

Abstract
To uncover the function of and interplay between the mammalian cytosine modifications 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC), new techniques and advances in current technology are needed. To this end, we have developed oxidative bisulfite sequencing (oxBs-seq), which can quantitatively locate 5mC and 5hmC marks at single-base resolution in genomic DNA. In bisulfite sequencing (BS-seq), both 5mC and 5hmC are read as cytosines and thus cannot be discriminated; however, in oxBS-seq, specific oxidation of 5hmC to 5-formylcytosine (5fC) and conversion of the newly formed 5fC to uracil (under bisulfite conditions) means that 5hmC can be discriminated from 5mC. a positive readout of actual 5mC is gained from a single oxBS-seq run, and 5hmC levels are inferred by comparison with a BS-seq run. Here we describe an optimized second-generation protocol that can be completed in 2 d.
INTRODUCTION
5mC is a well-known epigenetic mark in mammals, with important functions in development, transposon and gene silencing, genomic imprinting, X-chromosome inactivation and genome stability1. Aberrant DNA methylation has been implicated in many human diseases, including cancer, in which gross hypomethylation is accompanied by hypermethylation within certain CpG islands2,3. The recent discovery that 5mC can be oxidized to 5hmC by the ten-eleven translocation (TET) proteins has prompted wide interest4–6. The highest known levels of 5hmC are found in the brain and in embryonic stem cells7,8. Recent evidence suggests that 5hmC may act as an intermediate in active DNA demethylation, and it has also been implicated in pluripotency, development and disease9. Additional studies have demonstrated that the TET proteins can oxidize 5hmC further to 5fC and then subsequently to 5-carboxylcytosine (5caC; Fig. 1; refs. 6,10,11). Both 5fC and 5caC can be actively removed by thymine DNA glycosylase11–13.
Figure 1.
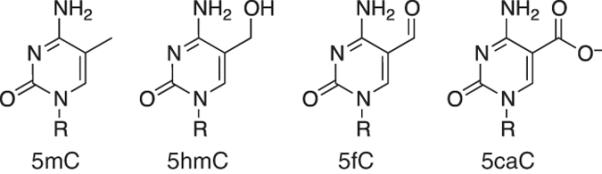
Structures of 5mC, 5hmC, 5fC and 5caC. R, DNA backbone.
Since the discovery of the TET enzymes and their ability to oxidize 5mC, many new techniques have been used to study these DNA modifications. Most techniques are antibody-based or involve chemical pull-down of DNA fragments, and their implementation offers limited resolution and quantitative information. Investigating the precise location and obtaining reliable quantitative data on all these marks are essential to ascertaining their function and relevance to normal biology and disease. Here we describe, in great depth, the oxBS-seq technique for quantitatively sequencing 5mC and 5hmC at single-base resolution14, as well as recent improvements to the workflow.
Development
The gold standard for 5mC detection has been BS-seq, a base-specific method in which treatment with sodium bisulfite deaminates cytosine bases (within single-stranded DNA (ssDNA)) to uracil. Converted cytosines are subsequently read as thymines in various sequencing methods that may include a PCR amplification step in the workflow. Methylated cytosines are resistant to bisulfite conversion and are read as cytosines at the sequencing step, which enables quantitative discrimination between unmodified cytosines and 5mC (ref. 15). BS-seq has been used extensively to map methylation patterns in the DNA of a variety of organisms. Treatment of 5hmC with bisulfite results in a stable methyl-sulfonate adduct that is also read as a cytosine when sequenced16,17. For this reason, 5mC and 5hmC are indistinguishable by bisulfite conversion alone as classic BS-seq methylation analyses will provide data on the location and levels of 5mC plus 5hmC rather than those of the actual 5mC. Resolving 5mC and 5hmC in genomic BS-seq data is likely to have important interpretive implications.
OxBS-seq discriminates between 5mC and 5hmC via a highly selective chemical oxidation of 5hmC to 5fC. The approach also exploits the difference in reactivity between 5hmC and 5fC toward bisulfite. In contrast to 5hmC, which does not deaminate, bisulfite treatment causes 5fC to be deformylated and deaminated to form uracil (which is read as thymine at the sequencing stage)14. Thus, the only base that is not deaminated and therefore read as a cytosine after oxBS-seq is 5mC. Consequently, this approach gives an accurate readout of all the 5mC present. Subsequent detection of 5hmC is achieved by performing BS-seq (which identifies 5hmC + 5mC) and by comparing its results with those of oxBS-seq (which identifies 5mC alone; Fig. 2).
Figure 2.
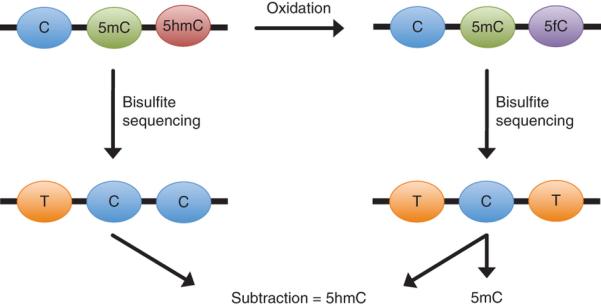
After bisulfite treatment of DNA, C reads as T, whereas 5mC and 5hmC read as C. With a specific oxidation of 5hmC to 5fC followed by bisulfite treatment, C and 5hmC read as T, whereas only 5mC reads as C, thereby giving a positive readout of 5mC. 5hmC can then be ascertained by the difference of these two outputs.
The water-soluble chemical oxidant used in oxBS-seq specifically and quantitatively oxidizes 5hmC within ssDNA, irrespective of sequence context or composition14. The oxidant does not oxidize 5mC, as previously demonstrated14.
Applications
The oxBS-seq workflow can be used to modify DNA in conjunction with a multitude of analytical tools that have already been developed for BS-seq, including DNA sequencing (Sanger sequencing, pyrosequencing, high-throughput sequencing) and methylation arrays (e.g., Illumina 450k arrays). When used in conjunction with next-generation sequencing, this method is platform agnostic and compatible with the analysis of 5mC and 5hmC at single-base resolution in both whole-genome or targeted-region formats. Besides whole-genome analysis, oxBS-seq is compatible with enrichment and targeting techniques, such as post-bisulfite locus-specific PCR and reduced-representation BS-seq14,18.
Comparisons
Other methods for the detection of 5hmC in the genome rely on the β-glucosyltransferase (βGT) from the T4 bacteriophage that glucosylates 5hmC. However, βGT is known to be an inefficient glucosylating agent on symmetrically hydroxymethylated CpGs19, a context that is typical of the majority of 5hmCs19.
5mC and 5hmC can be mapped using antibodies that are commercially available20. Other pull-down–based methods have been developed to detect 5hmC, using βGT followed by chemical attachment of biotin to the glucose moiety and subsequent pull-down and sequencing21. Implementation of these methods gives a low-resolution map with little quantitative information.
A recently published study has shown that a DNA modification–dependent enzyme, AbaSI, can digest DNA 11–13 bp away from a βGT-glucosylated 5hmC, when another cytosine is also present 9–11 bp away in the opposite direction22. This AbaSI-based method can detect 5hmC with relative quantitative reliability and single-base resolution in <40% of cases22.
TET-assisted BS-seq has been shown to quantitatively map 5hmC at single-base resolution19. This method uses βGT to block 5hmC sites, and then uses a recombinant mouse TET1 enzyme to convert 5mC to 5caC. Bisulfite treatment then converts 5caC to uracil, and the only base that is read as a cytosine is glucosylated 5hmC. However, this approach requires the use of highly active TET1 protein, which is expensive and difficult to purify. A potential drawback of this technique is that inefficiencies in 5hmC glucosylation or TET1-mediated 5mC oxidation can result in false-positive base calling of 5hmC. Even with a high efficiency of 5mC oxidation by TET1 (95%; ref. 23), at sites with 80–100% 5mC, only 95% will convert to uracil. In these conditions, the remaining 5% of unconverted 5mCs will be falsely identified as 5hmCs.
Third-generation sequencing methods have also been used to detect these epigenetic modifications. Single-molecule real-time (SMRT) sequencing can measure the kinetics of nucleotide incorporation during DNA synthesis. The kinetic signature of nucleotide incorporation is sensitive to the sequence environment of the incorporated base and can be used to discriminate between different types of base modification24. Because of the subtle differences in kinetic signature between 5mC and 5hmC relative to native cytosine, large depths of coverage are required to enable this type of analysis to be conducted, making this approach expensive on a (mammalian) genome-wide scale. An expansion to SMRT was performed with the glucosylation (using βGT), biotinylation and pull-down of 5hmC DNA, which gave a greatly increased kinetic signature in SMRT sequencing25. However, this method did not enable researchers to obtain quantitative data, as it involves a 5hmC enrichment step. Nanopore sequencing has also been shown to discriminate 5mCpG and 5hmCpG dinucleotides over C, A, G and T(pG) in synthetic DNA26. We currently wait for this technology to become broadly available.
Limitations
Because of the relatively low levels of 5hmC in the genome, high coverage is needed to sequence this base quantitatively. Furthermore, a subtraction is needed in order to obtain the actual levels of 5hmC, an operation that increases noise level and thus further enhances the need to increase coverage and the number of replicates. For instance, first, assume that a given position contains 84% 5mC and 11% 5hmC. A BS-seq experiment with 100× coverage would identify 5mC + 5hmC at 95% (i.e., 95 out of 100 reads are unconverted C to T). A matched oxBS-seq experiment with the same coverage would identify 5mC at 84% (i.e., 84 out of 100 reads are unconverted Cs to Ts). The level of 5hmC would then be ascertained from a subtraction of the BS-seq and the oxBS-seq at 11% (95–84%). In this case, a one-sided Fisher’s test applied to a contingency table of number of reads, converted and unconverted in the BS- and oxBS-seq experiment, would return a P value of ~0.01 for the null hypothesis (that is, when the proportion of reads methylated in the two experiments is equal). Therefore, it is advantageous to use genome enrichment methods to sequence parts of the genome with higher depth. See Supplementary Table 1 for more Fisher’s tests on different sequencing coverage. Alternatively, pooling data from neighboring CpGs can provide increased statistical power with a small compromise in resolution14.
Experimental design
An optimized second-generation oxBS-seq workflow has been developed that interfaces with next-generation sample preparation (Fig. 3).
Figure 3.
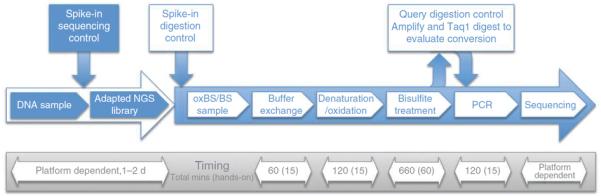
Workflow of oxBS-seq. NGS, next-generation sequencing.
DNA purification (before oxidation)
We have tested the protocol using between 100 ng and 1 μg of DNA starting material. As the protocol makes use of a chemical oxidant for the oxidation of the primary alcohol of 5hmC, it is of utmost importance that the DNA is well purified. The oxidation efficiency can be reduced if any other primary alcohol or buffer is present in the mixture (for instance, ethanol, Tris or phosphate). As is described in the protocol, we have come up with an easy way to purify DNA before the oxidation step.
DNA purification (after oxidation)
Most regular DNA purification methods (e.g., silica centrifuge columns) do not remove the oxidant after oxidation. However, the oxidant is retained on Sephadex columns, but recoveries of ssDNA from these columns (required for efficient conversion) are poor. We have found that polyacrylamide columns give the best balance of contaminant removal and ssDNA recovery.
Oxidant
Commercially available KRuO4 (Alfa Aesar) can be used for oxBS-seq as previously described14; however, Cambridge Epigenetix (CEGX) has formulated this oxidant as an easy-to-use solution that improves consistency and reproducibility in oxBS-seq, particularly when used by non-experts. For this study, we obtained a prototype 10× stock of oxidant from CEGX.
Bisulfite conversion
Most bisulfite treatment kits are optimized for the conversion of cytosine to uracil. However, 5fC does not convert to uracil as quickly as cytosine does14. This difference in reactivity means that, to fully convert 5fC to uracil, longer incubation times are required than those prescribed by most bisulfite kits. An optimized conversion is outlined in the PROCEDURE. This approach has little effect on the conversion of 5mC to T and the quantity of DNA required.
It is noteworthy that 5fC from commercial sources can convert to uracil at different rates. In fact, in DNA synthesized from commercial 5fC phosphoramidites, the extent of 5fC conversion to uracil often does not exceed 50%. However, in DNA produced via PCR and the incorporation of d5fCTP, 5fC converts to uracil more efficiently compared with the 5fC phosphoramidite, as measured by Sanger sequencing (Supplementary Fig. 1). We reason that this difference may be due to either phorphoramidites that are not fully deprotected after synthesis or a substantial proportion of the 5fCs being converted to the hydrate form. We therefore suggest that researchers exercise caution when using synthetic DNA containing 5fC.
Spike-in controls
We recommend the use of sequencing controls to accurately determine the conversion level of all three cytosine forms (unmodified, 5mC and 5hmC) after sequencing (Supplementary Table 2). Our spike-in controls were constructed by PCR, using primers that contained only 5mC, C, A and T. Thereafter, 5hmC could also be incorporated into the DNA using 5hmCTP. To thoroughly assess the power of the oxBS-seq workflow, we designed multiple strands with different densities of 5hmC in different sequence contexts.
Digestion controls, which we also recommend using (see PROCEDURE), were used as an immediate qualitative tool to measure the conversion of 5hmC to uracil before DNA sequencing (Supplementary Table 2). We designed the digestion controls in a very similar way to the sequencing controls, but with one primer containing 5mC and the other C to allow strand-specific amplification. This design choice meant that after bisulfite treatment, the strand containing C would convert to U and the primer site would not amplify, whereas the opposite strand containing 5mC would amplify. The control DNA strands also contained a 5hmC in a TaqI site in the center. If 5hmC is converted to uracil, then the TaqI site is destroyed and the enzyme will not cut; however, if the conversion has not reached completion, the TaqI site will still be present in some cases and the DNA will be digested.
Outcome from the optimized method
More ssDNA is recovered from the improved oxBS-seq method (Fig. 4). We now observe little loss of ssDNA compared with the first-generation oxBS-seq protocol14. The recovery of DNA from the oxBS-seq workflow is now comparable to that from BS-seq only. Therefore, no evidence exists for substantial amounts of DNA damage arising from the chemical treatment of DNA specific to the oxidation step. As a consequence, we are now able to successfully process as little as 100 ng of starting material through the oxBS-seq work-flow. Typical total DNA recovery from the preoxidation buffer exchange, starting with 1,000 ng input, is ~75% (~750 ng), which is followed by a further ~50% observed DNA loss after postoxidation cleanup (~375 ng recovered) as measured by a NanoDrop 1000 spectrophotometer (in double-stranded DNA (dsDNA) and ssDNA modes, respectively).
Figure 4.
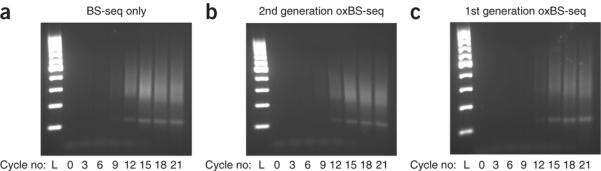
Efficacy of PCR amplification. (a–c) Gel electrophoresis experiments (in 2% (wt/vol) agarose gel) of PCR of Illumina libraries prepared from 1 μg of sonicated genomic DNA after undergoing the BS-only workflow (a); the oxBS-seq workflow described in the present protocol (b); and the previous version of the oxBS-seq workflow (c)14. From each 50-μl PCR solution prepared for use with Agilent Pfu Turbo Cx DNA polymerase, 5-μl aliquots of each PCR solution were removed at cycles 0, 3, 6, 9, 12, 15, 18 and 21 and placed in the wells of the agarose gels. Libraries analyzed in a and b show similar amplification, whereas three additional cycles are required for the library analyzed in c to achieve the same DNA intensity as a and b.
The ladder (L) is the Thermo Scientific GeneRuler 100-bp DNA ladder.
Data analysis overview
oxBS-seq data should be analyzed according to the same guidelines used for standard BS-seq data, including using the Bismark tool for alignment of bisulfite-modified reads27. The percentage of 5hmC at each site will then be inferred by subtracting the detected methylation in the oxBS-seq experiment from the methylation detected in a matched BS-seq experiment. The simplest way to test for a significant difference in methylation calls between BS-seq and oxBS-seq is to apply a Fisher’s test at each site of interest or at several sites mapping to the same region. False discovery thresholds might be identified from the distribution of negative percentages, as described in Booth et al.14.
MATERIALS
REAGENTS
Genomic DNA sample (100 ng–1 μg)
Illumina TruSeq DNA sample preparation kit (Illumina, cat. no. FC-121-2001)
Ampure XP beads (Beckman Coulter, cat. no. A63880)
Milli-Q water
Sodium hydroxide, 1.0 M (NaOH; Sigma-Aldrich, cat. no. 319511)
Oxidant solution (10× solution; Cambridge Epigenetix (CEGX)). Alternatively, in place of Steps 12–14 and 17, the oxidant and oxidation protocol can be prepared and used as described in http://www.sciencemag.org/content/suppl/2012/04/25/science.1220671.DC1/Booth-SOM.revision.1.pdf (ref. 14)
Hydroxymethyl dCTP (dhmCTP, Bioline, cat. no. BIO-39046)
dNTP mix, 10 mM (NEB, cat. no. N0447S)
Epitect bisulfite kit (Qiagen, cat. no. 59104)
P-6 saline–sodium citrate (SSC) Micro Bio-Spin columns (Bio-Rad, cat. no. 732-6200)
Pfu Turbo Cx hotstart DNA polymerase (Agilent, cat. no. 600410)
Agarose (Sigma-Aldrich, cat. no. A9539)
dATP, 100 mM (NEB, cat. no. N0446S)
dGTP, 100 mM (NEB, cat. no. N0446S)
dTTP, 100 mM (NEB, cat. no. N0446S)
DNA templates (Biomers; Supplementary Table 2)
DNA primers (Biomers; Supplementary Table 2)
DreamTaq DNA polymerase (Thermo Scientific, cat. no. EP0701)
KAPA HiFi uracil + ReadyMix (KAPA Biosystems, cat. no. KK2801)
GeneJet PCR purification kit (Thermo Scientific, cat. no. K0701)
Taqa1 restriction endonuclease (Taq1 RE; NEB, cat. no. R0149S)
Ethanol (Sigma-Aldrich, cat. no. 32221)
GelRed, 3× dye (Cambridge Bioscience, cat. no. 41001)
Ammonium acetate (Fisher, cat. no. A/3440/53)
Acetic acid (Sigma-Aldrich, cat. no. 320099)
HPLC-grade acetonitrile (Sigma-Aldrich, cat. no. 34851)
EQUIPMENT
Manual pipettes (Thermo Finnpipettes F1, cat. nos. 4641010, 4641060, 4641080 and 4641100)
Pipette tips (StarLabs, cat. nos. S1122-1830 and S1120-8810)
MiniSpin Plus microcentrifuge (Eppendorf, cat. no. 5453 000.011)
Thermomixer (Eppendorf, cat. no. 5355 000.011)
Thermal cycler (Bio-Rad, cat. no. 95-95002)
Magnetic rack (Invitrogen, cat. no. CS15024)
Microcentrifuge tubes, 1.5 ml (StarLabs, cat. no. S1615-5500)
Thin-walled PCR tubes, 0.2 ml (Corning, cat. no. 3745)
HPLC 1100 system (Agilent)
MiSeq (Illumina)
Gel electrophoresis equipment (Bio-Rad)
TapeStation (Agilent)
Qubit fluorimeter (Invitrogen)
Amicon Ultra 10-kDa filters (Millipore, cat. no. UFC503096)
Bioruptor (Diagenode, cat. no. UCD-200)
NanoDrop 1000 spectrophotometer (Thermo Scientific)
REAGENT SETUP
Ammonium acetate (500 mM), pH 5 Dissolve 38.5 g of ammonium acetate in 900 μl of Milli-Q water and adjust the pH to 5 with acetic acid. Bring the volume to 1 liter with Milli-Q water (Box 1). This reagent can be stored at room temperature (22 °C) for up to 1 month.
Box 1. Assaying the chemical oxidant activity on 5hmC with HPLC  20 h.
20 h.
Purchase oligonucleotide containing 5hmC.
Follow Steps 14–19 of the main PROCEDURE to denature, oxidize and purify the synthetic oligonucleotides DNA from step 1.
Digest the DNA to 2′-deoxynucleosides overnight according to the literature protocol28.
Purify the 2′-deoxynucleosides with Amicon Ultra 0.5-ml 10-kDa columns.
Analyze the 2′-deoxynucleosides by HPLC. We have optimized an HPLC protocol using an Agilent 1100 HPLC instrument with a flow of 1 ml min–1 over an Eclipse XDB-C18 3.5-μm, 3.0- × 150-mm column. The column temperature is maintained at 45 °C. Eluting buffers are Buffer A (500 mM ammonium acetate, pH 5), Buffer B (acetonitrile) and Buffer C (H2O). Buffer A is held at 1% throughout the whole run and the gradients for the remaining buffers are 0 min, 0.5% B; 2 min, 1% B; 8 min, 4% B; and 10 min, 95% B. There is an example of the expected results of this process in supplementary Figure 2.
PROCEDURE
Preparation of the sequencing and digestion spike-in controls  2 h
2 h
1∣ Amplify the sequencing and digestion controls using a synthetic single-stranded 100mer template. Prepare the PCR as described below. Mix the reaction briefly by vortexing.
| Component | Volume (μl) |
|---|---|
| Milli-Q water | 37.75 |
| Polymerase buffer, 10× | 5 |
| DATP, dGTP, dTTP mix (10 mM each) | 1 |
| dhmCTP (10 mM) | 1 |
| Control forward PCR primer (10 μM) | 2 |
| Control reverse PCR primer (10 μM) | 2 |
| Template (1 ng μl−1) | 1 |
| Taq polymerase (e.g., DreamTaq 5 U μl−1) | 0.25 |
| Total | 50 |
2∣ Amplify the reaction using the following thermocyling program.
| Segment | Temperature (°C) | Duration (s) | Iterations |
|---|---|---|---|
| Initial denaturation | 95 | 180 | 1 |
| Loop | |||
| Denaturation | 95 | 30 | 35 |
| Annealing | 57 | 30 | |
| Extension | 72 | 15 | |
| Store | 4 | Forever |
3∣ Clean up the amplicons using a PCR purification kit (e.g., Thermo GeneJet, Qiagen QIAquick).
4∣ Check that the desired PCR product has been obtained by running an electrophoresis experiment in 2% (wt/vol) agarose gel, and then measure the product’s concentration with a Qubit or TapeStation.
5∣ Calculate the mass of sequencing and digestion controls to add to the adapted library. You should aim to spike the control into a level of 0.5% (wt/wt). For example: adapted library amount = 1,000 ng; sequencing/digestion control (0.5% (wt/wt)) = (1,000 ng/100) × 0.5 = 5 ng; volume of 2 ng μl−1 sequencing/digestion control to add = 5 ng/2 ng μl−1 = 2.5 μl.
Preparation of genomic DNA for BS-seq and oxBS-seq  5 h
5 h
6∣ Sonicate 100 ng–1 μg of genomic DNA to the size that is needed for your application; for whole-genome Illumina sequencing, we recommend 100–400 bp. Please note, however, that if you intend to implement locus-specific PCR, you have to proceed through the workflow without sonicating the genomic DNA.
7∣ Add the appropriate volume of sequencing control to the sonicated DNA to achieve a 0.5% (wt/wt) spike-in.
8∣ If Illumina sequencing is being used, implement the Illumina TruSeq library preparation protocol according to the manufacturer’s directions. Please, however, do not implement the last step of AMPure XP bead purification, which will be replaced by Step 9 below. For locus-specific PCR, proceed directly to Step 9.
Purification of genomic DNA and spike-in sequencing and digestion controls  1 h
1 h
9∣ Purify DNA with AMPure XP beads (1:1 for sonicated genomic DNA, 1:1.6 for nonsonicated genomic DNA and 100-mer DNA). Wash the beads twice with 200 μl of 80% (vol/vol) ethanol; remove all traces of ethanol with a pipette and leave the DNA to air dry for 30 min. Elute the DNA with 22.5 μl of Milli-Q water or HPLC-quality water (not diethylpyrocarbonate (DEPC)-treated water). At this point, set aside a nonoxidized sample, which will be processed later, as described starting at Step 20.
 Make sure that all ethanol is removed at this stage, as the oxidation step in oxBS-seq is sensitive to any buffer and/or ethanol present.
Make sure that all ethanol is removed at this stage, as the oxidation step in oxBS-seq is sensitive to any buffer and/or ethanol present.
10∣ Add the appropriate volume of digestion control to the adapted library to achieve a 0.5% (wt/wt) spike-in.
11∣ Prepare a Bio-Rad Micro Bio-Spin P-6 SSC column for preoxidation purification via a modification of the manufacturer’s protocol: centrifuge the column at 1,000g for 120 s at room temperature. Wash the column three times by adding 500 μl of water to it each time and centrifuging it at 1,000g for 60 s at room temperature. Wash the column a final time with 500 μl of water and then centrifuge it at 1,000g for 120 s at room temperature. Add the sample DNA to the washed column. Centrifuge the column at 1,000g for 240 s at room temperature and retain the column eluate, which should be ~22 μl in volume (up to a maximum of 25 μl).
 It is essential to remove any last buffer and/or ethanol from the DNA before the oxidation step.
It is essential to remove any last buffer and/or ethanol from the DNA before the oxidation step.
 Purified DNA can be stored for 24 h at −20 °C before the oxidation is performed.
Purified DNA can be stored for 24 h at −20 °C before the oxidation is performed.
Preparation of the 1× oxidant solution  5 min
5 min
12∣ Take the 10× oxidant solution out of the freezer and thaw on ice.
13∣ Mix the thawed 10× oxidant by vortexing and centrifuging it at 1,000g for 5 s at room temperature. Return the thawed 10× stock to ice. To 18 μl of Milli-Q water placed in a new 1.5-ml microcentrifuge tube, add 2 μl of the 10× oxidant solution. Mix the solution briefly by vortexing and centrifuge at 1,000g for 5 s at room temperature. Keep this solution on ice.
The activity of this 1× oxidant on 5hmC can be ascertained by HPLC (Box 1 and supplementary Fig. 2).
Sample processing through the oxidation workflow  2 h
2 h
14∣ To denature the dsDNA, add to a new 1.5-ml microcentrifuge tube the buffer-exchanged dsDNA sample, 1 M NaOH and Milli-Q water as detailed below. Mix briefly by vortexing.
| Component | Volume per reaction |
|---|---|
| Buffer-exchanged dsDNA (1 μg) | Variable (maximum 21.75 μl) |
| NaOH, 1 M | 1.25 μl |
| Milli-Q water | Variable |
| Total | 23 μl |
 Please note that although the volumes of the individual components may vary, the combined volume of the dsDNA solution and of Milli-Q water must be 21.75 μl.
Please note that although the volumes of the individual components may vary, the combined volume of the dsDNA solution and of Milli-Q water must be 21.75 μl.
15∣ Incubate the DNA denaturing solution at 37 °C in a shaking incubator for 30 min.
16∣ Snap-cool the DNA denaturing solution by placing the microcentrifuge tube in an ice-water bath (0 °C) for 5 min.
Oxidation reaction  1 h
1 h
17∣ While the microcentrifuge tube is in the ice-water bath, add to the 23-μl solution of denatured DNA 2 μl of the freshly prepared 1× oxidant solution. Mix the reaction by briefly vortexing and centrifuging it at 1,000g for 5 s at room temperature; return the microcentrifuge tube to the ice-water bath.
18∣ Leave the oxidation reaction in the ice-water bath for 60 min. Briefly vortex the reaction twice, after 20 min and after 40 min, each time returning the reaction vessel (the 1.5-ml microcentrifuge tube) to the ice-water bath.
 The color of the oxidation reaction is indicative of a successful oxidation (supplementary Fig. 3).
The color of the oxidation reaction is indicative of a successful oxidation (supplementary Fig. 3).
The solution should remain orange after the 1-h incubation period. Any other color of the solution, green or brown/black, implies reaction of the oxidant with traces of contaminants in the oxidation solution (e.g., Tris or ethanol). If these colors are observed, researchers must re-start with a fresh sample of genomic DNA.

19∣ Prepare the Bio-Rad Micro Bio-Spin P-6 column with an initial centrifugation at 1,000g for 120 s at room temperature. Add the DNA oxidation reaction to the column and centrifuge at 1,000g for 480 s at room temperature and retain the column eluate.
 Once it is oxidized, the eluate containing the DNA sample can be safely stored at −20 °C for up to 24 h without any effect on the quality of the sample.
Once it is oxidized, the eluate containing the DNA sample can be safely stored at −20 °C for up to 24 h without any effect on the quality of the sample.
Bisulfite treatment  11 h
11 h
20∣ Treat the oxidized DNA sample and a nonoxidized sample of DNA (from Step 9 for a BS-seq) using the Qiagen EpiTect bisulfite kit according to the protocol ‘Sodium Bisulfite Conversion of Unmethylated Cytosines in DNA Isolated from FFPE Tissue Samples’, which is outlined in the Qiagen EpiTect handbook with slight modifications indicated below. Prepare the bisulfite reactions in 200-μl PCR tubes according to the instructions below and add each component in the order listed.
| Component | Volume per reaction (μl) |
|---|---|
| DNA solution | 30 |
| Bisulfite mix (dissolved) | 80 |
| DNA protect buffer | 30 |
| Total | 140 |
21∣ Perform the bisulfite DNA conversion using a thermal cycler. Program the thermal cycler according to the instructions below.
| Steps | Time (min) | Temperature (°C) |
|---|---|---|
| Denaturation | 5 | 95 |
| Incubation | 25 | 60 |
| Denaturation | 5 | 95 |
| Incubation | 85 | 60 |
| Denaturation | 5 | 95 |
| Incubation | 175 | 60 |
| Denaturation | 5 | 95 |
| Incubation | 25 | 60 |
| Denaturation | 5 | 95 |
| Incubation | 85 | 60 |
| Denaturation | 5 | 95 |
| Incubation | 175 | 60 |
| Hold | Indefinite | 20 |
22∣ Purify the DNA according to the Qiagen EpiTect bisulfite kit manufacturer’s instructions.
PCR of spike-in digestion control  2 h
2 h
23∣ Prepare the PCR as described below to amplify out the processed digestion control from the template recovered from the oxBS-seq and BS-seq workflow.
| Component | Volume (μl) |
|---|---|
| Milli-Q water | 30 |
| Pfu buffer (10×) | 5 |
| dNTP mix (10 mM) | 2 |
| Digestion control post-bisulfite forward primer DFP2 (10 μM) | 5 |
| Digestion control reverse primer DRP1 (10 μM) | 5 |
| Template | 2 |
| Pfu Turbo Cx DNA polymerase | 1 |
| Total | 50 |
24∣ Amplify the PCR using the thermocyling protocol outlined below.
| Segment | Temperature (°C) | Duration (s) | Iterations |
|---|---|---|---|
| Initial denaturation | 95 | 120 | 1 |
| Loop | |||
| Denaturation | 95 | 30 | 40 |
| Annealing | 50 | 30 | |
| Extension | 72 | 15 | |
| Store | 4 | Forever |
25∣ Purify the amplicons using a PCR purification kit (e.g., Thermo GeneJet, Qiagen QIAquick).

26∣ Check that the desired PCR product has been obtained by running an electrophoresis experiment on a 2% (wt/vol) agarose gel, and then measure the product’s concentration with a Qubit or TapeStation.
Taq1 digestion of the oxBS-seq–processed digestion control amplicon  20 h
20 h
27∣ Prepare digestion reaction mixtures of the amplicon recovered from the previous step with Taq1 mixes as described below. The extent of digestion correlates with the level of oxidation and bisulfite conversion achieved during the oxBS-seq workflow. Please note that we recommend running a digestion negative control (without Taq1) for all samples. A single cutting control digest is required per set of digestions.
| Component | Digestion control (volume) |
Digestion negative control (volume) |
Cutting control (volume) |
|---|---|---|---|
| Taq1 buffer (10×) | 2 μl | 2 μl | 2 μl |
| Taq1 RE (20 U μl−1) | 40 U | — | 40 U |
| BSA | 0.2 μl | 0.2 μl | 0.2 μl |
| Processed digestion control amplicon DNA (10 ng μl−1) |
10 μl | 10 μl | — |
| Cutting control DNA (20 ng μl−1) | — | — | 5 μl |
| Milli-Q water | 5.8 μl | 7.8 μl | 10.8 μl |
| Total | 20 μl | 20 μl | 20 μl |
28∣ Incubate the Taq1 digestion mix for 18 h at 65 °C.
29∣ Denature the samples by incubating them at 80 °C for 20 min.
30∣ To check for the quality of the products, run the digestion mixes on a 2% (wt/vol) agarose gel or other gel electrophoresis systems (Bioanalyzer, TapeStation and so on). Example of results are shown in supplementary Figure 4.
31∣ (Optional) The digestion control amplicons can also be sent for Sanger sequencing with commercial companies (Source BioScience) to qualitatively determine the efficiency of bisulfite-based conversion of 5mC and 5hmC to cytosine. This step needs to be implemented only if you wish to attain a visual readout of efficiency.
PCR amplification and purification of Illumina libraries  2 h
2 h
32∣ Prepare a PCR mix outlined below using the Agilent Pfu Turbo Cx DNA polymerase with the Illumina library prepared by treating the DNA samples subjected to oxBS-seq and BS-seq.
| Component | Volume (μl) |
|---|---|
| Milli-Q water | 38.5 |
| Pfu buffer (10×) | 5 |
| dNTP (10 mM) | 2 |
| TruSeq PCR primer cocktail | 1.5 |
| Bisulfite-converted DNA | 2 |
| Pfu Turbo Cx DNA polymerase | 1 |
| Total | 50 |
33∣ Amplify the DNA using the thermal cycling regime outlined below.
| Segment | No. of cycles | Temperature (°C) | Duration (s) |
|---|---|---|---|
| 1 | 1 | 95 | 120 |
| 2 | 12–15 | 95 | 30 |
| 60 | 20 | ||
| 72 | 45 | ||
| 3 | 1 | 4 | Forever |
34∣ Purify the PCRs with 50 μl of AMPure XP beads according to the manufacturer’s instructions, and then elute the DNA in 22.5 μl of water. 
35∣ Accurately quantify the library concentration by using quantitative PCR (qPCR) or the Qubit, and then measure the size of the DNA with the TapeStation or Bioanalyzer. Sequence the DNA on an Illumina MiSeq or HiSeq instrument.
36∣ After sequencing, check the conversion efficiency of the sequencing controls (see ANTICIPATED RESULTS). 
Analysis of specific loci  7 h
7 h
 Nonsonicated genomic DNA that has undergone the oxBS-seq and BS-seq protocol can be analyzed at specific loci by implementing the steps in this section.
Nonsonicated genomic DNA that has undergone the oxBS-seq and BS-seq protocol can be analyzed at specific loci by implementing the steps in this section.
37∣ Design primers for specific loci using online-based tools (e.g., MethPrimer, http://www.urogene.org/methprimer, methPrimerDB http://medgen.ugent.be/methprimerdb, MethylPrimerExpress http://www.appliedbiosystems.com/ or BiSearch http://bisearch.enzim.hu/). To PCR regions designed from these tools, we recommend using the KAPA HiFi uracil+ enzyme with each set of primers. Optimization of the exact annealing temperature will be needed for each primer set.
38∣ Prepare two PCR mixes as outlined below with the nonsonicated genomic DNA that has undergone oxBS-seq or BS-seq, respectively.
| Component | Volume (μl) |
|---|---|
| Milli-Q water | 5.5 |
| Designed forward primer (10 μM) | 2.5 |
| Designed reverse primer (10 μM) | 2.5 |
| Bisulfite-converted DNA | 2 |
| KAPA HiFi uracil+ master mix (2×) | 12.5 |
| Total | 25 |
39∣ Amplify the DNAs using the thermal cycling regime outlined below.
| Segment | No. of cycles | Temperature (°C) | Duration (s) |
|---|---|---|---|
| 1 | 1 | 95 | 180 |
| 2 | 35 | 98 | 20 |
| 50–60 | 15 | ||
| 72 | 60 | ||
| 3 | 1 | 4 | Forever |
40∣ Purify the amplicons using a PCR purification kit (e.g., Thermo GeneJet, Qiagen QIAquick).
41∣ Accurately quantify the library concentration with a TapeStation or Bioanalyzer.
42∣ Sequence the DNA fragments obtained. Because of the need for high-sequencing coverage, we recommend pooling PCR fragments and sequencing this DNA pool with Illumina sequencing, according to an Illumina TruSeq Library preparation.
We also recommend adding the PCR product of the spike-in digestion control to this mix to get an accurate quantification of conversion efficiency. However, Sequenom and pyrosequencing technologies are also applicable.
 ing advice
ing advice
Troubleshooting advice can be found in Table 1.
Table 1.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 18 | Oxidation reaction turns green after incubation |
DNA contamination with buffer or ethanol |
Repeat the experiment and make sure to go through the exact DNA purification procedure |
| 25 | Little or no PCR product for digestion control |
Gross loss of template because of incorrect purification of either oxidation or bisulfite |
Repeat the experiment and make sure to go through the exact DNA purification procedure |
| 34 | Little or no PCR product for the whole-genome library |
Gross loss of template because of incorrect purification of either oxidation or bisulfite |
Repeat the experiment and make sure to go through the exact DNA purification procedure Ensure that the pH of the bisulfite solution is correct |
| 36 | Low conversion rate of 5mC and 5hmC to cytosine |
DNA not denatured properly | Ensure that the NaOH concentration is correct and that the denaturation mix is vortexed |
| Oxidation of 5hmC to 5fC failed | Ensure that the oxidant has not precipitated and that it is red/orange |
||
| Bisulfite conversion failed | The bisulfite incubation time was not long enough; check the correct length of bisulfite incubation |

Steps 1–5, preparation of spike-in controls: 2 h
Steps 6–8, preparation of genomic DNA: 5 h
Steps 9–11, purification of genomic DNA: 1 h
Steps 12 and 13, preparation of oxidant solution: 5 min
Steps 14–16, sample processing through the oxidation workflow: 2 h
Steps 17–19, oxidation reaction: 1 h
Steps 20–22, bisulfite treatment: 11 h
Steps 23–26, PCR of spike-in digestion control: 2 h
Steps 27–31, Taq1 digestion: 20 h
Steps 32–36, preparation of DNA for whole-genomic sequencing: 2 h
Steps 37–42, locus-specific sequencing: 7 h
Box 1, HPLC analysis of oxidant activity for 5hmC: 20 h
ANTICIPATED RESULTS
For successful oxBS-seq, the control DNA should give a cytosine-to-uracil conversion efficiency of >99%, and an overall conversion efficiency of 5hmC to uracil of >85% is needed to achieve accurate quantification of 5mC and 5hmC. On average, a 5hmC-to-uracil conversion efficiency of 92% has been obtained in our experiments. Conversion efficiencies below these thresholds are a sign of a problem with either the oxidation or bisulfite conversion steps of the workflow.
For the Taq1 digestion, if the DNA has been digested, it means that the 5hmC in the Taq1 site has not converted to uracil, as Taq1 still recognizes the digestion motif (BS-seq). However, if the DNA has not been digested, it means that the 5hmC has converted to uracil, as the Taq1 no longer recognizes the digestion motif (oxBS-seq).
Supplementary Material
ACKNOWLEDGMENTS
We thank the UK Biotechnology and Biological Sciences Research Council for funding this work. S.B. and W.R. are recipients of Wellcome Trust Senior Investigator Awards. CEGX is a Syncona Partners–funded company.
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
COMPETING FINANCIAL INTERESTS The authors declare competing financial interests: details are available in the online version of the paper.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 3.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat. Rev. Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 4.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Globisch D, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munzel M, et al. Quantification of the sixth DNA base hydroxymethylcytosine in the brain. Angew. Chem. Int. Ed. 2010;49:5375–5377. doi: 10.1002/anie.201002033. [DOI] [PubMed] [Google Scholar]
- 9.Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 2012;13:7–13. doi: 10.1038/nrg3080. [DOI] [PubMed] [Google Scholar]
- 10.Pfaffeneder T, et al. The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chem. Int. Ed. Engl. 2011;50:7008–7012. doi: 10.1002/anie.201103899. [DOI] [PubMed] [Google Scholar]
- 11.He YF, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L, et al. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 2012;8:328–330. doi: 10.1038/nchembio.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Booth MJ, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–937. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- 15.Frommer M, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayatsu H, Shiragami M. Reaction of bisulfite with the 5-hydroxymethyl group in pyrimidines and in phage DNAs. Biochemistry. 1979;18:632–637. doi: 10.1021/bi00571a013. [DOI] [PubMed] [Google Scholar]
- 17.Huang Y, et al. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS ONE. 2010;5:e8888. doi: 10.1371/journal.pone.0008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meissner A, et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu M, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ficz G, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 21.Pastor WA, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Z, et al. High-resolution enzymatic mapping of genomic 5-hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep. 2013;3:567–576. doi: 10.1016/j.celrep.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu M, et al. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat. Protoc. 2012;7:2159–2170. doi: 10.1038/nprot.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flusberg BA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods. 2010;7:461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song CX, et al. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nat. Methods. 2011;9:75–77. doi: 10.1038/nmeth.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wallace EV, et al. Identification of epigenetic DNA modifications with a protein nanopore. Chem. Commun. 2010;46:8195–8197. doi: 10.1039/c0cc02864a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quinlivan EP, Gregory JF. III DNA digestion to deoxyribonucleoside: a simplified one-step procedure. Anal. Biochem. 2008;373:383–385. doi: 10.1016/j.ab.2007.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.