For bands 7 and 8, there was no matching signal in the known databases.
Table 1.
List of proteins identified by q-TOF MS
| Band # | Protein name | Mascot # | Mass (Da) | Species | Matching Score | Matching peptides |
|---|---|---|---|---|---|---|
|
| ||||||
| 1 | MG53 (Trim72) | gi∣45500997 | 41411 | Mouse | 65 | 632.3 |
| RWALGVMAADASRR | ||||||
|
| ||||||
| 2 | Chaperone protein htpG | HTPG_ECOHS | 71378 | E. coli | 46 | 635.3 |
| RALSNPDLYEGDGELRVR.V | ||||||
|
| ||||||
| 3 | Chain A, Crystal Structure Of C418a, C419a Mutant Of Pf1 | gi∣6730181 | 85279 | E. coli | 98 | 1033.2 |
| KYGYDISGPATNAQEAIQWTYFGYLAAVKS | ||||||
|
| ||||||
| 4 | SERCA1a (Chain A of Ca2+-ATPase) | gi∣18159010 | 110787 | Rabbit | 114 | 758.9; 781.4 |
| KVGEATETALTTLVEKM | ||||||
|
| ||||||
| Pyruvate dehydrogenase subunit E1 | gi∣15799798 | 99948 | E. coli | 66 | 752.4 | |
| RAQYLIDQLLAEARK | ||||||
|
| ||||||
| Bifunctional aconitate hydratase 2/2-methylisocitrate dehydratase | gi∣15602069 | 95141 | Pasteurella | 71 | 841.4 | |
| KGFPLAYVGDVVGTGSSRK | ||||||
|
| ||||||
| 5 | SERCA1a (Chain A of Ca2+-ATPase) | gi∣18159010 | 110787 | Rabbit | 82 | 758.9; 781.4 |
| KVGEATETALTTLVEKM | ||||||
|
| ||||||
| 6 | Heat shock protein 90 | gi∣15800202 | 71374 | E. coli | 69 | 743.4 |
| RGLIDSSDLPLNVSRE | ||||||
|
| ||||||
| 7 | No matching signal | |||||
|
| ||||||
| 8 | No matching signal | |||||
|
| ||||||
| 9 | Tryptophanase | gi∣41936 | 53098 | E. coli | 87 | 551.3; 993.5; 1103.9 |
| RGIEEVGPNNVPYIVATITSNSAGGQPVSLANLKA | ||||||
The binding of MG53 to SERCA1a was directly accessed in rabbit skeletal muscle tissue and mouse primary skeletal myotubes. MG53 was co-immunoprecipitated with SERCA1a in the triad vesicle sample from rabbit skeletal muscle (Fig. 2A). Immunocytochemistry with anti-MG53 and anti-SERCA1a antibodies in mouse primary skeletal myotubes showed a co-localization of MG53 and SERCA1a near the nucleus (Fig. 2B). Therefore, based on the three different approaches (binding assay and qTOP MS, co-immunoprecipitation, and immunocytochemistry), we suggest that, with the exception of the proteins that mediate membrane repair, SERCA1a represents the first identification of a protein that binds directly to MG53 in skeletal muscle.
Figure 2. Co-immunoprecipitation and co-localization of full-length MG53 with SERCA1a in rabbit skeletal muscle and in mouse primary skeletal myotubes.
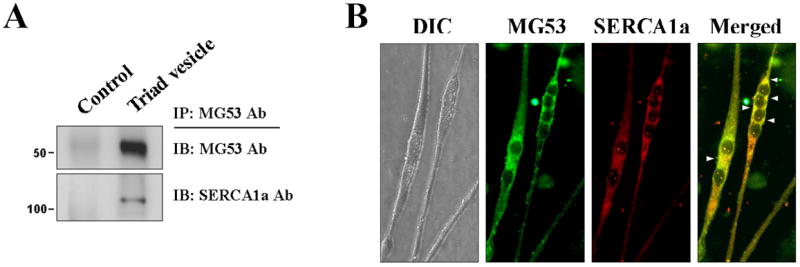
(A) MG53 was co-immunoprecipitated with SERCA1a in the triad vesicle sample from rabbit skeletal muscle. Control indicates a reaction without anti-MG53 antibody. (B) MG53 was co-localized with SERCA1a in the mouse primary skeletal myotubes (indicated by arrowheads). DIC, differential interference contrast microscopy. At least three independent experiments were conducted.
3.2. MG53 attenuates SERCA1a activity in skeletal myotubes
To examine how MG53 is functionally related to SERCA1a, we performed a knockdown of MG53 in mouse primary skeletal myotubes using siRNA transfections. Two different siRNAs were used to knockdown MG53 (Supplementary Material 3), and qPCR results showed that siRNA #2 more effectively reduced the mRNA level of MG53 (86.45±6.67 % reduction) compared with the untransfected control (Fig. 3A). A scrambled siRNA was used as the negative control. Immunoblot assay with anti-MG53 antibody using the lysate of the MG53 knockdown myotubes by siRNA #2 showed that MG53 expression was reduced by up to 80% (Fig. 3B), suggesting the successful knockdown of MG53 in mouse primary skeletal myotubes. On the other hand, the MG53 knockdown myotubes showed hampered differentiations to myotubes, with thinner and shorter myotube formations than those found in either the untransfected or the scrambled siRNA-transfected control (Fig. 3C). This suggests that MG53 is not indispensable but is needed to facilitate the differentiation of mouse primary skeletal myoblasts to myotubes. In accordance with this, severely hampered differentiation has been reported in an MG53 knockdown of the C2C12 cell line [9].
Figure 3. Knockdown of MG53 in mouse primary skeletal myotubes and 45Ca2+-uptake into the SR.
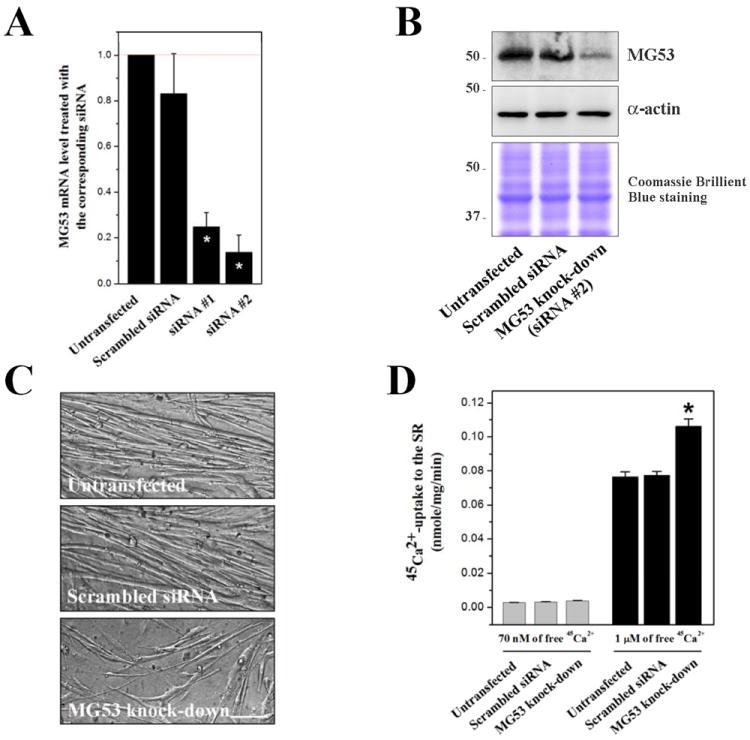
(A) qPCR results show the reduced mRNA levels of MG53 by the siRNA transfections. siRNA #2 knocked down MG53 more effectively. A scrambled siRNA was used as a negative control. The values were normalized to the value from the untransfected control. The results are presented as the mean ± S.E. of duplicated experiments. (B) The lysate of the MG53 knockdown myotubes by siRNA #2 (50 μg of total protein) was subjected to immunoblot analysis with anti-MG53 antibody. α-actin and Coomassie Brilliant Blue staining were loading controls. The expression of MG53 protein was significantly reduced by up to 80%. Three independent experiments were conducted. (C) The MG53 knockdown myotubes show thinner and shorter myotube formations compared with the untransfected or the scrambled siRNA-transfected control. The bar represents 100 μm. (D) Oxalate-supported 45Ca2+-uptake into the SR using the homogenate of the MG53 knockdown myotubes was measured at 70 nM or 1 μM of free 45Ca2+. The MG53 knockdown myotubes showed a significantly increased 45Ca2+-uptake only at 1 μM of free 45Ca2+. The results are presented as the mean ± S.E. of five independent experiments. *Significant difference versus untransfected control (p < 0.05).
Ca2+-uptake from the myoplasm to the SR by SERCA1a is an important event for skeletal muscle relaxation [3]. Therefore, the Ca2+-uptake activity of SERCA1a was examined in MG53 knockdown myotubes using an oxalate-supported 45Ca2+-uptake assay. The Ca2+-uptake activity of SERCA1a was not changed at a resting myoplasmic Ca2+ concentration (70 nM of free 45Ca2+ ([21; 22]), Fig. 3D (left-hand)), suggesting that MG53 is not involved in the regulation of SERCA1a activity in the resting state of skeletal muscle. At a higher myoplasmic Ca2+ concentration, such as that found during skeletal muscle contraction (1 μM of free 45Ca2+ [1; 25]), Ca2+-uptake activity of SERCA1a was significantly increased (more than 35% increase compared with the untransfected control, Fig. 3D (right-hand)), suggesting that MG53 is an attenuator of SERCA1a activity at micro-molar Ca2+ concentrations in skeletal muscle. There are two possible explanations for the role of MG53 at micro-molar Ca2+ concentration. One is that MG53 attenuates SERCA1a activity during skeletal muscle contraction in order to maximize the myoplasmic Ca2+ concentration for contraction. The other is that MG53 is needed to maintain a moderate Ca2+-uptake to the SR by attenuating SERCA1a activity during skeletal muscle relaxation, and to subsequently moderate the rate of muscle relaxation and contraction.
3.3. MG53 shares functional and structural homologies with sarcolipin
The structure-function relationship of SERCA1 is well-understood, but the regulatory aspects are best understood for SERCA2a, which is the major isoform in cardiac and slow-twitch skeletal muscle [26]. Phospholamban (PLN) binds to SERCA2a, and the unphosphorylated form of PLN results in decreases in SERCA2a activity and in the rate of muscle relaxation and contraction [27]. However, PLN is absent from fast-twitch skeletal muscle that expresses SERCA1a as its major SERCA isoform [28], which suggests that PLN is not an in-situ regulator of SERCA1.
On the other hand, sarcolipin, a small protein composed of 31 amino acids, is thought to be a PLN-like protein in skeletal muscle [29]. Sarcolipin knockout mice have shown increased SERCA1a activity in skeletal muscle (in soleus and red gastrocnemius) compared with wild-type mice [30], and MG53 knockdown myotubes also showed an increase in SERCA1a activity in the present study. Therefore, MG53 seems to share a functional homology with sarcolipin. The 3D structure of sarcolipin obtained from solution and solid-state nuclear magnetic resonance spectroscopy (PDB ID: 1JDM) showed an alpha-helix (Supplementary Material 5A, upper panel) that participated in binding to SERCA1a [31], and the helix was accurately predicted by a prediction program for protein secondary structures (Jpred3 [32], GenBank accession number: NM_025540.2) (Supplementary Material 5A, bottom panel). The TRIM domain of mouse MG53 was also predicted, with a high degree of confidence, to have two alpha-helixes (Supplementary Material 5B, GenBank accession number: NM_001079932). This structural homology suggests the possibility that the helixes in the TRIM domain may play the same role in SERCA1a regulation that the alpha-helix in sarcolipin plays.
3.4. The binding of MG53 to SERCA1a could be mediated by unique ways
Considering that the regulation of SERCA2a activity by PLN depends on the phosphorylation status of PLN [27], although sarcolipin has no obvious phosphorylation site [33], we searched phosphorylation sites in the TRIM and PRY domains of mouse MG53 using in-silico tools. A NetPhos program based on the primary sequences of proteins [34] suggested that 11 serines and 2 threonines in the TRIM and PRY domains were possible phosphorylation sites. Among them, serine at 307 in the PRY domain was also predicted with a high degree of confidence using a Phos3D program that is based on the spatial contexts of the known 3D protein structures [35] (emphasized in Supplementary Material 5C). Interestingly, the serine at 307 in mouse MG53 is a unique amino acid compared with other proteins that contain a PRY domain (mainly aspartic acid in other proteins, Supplementary Material 6).
The 3D structures of several proteins containing the TRIM domain have been revealed. However, these are not useful in predicting SERCA1a binding sites in the TRIM domain of mouse MG53 because they have a very low identity among amino acid sequences. The 3D structure of the PRY domain of human MG53 has been revealed as a complex with the SPRY domain (Supplementary Material 5C, PDB ID: 3kb5, GenBank accession number: NM_001008274.3, ‘91% identity in amino acid sequence with mouse MG53’). In the PRY and SPRY complex, the prominent binding pocket for protein-protein interactions is mainly composed of the SPRY domain ([10], indicated by a transparent red circle in Supplementary Material 5C). The predicted phosphorable serine at 307 is irrelevant to the prominent binding pocket on the SPRY domain. In addition, the structure of the PRY and SPRY domains of human MG53 differs from those of other PRY- and/or SPRY-containing proteins [10]. Therefore, based on the prediction of phosphorylation sites and the structural considerations above, the binding of MG53 to SERCA1a via its TRIM and PRY domains could be mediated by different ways from the binding by other proteins containing a TRIM and/or a PRY domain.
3.5. MG53 could be a new candidate for the diagnosis and treatment of patients with Brody syndrome
Brody disease is an inherited myopathy due to mutations in the SERCA1a gene, and to the subsequent reduction in SERCA1a activity [36]. However, some groups of patients with Brody disease show reduced SERCA1a activity ‘without mutations in the SERCA1a gene,’ although they suffered from exercise-induced muscle stiffness and delayed muscle relaxation (so called Brody syndrome) [37; 38]. This suggests that SERCA1 is not the only candidate protein for the cause of the Brody disease and Brody syndrome. Sarcolipin was a prime candidate, but unfortunately, as with the SERCA1a gene, no sarcolipin mutation has been found in patients with Brody syndrome [33]. The present study showed that MG53 attenuates SERCA1a activity by direct binding. MG53 knockout mice have shown progressive myopathy [8], and MG53 has ameliorated the pathology of muscular dystrophy in animal models [15; 16]. Therefore, it is possible that MG53 is a new candidate for the diagnosis and treatment of patients with Brody syndrome.
Supplementary Material
HIGHLIGHTS.
SERCA1a is the first MG53-binding protein to be identified in skeletal muscle with the exception of the proteins that mediate membrane repair.
MG53 attenuates SERCA1a activity in skeletal muscle.
MG53 could be a candidate for the diagnosis/treatment of patients with Brody syndrome.
Acknowledgments
This work was supported by the Mid-career Researcher Program (2012-0005435) and by the Basic Science Research Program (2011-0026752) through NRF grants funded by the MEST, Korea.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lee EH. Ca2+ channels and skeletal muscle diseases. Prog Biophys Mol Biol. 2010;103:35–43. doi: 10.1016/j.pbiomolbio.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Lee EH, Kim DH, Allen PD. Interplay between intra- and extracellular calcium ions. Mol Cells. 2006;21:315–29. [PubMed] [Google Scholar]
- 3.Shamoo AE, MacLennan DH. A Ca2+-dependent and -selective ionophore as part of the Ca2+ plus Mg2+-dependent adenosinetriphosphatase of sarcoplasmic reticulum. Proc Natl Acad Sci USA. 1974;71:3522–6. doi: 10.1073/pnas.71.9.3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brandl CJ, deLeon S, Martin DR, MacLennan DH. Adult forms of the Ca2+-ATPase of sarcoplasmic reticulum. Expression in developing skeletal muscle. J Biol Chem. 1987;262:3768–74. [PubMed] [Google Scholar]
- 5.Brandl CJ, Green NM, Korczak B, MacLennan DH. Two Ca2+-ATPase genes: homologies and mechanistic implications of deduced amino acid sequences. Cell. 1986;44:597–607. doi: 10.1016/0092-8674(86)90269-2. [DOI] [PubMed] [Google Scholar]
- 6.Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, Guffanti A, Minucci S, Pelicci PG, Ballabio A. The tripartite motif family identifies cell compartments. EMBO J. 2001;20:2140–51. doi: 10.1093/emboj/20.9.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meroni G, Diez-Roux G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. Bioessays. 2005;27:1147–57. doi: 10.1002/bies.20304. [DOI] [PubMed] [Google Scholar]
- 8.Cai C, Masumiya H, Weisleder N, Matsuda N, Nishi M, Hwang M, Ko JK, Lin P, Thornton A, Zhao X, Pan Z, Komazaki S, Brotto M, Takeshima H, Ma J. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol. 2009;11:56–64. doi: 10.1038/ncb1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai C, Masumiya H, Weisleder N, Pan Z, Nishi M, Komazaki S, Takeshima H, Ma J. MG53 regulates membrane budding and exocytosis in muscle cells. J Biol Chem. 2009;284:3314–22. doi: 10.1074/jbc.M808866200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park EY, Kwon OB, Jeong BC, Yi JS, Lee CS, Ko YG, Song HK. Crystal structure of PRY-SPRY domain of human TRIM72. Proteins. 2010;78:790–5. doi: 10.1002/prot.22647. [DOI] [PubMed] [Google Scholar]
- 11.Hwang M, Ko JK, Weisleder N, Takeshima H, Ma J. Redox-dependent oligomerization through a leucine zipper motif is essential for MG53-mediated cell membrane repair. Am J Physiol Cell Physiol. 2011;301:C106–14. doi: 10.1152/ajpcell.00382.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu H, Lin P, De G, Choi KH, Takeshima H, Weisleder N, Ma J. Polymerase transcriptase release factor (PTRF) anchors MG53 protein to cell injury site for initiation of membrane repair. J Biol Chem. 2011;286:12820–4. doi: 10.1074/jbc.C111.221440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin P, Zhu H, Cai C, Wang X, Cao C, Xiao R, Pan Z, Weisleder N, Takeshima H, Ma J. Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J. 2012;26:1875–83. doi: 10.1096/fj.11-188599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cai C, Weisleder N, Ko JK, Komazaki S, Sunada Y, Nishi M, Takeshima H, Ma J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J Biol Chem. 2009;284:15894–902. doi: 10.1074/jbc.M109.009589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weisleder N, Takizawa N, Lin P, Wang X, Cao C, Zhang Y, Tan T, Ferrante C, Zhu H, Chen PJ, Yan R, Sterling M, Zhao X, Hwang M, Takeshima M, Cai C, Cheng H, Takeshima H, Xiao RP, Ma J. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci Transl Med. 2012;4:139ra85. doi: 10.1126/scitranslmed.3003921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He B, Tang RH, Weisleder N, Xiao B, Yuan Z, Cai C, Zhu H, Lin P, Qiao C, Li J, Mayer C, Ma J, Xiao X. Enhancing muscle membrane repair by gene delivery of MG53 ameliorates muscular dystrophy and heart failure in delta-Sarcoglycan-deficient hamsters. Mol Ther. 2012;20:727–35. doi: 10.1038/mt.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee CS, Yi JS, Jung SY, Kim BW, Lee NR, Choo HJ, Jang SY, Han J, Chi SG, Park M, Lee JH, Ko YG. TRIM72 negatively regulates myogenesis via targeting insulin receptor substrate-1. Cell Death Differ. 2010;17:1254–65. doi: 10.1038/cdd.2010.1. [DOI] [PubMed] [Google Scholar]
- 18.Lee EH, Rho SH, Kwon SJ, Eom SH, Allen PD, Kim DH. N-terminal region of FKBP12 is essential for binding to the skeletal ryanodine receptor. J Biol Chem. 2004;279:26481–8. doi: 10.1074/jbc.M309574200. [DOI] [PubMed] [Google Scholar]
- 19.Saito A, Seiler S, Chu A, Fleischer S. Preparation and morphology of sarcoplasmic reticulum terminal cisternae from rabbit skeletal muscle. J Cell Biol. 1984;99:875–85. doi: 10.1083/jcb.99.3.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woo JS, Kim DH, Allen PD, Lee EH. TRPC3-interacting triadic proteins in skeletal muscle. Biochem J. 2008;411:399–405. doi: 10.1042/bj20071504. [DOI] [PubMed] [Google Scholar]
- 21.Woo JS, Cho CH, Lee KJ, Kim DH, Ma J, Lee EH. Hypertrophy in skeletal myotubes induced by junctophilin-2 mutant, Y141H, involves an increase in store-operated Ca2+ entry via Orai1. J Biol Chem. 2012;287:14336–48. doi: 10.1074/jbc.M111.304808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woo JS, Hwang JH, Ko JK, Weisleder N, Kim DH, Ma J, Lee EH. S165F mutation of junctophilin 2 affects Ca2+ signalling in skeletal muscle. Biochem J. 2010;427:125–34. doi: 10.1042/BJ20091225. [DOI] [PubMed] [Google Scholar]
- 23.Rando TA, Blau HM. Methods for myoblast transplantation. Methods Cell Biol. 1997;52:261–72. doi: 10.1016/s0091-679x(08)60382-9. [DOI] [PubMed] [Google Scholar]
- 24.Park KS, Kim TK, Kim DH. Cyclosporin A treatment alters characteristics of Ca2+-release channel in cardiac sarcoplasmic reticulum. Am J Physiol. 1999;276:H865–72. doi: 10.1152/ajpheart.1999.276.3.H865. [DOI] [PubMed] [Google Scholar]
- 25.Sandow A. Excitation-contraction coupling in skeletal muscle. Pharmacol Rev. 1965;17:265–320. [PubMed] [Google Scholar]
- 26.Brini M, Carafoli E. Calcium pumps in health and disease. Physiol Rev. 2009;89:1341–78. doi: 10.1152/physrev.00032.2008. [DOI] [PubMed] [Google Scholar]
- 27.Kim HW, Steenaart NA, Ferguson DG, Kranias EG. Functional reconstitution of the cardiac sarcoplasmic reticulum Ca2+-ATPase with phospholamban in phospholipid vesicles. J Biol Chem. 1990;265:1702–9. [PubMed] [Google Scholar]
- 28.Arai M, Otsu K, MacLennan DH, Periasamy M. Regulation of sarcoplasmic reticulum gene expression during cardiac and skeletal muscle development. Am J Physiol. 1992;262:C614–20. doi: 10.1152/ajpcell.1992.262.3.C614. [DOI] [PubMed] [Google Scholar]
- 29.Odermatt A, Becker S, Khanna VK, Kurzydlowski K, Leisner E, Pette D, MacLennan DH. Sarcolipin regulates the activity of SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+-ATPase. J Biol Chem. 1998;273:12360–9. doi: 10.1074/jbc.273.20.12360. [DOI] [PubMed] [Google Scholar]
- 30.Tupling AR, Bombardier E, Gupta SC, Hussain D, Vigna C, Bloemberg D, Quadrilatero J, Trivieri MG, Babu GJ, Backx PH, Periasamy M, MacLennan DH, Gramolini AO. Enhanced Ca2+ transport and muscle relaxation in skeletal muscle from sarcolipin-null mice. Am J Physiol Cell Physiol. 2011;301:C841–9. doi: 10.1152/ajpcell.00409.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mascioni A, Karim C, Barany G, Thomas DD, Veglia G. Structure and orientation of sarcolipin in lipid environments. Biochemistry. 2002;41:475–82. doi: 10.1021/bi011243m. [DOI] [PubMed] [Google Scholar]
- 32.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Odermatt A, Taschner PE, Scherer SW, Beatty B, Khanna VK, Cornblath DR, Chaudhry V, Yee WC, Schrank B, Karpati G, Breuning MH, Knoers N, MacLennan DH. Characterization of the gene encoding human sarcolipin (SLN), a proteolipid associated with SERCA1: absence of structural mutations in five patients with Brody disease. Genomics. 1997;45:541–53. doi: 10.1006/geno.1997.4967. [DOI] [PubMed] [Google Scholar]
- 34.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–62. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 35.Durek P, Schudoma C, Weckwerth W, Selbig J, Walther D. Detection and characterization of 3D-signature phosphorylation site motifs and their contribution towards improved phosphorylation site prediction in proteins. BMC Bioinformatics. 2009;10:117. doi: 10.1186/1471-2105-10-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brody IA. Muscle contracture induced by exercise. A syndrome attributable to decreased relaxing factor. N Engl J Med. 1969;281:187–92. doi: 10.1056/NEJM196907242810403. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Fujii J, Phillips MS, Chen HS, Karpati G, Yee WC, Schrank B, Cornblath DR, Boylan KB, MacLennan DH. Characterization of cDNA and genomic DNA encoding SERCA1, the Ca2+-ATPase of human fast-twitch skeletal muscle sarcoplasmic reticulum, and its elimination as a candidate gene for Brody disease. Genomics. 1995;30:415–24. doi: 10.1006/geno.1995.1259. [DOI] [PubMed] [Google Scholar]
- 38.Voermans NC, Laan AE, Oosterhof A, van Kuppevelt TH, Drost G, Lammens M, Kamsteeg EJ, Scotton C, Gualandi F, Guglielmi V, van den Heuvel L, Vattemi G, van Engelen BG. Brody syndrome: A clinically heterogeneous entity distinct from Brody disease: A review of literature and a cross-sectional clinical study in 17 patients. Neuromuscul Disord. 2012 doi: 10.1016/j.nmd.2012.03.012. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.