

Abstract
Spatial and temporal organization of the genome represents an additional step in the regulation of nuclear functions. The nuclear lamina, a polymeric meshwork formed by lamins (A/C and B type) and lamin-associated proteins, plays a key role in the maintenance of genome localization, structure and function. Specifically, mutations in the LMNA gene encoding lamins A/C or changes in its expression, either upregulation or silencing, are associated with defects in DNA replication, transcription and repair, as well as alterations in epigenetic modifications of chromatin. These data, together with the fact that defects in A-type lamins are associated with a whole variety of degenerative disorders, premature aging syndromes and cancer, support the notion that these proteins operate as caretakers of the genome. However, our understanding of their functions is limited due to the lack of well-defined mechanisms behind the genomic instability observed in lamin-related diseases. Here, we summarize our recent discovery of new pathways that are affected by the loss of A-type lamins. In particular, we found that A-type lamins control transcription and degradation of proteins with key roles in cell cycle regulation and DNA double-strand breaks (DSBs) repair by nonhomologous end-joining (NHEJ) and homologous-recombination (HR). Importantly, the proteins regulated by A-type lamins—Rb family members, 53BP1, BRCA1 and RAD51— exert tumor suppressor functions, with their loss being associated with cancer susceptibility. Moreover, our studies revealed novel pathways that contribute to genomic instability and that can be activated in disease states independent of the status of A-type lamins.
Keywords: A-type lamins, DNA repair, cell cycle, proteases, vitamin D
Introduction
Lamin-related diseases are characterized by the presence of nuclear deformation, epigenetic alterations of chromatin and chromosomal aberrations.1,2 Most of the data on genomic instability has resulted from the study of cells from patients with Hutchinson-Gilford Progeria Syndrome (HGPS) and from mouse models of progeria. The progeria phenotype arises from mutations that alter the normal processing and maturation of lamin A.3,4 Accumulation of unprocessed lamin A species at the nuclear lamina causes the characteristic nuclear defects that lead to cell toxicity.5 Interestingly, progeria cells accumulate DNA DSBs,6,7 similar to cells of aged individuals,7,8 indicating a compromised DNA repair system. However, no clear defects in repair proteins themselves or in the activation of the DNA damage response (DDR) pathway have been observed in progeria cells.9 One exception is the observed accumulation of the protein XPA (Xeroderma pigmentosum group A) at DSBs10 which was associated with impaired recruitment of key DNA repair factors such as Rad50, Rad51 and 53BP1 to the breaks. Binding of XPA also activates ATM- and ATR-dependent signaling cascades that arrest the cell cycle. However, depletion of XPA in progeria cells only partially restored the recruitment of DNA repair factors to DSBs, indicating that additional mechanisms contribute to the DNA repair deficiencies in these cells. Interestingly, a recent report demonstrated the absence of the nuclear DNA-PK holoenzyme in premature as well as physiological aging.11 These studies have started to shed some light into putative molecular mechanisms that could be impacted upon by alterations in A-type lamins function. We have undertaken a loss-of-function approach to gain a deeper understanding of the role that A-type lamins play in the maintenance of genomic stability. We found that loss of A-type lamins leads to the downregulation of a number of factors with key roles in cell cycle regulation, e.g., Rb family members and DNA DSBs repair, e.g., 53BP1, BRCA1 and RAD51. We will summarize here the molecular mechanisms behind the regulation of these factors by A-type lamins and their significance for understanding aging-related diseases.
Mechanisms of DNA DSBs Repair
Repair of damaged DNA is critical for maintenance of genomic stability. Among the various types of DNA damage, DSBs are the most deleterious, leading to mutations, loss of genomic material and translocations if not properly repaired. The two major pathways of DSBs repair, homologous recombination (HR) and classic nonhomologous end-joining (C-NHEJ) are considered to compete for repair substrate and be mutually exclusive.12-14 HR is error-free and requires both resection of the 5′ DNA ends around the break and the presence of a homologous template. In contrast, C-NHEJ involves end ligation of damaged DNA and requires neither extensive resection nor homologous templates. C-NHEJ is a fast and error-prone mechanism which can cause translocations and/or loss of genetic material. While C-NHEJ is the predominant repair mechanism in G0/G1 stages of the cell cycle, when the lack of the sister chromatid prevents HR from being activated, the slower HR repair mechanism has traditionally been thought to dominate during S and G2 phases of the cell cycle. However, recent evidence15 has challenged the notion of HR dominance in S/G2, suggesting that the need for rapid DNA damage repair makes NHEJ the preferred pathway even when HR is possible. According to this data, it is only when the damage cannot be repaired by NHEJ that end-resection is promoted and additional mechanisms undertake DNA repair. Besides HR and C-NHEJ, a less understood pathway, alternative nonhomologous end-joining (A-NHEJ),16,17 is sometimes used as a backup repair pathway. A-NHEJ involves processing of DNA by end-resection to reveal regions of short microhomology which are then ligated. In contrast to HR, resected DNA is not filled in during A-NHEJ, making it a potentially more deleterious process than both C-NHEJ and HR. In line with this notion, A-NHEJ is associated with high frequencies of chromosomal translocations and genomic instability.
Lamins Role in DNA DSBs Repair by NHEJ
Our studies have provided strong evidence for a role of A-type lamins in the maintenance of DNA repair mechanisms and telomere homeostasis. We found that loss of A-type lamins leads to sustained DNA damage signaling as measured by formation of ©H2AX foci, as well as an increase in aneuploidy, chromosomal abnormalities and telomere shortening/loss.18-21 Furthermore, loss of these structural proteins was associated with alterations in the nuclear organization of chromosomes, such that the distribution of telomeres was shifted toward the periphery of the nucleus.18,21 Despite the increase in genomic instability, and similar to what was reported in progeria cells, loss of A-type lamins does not impair activation of the DNA damage response (DDR) when cells are exposed to ionizing radiation.19 ATM-dependent phosphorylation of H2AX (©H2AX) and p53 at Ser15 was not affected in Lmna-KO mouse embryonic fibroblasts (MEFs), and the kinetics of formation and resolution of ©H2AX ionizing radiation-induced foci (IRIF) was indistinguishable between lamins-deficient and -proficient cells. In contrast, lamins-deficient cells show defective accumulation of 53BP1 at IRIF at all post-irradiation times tested.19 Importantly, we found that this deficiency is due to a marked decrease in the global levels of the 53BP1 protein, and not to failed recruitment, since 53BP1 IRIF formed although at a much lower intensity. These results are highly relevant, since 53BP1 is an important player in long-range end-joining processes such as class-switch and V(D)J recombination, as well as in the joining of dysfunctional telomeres.22-25 In addition, 53BP1 is thought to play a role in the repair of short-range DNA DSBs by binding to the breaks, inhibiting end-resection and facilitating the recruitment of the NHEJ DNA repair machinery.26,27 These data, together with the fact that 53BP1-deficient cells exhibit increased genomic instability and radiosensitivity28-31 suggested that the loss of 53BP1 could be responsible for the DNA repair deficiencies observed in Lmna-KO cells. Consistent with our hypothesis, we found that lamins-deficient cells treated with ionizing radiation exhibit profound defects in the fast phase of DNA DSBs repair.19,32 Fast repair is traditionally associated with C-NHEJ, since similar defects are observed upon depletion or mutation of essential factors in this process such as DNA-PK, Ku80, XRCC4 and DNA ligase IV.16 Furthermore, lamins- deficient cells are defective in the processing of dysfunctional telomeres by NHEJ. Importantly, we found that reconstitution of 53BP1 in lamins-deficient cells rescues the defects in NHEJ of DNA DSBs and dysfunctional telomeres.19 Overall, these results revealed that 53BP1 deficiency is a major contributor of the DNA repair phenotype observed in lamins-deficient cells. This is a critical observation, since many studies rely on foci formation to determine whether a step in the DDR is functional. Our results indicate that it is important to monitor the levels of DDR proteins at DSBs when assessing deficiencies in DNA repair.
How are the Levels of 53BP1 Regulated by A-Type Lamins?
During our exploration of mechanisms by which A-type lamins affect DNA DSBs repair we discovered a role for cathepsin L (CTSL) in the stability of 53BP1 protein. CTSL is a cysteine protease from the papain family that is ubiquitously expressed in mouse and human tissues. Like many other proteases, it is synthesized as a zymogen which undergoes autoproteolytic processing within the lysosomal/endosomal compartment to release the mature active form.33 Though its activity is enhanced by the low pH at the lysosome, CTSL can also be found in other cellular organelles, where it can selectively process other targets at less acidic or even neutral environments. CTSL can be secreted to the extracellular matrix where it is known to degrade some of its components under physiological conditions, i.e., favoring bone resorption in osteoclasts.34 Increased extracellular CTSL has been reported in numerous types of cancer and is often associated with increased invasiveness and metastasis.35-37 More recently, CTSL was found inside the nucleus, where in a more regulated fashion, it processes specific nuclear components such as histone H3 tails during stem cell differentiation and the transcription factor CDP/Cux during cell cycle progression.38,39
The first link between CTSL and A-type lamins was established in a mouse model of progeria. In particular, mice lacking Zmpste24, a metalloprotease that participates in the maturation of lamin A, exhibit a drastic increase in the levels of CTSL mRNA.40 Although this suggested a relationship between CTSL and the aging phenotype, no association was established between CTSL and the increase in genomic instability displayed by these mice. Our studies showed that Lmna-KO cells exhibit a marked increase in the levels of CTSL mRNA and protein, indicating that loss of A-type lamins induces transcriptional upregulation of CTSL.32 Furthermore, we demonstrated that the increase in CTSL is directly responsible for the downregulation of 53BP1 protein levels. Depletion of CTSL via lentiviral transduction with a specific shRNA restored 53BP1 protein levels and rescued NHEJ defects in lamins-deficient cells. Moreover, transduction with both 53BP1 and CTSL shRNAs in this context prevented restoration of NHEJ, demonstrating that the recovery is brought about by stabilization of 53BP1.
The regulatory effect of CTSL on 53BP1, and thus in DNA repair is not restricted to lamins-deficient cells. Overexpression of CTSL in wild-type cells is sufficient to lower the levels of 53BP1 and impair repair by NHEJ.32 This is a very relevant result because a great variety of tumors present with high CTSL expression. Our studies suggest that in addition to the previously reported effects of CTSL upregulation on the degradation of extracellular matrix components and cell- adhesion molecules, CTSL upregulation in cancer could inhibit mechanisms of DNA repair. Future studies need to determine whether upregulation of CTSL activity is a novel mechanism contributing to genomic instability in cancer, which could be targeted with therapeutic purposes.
Lamins Role in DNA DSBs Repair by HR
Loss of 53BP1 favors repair of DNA DSBs by HR.27,41 However, despite decreased 53BP1 levels, HR is suppressed upon depletion of A-type lamins.19 This inhibition of HR is explained by the significant reduction in expression of two key factors in this process, BRCA1 and RAD51. In contrast to the CTSL-mediated degradation of 53BP1, decreased levels of BRCA1 and RAD51 are brought about by transcriptional gene repression.19 Previous reports had demonstrated transcriptional repression of BRCA1 and RAD51 under certain stressful conditions, such as hypoxia or PARP inhibition, via formation of p130/E2F4 complexes at E2F sites within their promoters.42,43 Interestingly, in the context of lamin A/C-deficiency, we also find that repression of BRCA1 and RAD51 genes is linked to the status of the Rb family of tumor suppressors, pRb, p107 and p130, such that repression of BRCA1 and RAD51 requires p130 and occurs in the context of pRb and p107 deficiency. Furthermore, co-immunoprecipitation studies in cells depleted of A-type lamins showed an increase in p130/E2F4 complexes.19 These data suggest activation of a similar repressive mechanism in lamins-deficient cells, where altering the balance of the pocket proteins might favor association of p130 with E2F4, leading to transcriptional inhibition of responsive promoters. However, we cannot rule out the possibility that loss of A-type lamins leads to alterations in the nuclear localization of BRCA1 and RAD51 genes, which might contribute to their transcriptional repression.
It is well established that pocket proteins associate with lamins and that loss of A-type lamins leads to increased degradation of pRb and to a lesser extent p107.44,45 This is thought to occur partly through the ability of A-type lamins to regulate the sub-nuclear localization of these proteins. However, the specific mechanism by which pRb and p107 are targeted for degradation remains quite elusive, being independent of both MDM2 and gankyrin, a component of the 19S proteasome subunit which is overexpressed in Lmna-KO cells.46 Given our recent findings that CTSL promotes the degradation of 53BP1, we speculated that this protease could be the missing link between A-type lamins and pRb/p107 degradation. We envisioned a scenario where CTSL-mediated degradation of pRb and p107 alters the balance between the pocket family proteins, leading to increased formation of p130/E2F4 complexes, which can in turn mediate transcriptional repression of BRCA1 and RAD51, inhibiting HR. To test our model we overexpressed CTSL in wild-type MEFs via retroviral transduction and monitored the levels of Rb family members, BRCA1 and RAD51. Indeed, we found that upregulation of CTSL is associated with a substantial decrease in pRb and p107, with little or no effect on p130, mirroring the phenotype observed in lamins-deficient cells (Fig. 1). These results demonstrate a novel role for CTSL in the regulation of the Rb family of tumor suppressors. However, altering the levels of these proteins was not sufficient to induce transcriptional repression of BRCA1 or RAD51 in MEFs (data not shown). These data suggest that lamins have additional roles in the regulation of transcription of these genes independently of the CTSL-mediated degradation of Rb family proteins.
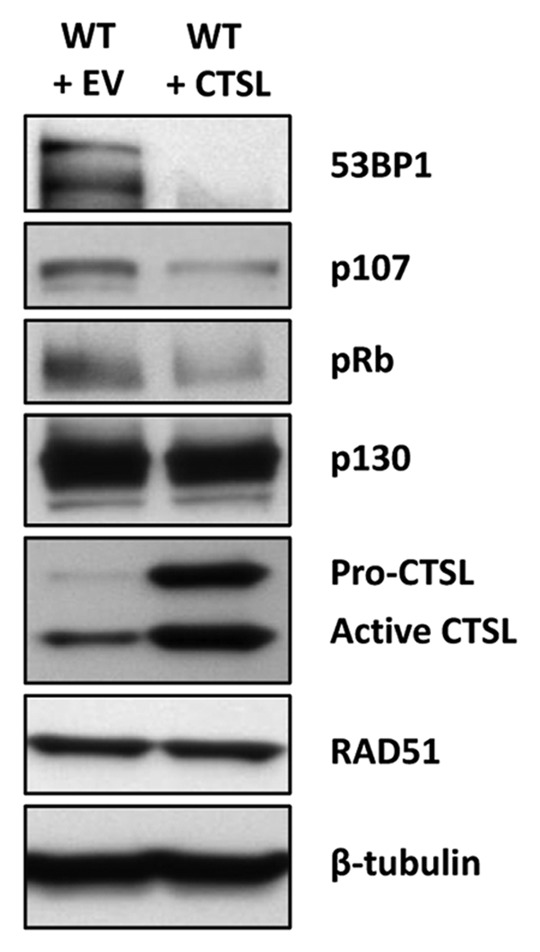
Figure 1. Regulation of Rb family members by CTSL. Blots showing that overexpression of CTSL via retroviral transduction of wild-type MEFs leads to downregulation of the pocket proteins pRb and p107 but has no effect in the levels of p130. As a control for increased CTSL activity, we show decreased levels of 53BP1 in the CTSL overexpressing cells.
Overall, our studies revealed that A-type lamins regulate HR indirectly by impacting on transcription of key players in this process. On one hand, loss of A-type lamins leads to transcriptional upregulation of CTSL, which in turns degrades pRb and p107. We speculate that the loss of these two proteins might favor the formation of p130/E2F4 complexes, which provide a permissive environment to repress transcription from BRCA1 and RAD51 gene promoters. However, loss of A-type lamins might induce some other changes, perhaps epigenetic modifications at the promoter, which result in shut down of gene expression.
Vitamin D Inhibition of CTSL Reduces Genomic Instability
Vitamin D is a liposoluble steroid prohormone present in certain foods but mainly obtained from the conversion in the skin of 7-dehydrocholecalcipherol to vitamin D3 by the action of UV light. Vitamin D3 is then hydroxylated in the liver, producing 25OH vitamin D3, and later in the kidney, where the enzyme 1-〈 hydroxylase produces 1,25(OH)2D3 or calcitriol, the active vitamin D metabolite (hereinafter vitamin D). Vitamin D exerts its actions through the vitamin D receptor (VDR), a member of the nuclear receptor superfamily.47 Ligand-bound VDR is translocated to the nucleus where it associates with the retinoic-X receptor (RXR). The VDR/RXR heterodimer can interact with a number of co-activators and/or co-repressors, regulating transcription of multiple target genes. Vitamin D is a pleiotropic hormone, with a major role in the regulation of bone and mineral metabolism. In addition, vitamin D regulates proliferation, inhibits invasion and promotes apoptosis or differentiation depending on the cellular context.48
Recently, it was shown that calcitriol inhibits invasion in colon cancer cells by activating the transcription of cystatin D, an endogenous cysteine protease inhibitor of cathepsins S, H and L.49 Similarly, the vitamin D analog EB1089 upregulates the expression of cystatins E/M, other endogenous cathepsin L inhibitors, in squamous carcinoma cells.50 These observations prompted us to investigate whether vitamin D could be used to counteract the effects of CTSL upregulation in lamins-deficient cells.
We found that treatment with vitamin D restored 53BP1 protein levels and DNA repair by NHEJ in both cells exogenously overexpressing CTSL and Lmna-KO cells.32 Furthermore, vitamin D ameliorated genomic instability, decreasing the levels of unrepaired DNA damage (determined as ©H2AX foci) as well as the nuclear abnormalities characteristic of lamins-deficient cells. Previous studies in MDA-MB-231 breast cancer cells have shown that the expression of CTSL is inhibited by vitamin D.51 Interestingly, in our hands vitamin D did not alter the levels of CTSL, but rather inhibited its activity, suggesting a cystatin-mediated mechanism. Therefore, we decided to investigate whether inhibition of CTSL by vitamin D could be mediated by a cystatin family member. Since there is no mouse ortholog for cystatin D, we performed a promoter analysis for vitamin D responsive elements (VDRE) in the 13 cystatin genes encoded by the mouse genome. We used the Genomatix software and ElDorado mouse genome database. In the initial analysis we selected 4 cystatin genes which contain at least two RXR/VDR heterodimer binding sites (Fig. 2A). One of them, cystatin B (also known as Stefin B) contains four VDR responding elements (human cystatin D contains five elements) and is downregulated in lamin deficient-cells. Moreover, treatment with vitamin D increases the expression of cystatin B (Fig. 2B), suggesting a role in the regulation of CTSL activity. Nevertheless, additional studies will be necessary to fully determine which cystatin(s) is responsible for the vitamin D-dependent inhibition of CTSL activity.

Figure 2. Vitamin D-dependent inhibition of CTSL activity may be mediated by cystatins. (A) Promoter analysis for vitamin D responding elements (VDREs) in the 13 cystatins encoded in the mouse genome resulted in four candidate genes containing at least two RXR/VDR heterodimer binding sites. (B) qRT-PCR experiments show that cystatin B expression is downregulated in lamins-deficient cells. Treatment with vitamin D 10−7 M for 48 h increases cystatin B expression in both wild-type and Lmna-KO MEFs. Values are expressed as mean ± SEM. N, the number of independent experiments; *, p value of statistical significance (P ≤ 0.05); R. U., relative units (normalization to GAPDH).
Conclusions
Our findings support a fundamental role for CTSL in the regulation of cell cycle progression and DNA repair, both in lamin-deficient and -proficient cells. Upregulation of CTSL, a hallmark of a variety of cancers, is directly linked to degradation of Rb family members and 53BP1 and possibly indirectly to transcriptional downregulation of BRCA1 and RAD51 (Fig. 3). Thus, inhibition of CTSL activity via treatment with vitamin D or specific inhibitors could represent a novel approach in the management of cancer and other age-related diseases that course with increased genomic instability and defects in DNA repair.

Figure 3. Proposed model for the regulation of DNA repair mechanisms by A-type lamins. The loss of A-type lamins upregulates CTSL expression, resulting in elevated protein levels both in the nucleus and in the lysosomes. CTSL processes 53BP1, which then accumulates in the cytoplasm and is targeted to degradation by the lysosomal pathway and/or the proteasome. Loss of 53BP1 impairs DNA repair by NHEJ. On the other hand, CTSL degrades the pocket family proteins pRB and p107, favoring the formation of p130/E2F4 repression complexes, which in turn inhibit RAD51 and BRCA1 gene expression and thereby impair DNA repair by HR.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/18201
References
- 1.Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev. 2006;86:967–1008. doi: 10.1152/physrev.00047.2005. [DOI] [PubMed] [Google Scholar]
- 2.Prokocimer M, Davidovich M, Nissim-Rafinia M, Wiesel-Motiuk N, Bar DZ, Barkan R, Meshorer E, Gruenbaum Y. Nuclear lamins: key regulators of nuclear structure and activities. J Cell Mol Med. 2009;13:1059–85. doi: 10.1111/j.1582-4934.2008.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 4.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–8. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101:8963–8. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, et al. Genomic instability in laminopathy-based premature aging. Nat Med. 2005;11:780–5. doi: 10.1038/nm1266. [DOI] [PubMed] [Google Scholar]
- 7.Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–63. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120:497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 9.Musich PR, Zou Y. Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A. Aging (Albany NY) 2009;1:28–37. doi: 10.18632/aging.100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Wang Y, Rusinol AE, Sinensky MS, Liu J, Shell SM, Zou Y. Involvement of xeroderma pigmentosum group A (XPA) in progeria arising from defective maturation of prelamin A. FASEB J. 2008;22:603–11. doi: 10.1096/fj.07-8598com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, Yang SL, Panopoulos AD, Suzuki K, Kurian L, Walsh C, et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011;472:221–5. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–68. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mao Z, Bozzella M, Seluanov A, Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst) 2008;7:1765–71. doi: 10.1016/j.dnarep.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G, Löbrich M, et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011;30:1079–92. doi: 10.1038/emboj.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iliakis G, Wang H, Perrault AR, Boecker W, Rosidi B, Windhofer F, Wu W, Guan J, Terzoudi G, Pantelias G. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet Genome Res. 2004;104:14–20. doi: 10.1159/000077461. [DOI] [PubMed] [Google Scholar]
- 17.Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–82. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalez-Suarez I, Redwood AB, Perkins SM, Vermolen B, Lichtensztejin D, Grotsky DA, Morgado-Palacin L, Gapud EJ, Sleckman BP, Sullivan T, et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009;28:2414–27. doi: 10.1038/emboj.2009.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Redwood AB, Perkins SM, Vanderwaal RP, Feng Z, Biehl KJ, Gonzalez-Suarez I, Morgado-Palacin L, Shi W, Sage J, Roti-Roti JL, et al. A dual role for A-type lamins in DNA double-strand break repair. Cell Cycle. 2011;10:2549–60. doi: 10.4161/cc.10.15.16531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez-Suarez I, Gonzalo S. Crosstalk between chromatin structure, nuclear compartmentalization, and telomere biology. Cytogenet Genome Res. 2008;122:202–10. doi: 10.1159/000167805. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez-Suarez I, Redwood AB, Gonzalo S. Loss of A-type lamins and genomic instability. Cell Cycle. 2009;8:3860–5. doi: 10.4161/cc.8.23.10092. [DOI] [PubMed] [Google Scholar]
- 22.Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, Kruhlak MJ, Callen E, Livak F, Nussenzweig MC, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–33. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol. 2004;5:481–7. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- 24.Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F, et al. 53BP1 is required for class switch recombination. J Cell Biol. 2004;165:459–64. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–8. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med. 2010;207:855–65. doi: 10.1084/jem.20100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakamura K, Sakai W, Kawamoto T, Bree RT, Lowndes NF, Takeda S, Taniguchi Y. Genetic dissection of vertebrate 53BP1: a major role in non-homologous end joining of DNA double strand breaks. DNA Repair (Amst) 2006;5:741–9. doi: 10.1016/j.dnarep.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Morales JC, Xia Z, Lu T, Aldrich MB, Wang B, Rosales C, Kellems RE, Hittelman WN, Elledge SJ, Carpenter PB. Role for the BRCA1 C-terminal repeats (BRCT) protein 53BP1 in maintaining genomic stability. J Biol Chem. 2003;278:14971–7. doi: 10.1074/jbc.M212484200. [DOI] [PubMed] [Google Scholar]
- 30.Ward IM, Minn K, van Deursen J, Chen J. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol. 2003;23:2556–63. doi: 10.1128/MCB.23.7.2556-2563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002;298:1435–8. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez-Suarez I, Redwood AB, Grotsky DA, Neumann MA, Cheng EH, Stewart CL, Dusso A, Gonzalo S. A new pathway that regulates 53BP1 stability implicates cathepsin L and vitamin D in DNA repair. EMBO J. 2011;30:3383–96. doi: 10.1038/emboj.2011.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katunuma N. Mechanisms and regulation of lysosomal proteolysis. Revis Biol Celular. 1989;20:35–61. [PubMed] [Google Scholar]
- 34.Reiser J, Adair B, Reinheckel T. Specialized roles for cysteine cathepsins in health and disease. J Clin Invest. 2010;120:3421–31. doi: 10.1172/JCI42918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gocheva V, Joyce JA. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle. 2007;6:60–4. doi: 10.4161/cc.6.1.3669. [DOI] [PubMed] [Google Scholar]
- 36.Jedeszko C, Sloane BF. Cysteine cathepsins in human cancer. Biol Chem. 2004;385:1017–27. doi: 10.1515/BC.2004.132. [DOI] [PubMed] [Google Scholar]
- 37.Miyamoto K, Iwadate M, Yanagisawa Y, Ito E, Imai J, Yamamoto M, Sawada N, Saito M, Suzuki S, Nakamura I, et al. Cathepsin L is highly expressed in gastrointestinal stromal tumors. Int J Oncol. 2011;39:1109–15. doi: 10.3892/ijo.2011.1127. [DOI] [PubMed] [Google Scholar]
- 38.Duncan EM, Muratore-Schroeder TL, Cook RG, Garcia BA, Shabanowitz J, Hunt DF, Allis CD. Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell. 2008;135:284–94. doi: 10.1016/j.cell.2008.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goulet B, Baruch A, Moon NS, Poirier M, Sansregret LL, Erickson A, Bogyo M, Nepveu A. A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol Cell. 2004;14:207–19. doi: 10.1016/S1097-2765(04)00209-6. [DOI] [PubMed] [Google Scholar]
- 40.Varela I, Cadiñanos J, Pendás AM, Gutiérrez-Fernández A, Folgueras AR, Sánchez LM, Zhou Z, Rodríguez FJ, Stewart CL, Vega JA, et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005;437:564–8. doi: 10.1038/nature04019. [DOI] [PubMed] [Google Scholar]
- 41.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–95. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bindra RS, Glazer PM. Repression of RAD51 gene expression by E2F4/p130 complexes in hypoxia. Oncogene. 2007;26:2048–57. doi: 10.1038/sj.onc.1210001. [DOI] [PubMed] [Google Scholar]
- 43.Hegan DC, Lu Y, Stachelek GC, Crosby ME, Bindra RS, Glazer PM. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc Natl Acad Sci U S A. 2010;107:2201–6. doi: 10.1073/pnas.0904783107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson BR, Nitta RT, Frock RL, Mounkes L, Barbie DA, Stewart CL, Harlow E, Kennedy BK. A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc Natl Acad Sci U S A. 2004;101:9677–82. doi: 10.1073/pnas.0403250101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nitta RT, Jameson SA, Kudlow BA, Conlan LA, Kennedy BK. Stabilization of the retinoblastoma protein by A-type nuclear lamins is required for INK4A-mediated cell cycle arrest. Mol Cell Biol. 2006;26:5360–72. doi: 10.1128/MCB.02464-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nitta RT, Smith CL, Kennedy BK. Evidence that proteasome-dependent degradation of the retinoblastoma protein in cells lacking A-type lamins occurs independently of gankyrin and MDM2. PLoS One. 2007;2:e963. doi: 10.1371/journal.pone.0000963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Demay MB. Mechanism of vitamin D receptor action. Ann N Y Acad Sci. 2006;1068:204–13. doi: 10.1196/annals.1346.026. [DOI] [PubMed] [Google Scholar]
- 48.Plum LA, DeLuca HF. Vitamin D, disease and therapeutic opportunities. Nat Rev Drug Discov. 2010;9:941–55. doi: 10.1038/nrd3318. [DOI] [PubMed] [Google Scholar]
- 49.Alvarez-Díaz S, Valle N, García JM, Peña C, Freije JM, Quesada V, Astudillo A, Bonilla F, López-Otín C, Muñoz A. Cystatin D is a candidate tumor suppressor gene induced by vitamin D in human colon cancer cells. J Clin Invest. 2009;119:2343–58. doi: 10.1172/JCI37205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin R, Nagai Y, Sladek R, Bastien Y, Ho J, Petrecca K, Sotiropoulou G, Diamandis EP, Hudson TJ, White JH. Expression profiling in squamous carcinoma cells reveals pleiotropic effects of vitamin D3 analog EB1089 signaling on cell proliferation, differentiation, and immune system regulation. Mol Endocrinol. 2002;16:1243–56. doi: 10.1210/me.16.6.1243. [DOI] [PubMed] [Google Scholar]
- 51.Swami S, Raghavachari N, Muller UR, Bao YP, Feldman D. Vitamin D growth inhibition of breast cancer cells: gene expression patterns assessed by cDNA microarray. Breast Cancer Res Treat. 2003;80:49–62. doi: 10.1023/A:1024487118457. [DOI] [PubMed] [Google Scholar]