

Abstract
Human lymphoblastoid cell lines (LCLs), generated through Epstein–Barr Virus (EBV) transformation of B-lymphocytes (B-cells), are a commonly used model system for identifying genetic influences on human diseases and on drug responses. We have previously used LCLs to examine the cellular effects of genetic variants that modulate the efficacy of statins, the most prescribed class of cholesterol-lowering drugs used for the prevention and treatment of cardiovascular disease. However, statin-induced gene expression differences observed in LCLs may be influenced by their transformation, and thus differ from those observed in native B-cells. To assess this possibility, we prepared LCLs and purified B-cells from the same donors, and compared mRNA profiles after 24 h incubation with simvastatin (2 µm) or sham buffer. Genes involved in cholesterol metabolism were similarly regulated between the two cell types under both the statin and sham-treated conditions, and the statin-induced changes were significantly correlated. Genes whose expression differed between the native and transformed cells were primarily implicated in cell cycle, apoptosis and alternative splicing. We found that ChIP-seq signals for MYC and EBNA2 (an EBV transcriptional co-activator) were significantly enriched in the promoters of genes up-regulated in the LCLs compared with the B-cells, and could be involved in the regulation of cell cycle and alternative splicing. Taken together, the results support the use of LCLs for the study of statin effects on cholesterol metabolism, but suggest that drug effects on cell cycle, apoptosis and alternative splicing may be affected by EBV transformation.
This dataset is now uploaded to GEO at the accession number GSE51444
INTRODUCTION
Human lymphoblast cell lines (LCLs), created by transforming B-lymphocytes with Epstein–Barr Virus (EBV), have been used for functional analysis of common genetic variation by testing for associations of genome-wide SNPs with measures of gene expression (eQTLs), chromatin state and mRNA transcript structure as well as the effect of copy number variation on gene expression (1–6). In addition, LCLs have been used to identify genetic variation associated with or linked to human diseases (7) and more recently have become a model system for pharmacogenetic studies such as investigation of genetic effects on cellular survival in response to radiation and chemotherapeutic drugs (8).
We have recently used human-derived LCLs to identify single nucleotide variants affecting transcriptional response to simvastatin exposure. Simvastatin is a member of a class of drugs that inhibit HMG-CoA reductase (HMGCR), the key regulatory enzyme in the cholesterol biosynthesis pathway, and are widely prescribed to lower low-density lipoprotein (LDL) cholesterol and risk for cardiovascular disease (9). The physiologic relevance of LCLs for use in these studies has been demonstrated by showing that the magnitude of genetically influenced alternative splicing of the HMGCR gene in LCLs in vitro is significantly correlated with the in vivo plasma LDL cholesterol response to simvastatin treatment of the individuals from whom the LCLs were derived (10). In addition, we have successfully used the LCL model system to functionalize haplotypes and SNPs in candidate genes (9,11) and to identify novel genes and pathways not previously implicated in statin effects on LDL cholesterol (12).
EBV is known to infect and transform B-lymphocytes by binding to the CD21 receptor on the cell surface (13). Upon infection it alters the cell cycle (14), impacting both the expression levels and methylation status of hundreds of genes (15–17). Of the recent studies performed to directly compare gene expression profiles in LCLs and B-cells (17, 18), the experiment performed by Calıskan et al. (15) is the most comprehensive. Examining matched B-cells and replicate LCLs derived from six donors, the study noted that replicate LCLs generated from the same individual clustered with each other, but clustering with progenitor B-cells was not shown. They also reported that while transformation affected the methylation profile as well as expression levels of genes involved in cell cycle and immune response, these changes were small in magnitude. Other studies assessing the validity of LCLs include that of Ding et al., who reported that ∼70% of eQTLs were common to both fibroblasts and LCLs (19), suggesting that EBV transformation has limited impact on genetic regulation of gene expression. However, in a much less well-powered study, Fairfax et al. (20) reported that only 9.8% of eQTLs identified in LCLs were also observed in B-cells. Thus, these findings emphasize the importance of understanding expression differences in B-cells compared with LCLs, which to date, have not been examined in the context of drug response.
In the present study, we sought to determine the effect of EBV transformation on the transcriptional response to statin exposure by comparing statin-induced changes in the transcriptomes of LCLs and native B-cells derived from the same individuals.
RESULTS
B-cells and LCLs clustering
Hierarchical clustering analysis of the genome-wide expression data that passed all quality controls (15 statin and sham-treated B-cells, and 11 statin and sham-treated LCLs) demonstrated a distinct clustering by cell type irrespective of treatment status (Supplementary Material, Fig. S1). Within each cell type the samples further clustered by donor individual, but not by treatment status.
We then adjusted for statin response of EBV transformation and repeated the analysis with the six samples with a complete set of matched arrays (statin and sham treated B-cells and LCLs). As shown in Figure 1, we demonstrate significant clustering of B-cells and LCLs from five of these individuals irrespective of treatment (statin or sham) status. The mismatched cluster (individuals S_068 and S_289) had very weak bootstrap support as indicated by numerals on branches (Fig. 1). We performed a similar analysis using genes whose expression was significantly changed (P < 0.05) by statin treatment in both B-cell and LCLs. Although the grouping by individual was imperfect, the majority of B-cells and LCLs clustered by individual (Supplementary Material, Fig. S2). We did not observe clustering by individual for variation of the difference of expression levels between statin versus sham treatment (data not shown).
Figure 1.
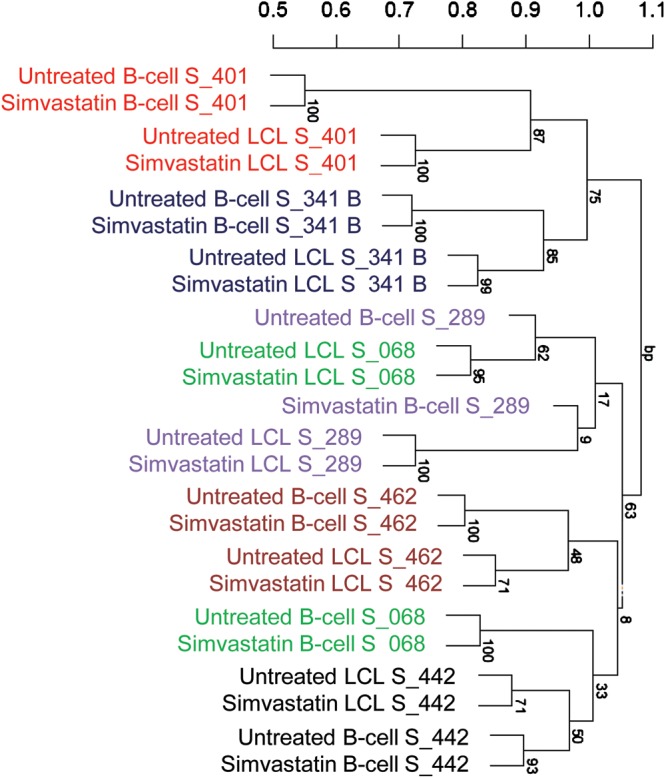
Clustering of B-cells and LCLs by individual. Expression array values for B-cells and LCLs were quantile normalized, adjusted for treatment status and clustered using pvclust in R. Individual IDs designated with S# are shown to co-cluster. Numbers on the branches are boot strap support (n = 1000).
We then conducted a principal component analysis using the 8000 most highly expressed genes to assess the proportion of variation explained by EBV treatment as well as other confounders. The first component explained 74% of the variance and correlated highly with EBV treatment status (P = 2.2E−16). The next four components correlated with individual identities (0.005 < P < 0.1) and together explained 18% of the variance. Components 6–10 were not correlated with a known covariate. Finally, components 11 and 12 together explained 1.1% of the variance and were highly correlated with statin treatment status (P < 0.001) (Supplementary Material, Table S1).
Comparison of mRNA expression between B-cells and LCLs
Comparison of transcripts between B-cells and LCLs (Supplementary Material, Table S2) identified 10 304 significantly differentially expressed genes (q < 0.05). Of these, 5491 were more highly expressed in LCLs than in B-cells, and the remainder showed greater expression in B-cells. Genes with ≥2-fold higher expression in LCLs compared with B-cells were enriched for the Gene Ontology (GO) biological process term ‘cell cycle’ (q = 1.3E−39, Fig. 2A), consistent with their transformation (Supplementary Material, Table S3). Genes with a smaller magnitude of up-regulation (changes between 1.32- and 2.0-fold) were enriched for the GO term ‘RNA processing’ (q = 4.4E−14) (Supplementary Material, Table S4).
Figure 2.

Global gene expression and methylation in B-cells versus LCLs. (A) Genome-wide gene expression was quantified by 38 arrays in statin and sham-treated B-cells and LCL (7 B-cell and 12 LCL) were analyzed using limma. The genes changed in response to statin treatment (q < 0.05) were classified into ‘high response’ (>2-fold) and ‘low response’ ( < 2-fold) groups, and subject to GO term analysis using DAVID. (B) Overlap of genes up-regulated in LCLs and EBNA2 ChIP-seq targets. (C) ChIP-seq and expression profiling of SFRS3. The peak diagram depicts the statistically significant (P = 9.77E−08) EBNA2 ChIP-seq peak found in the SFRS3 promoter. The H3K27Ac ChIP-seq ENCODE track, which marks promoter regions, is shown for reference. The boxplot depicts normalized values of SFRS3 expression in B-cells (n = 7) versus LCLs (n = 12). (D) Violin plots of distribution of SUMI sites in B-cells and transformed LCLs. Genome-wide methylation in B-cells and LCLs derived from the same donor (n = 4 matched pairs) was quantified by MetMap. Y-axis: 1 is completely methylated and 0 is completely un-methylated. X-axis: paired samples.
The most down-regulated genes in LCLs compared with B-cells (≥2-fold lower expression) were enriched for the GO terms ‘regulation of apoptosis’ (q = 3.3E−5) and ‘B-cell activation’ (q = 3.8E−05) (Supplementary Material, Table S5). Genes with a more modest reduction between the two cell types (between 1.31- and 2.0-fold lower) were enriched for GO terms ‘mRNA metabolic processing’ (q = 0.03) (Supplementary Material, Table S6). Given our interest in the use of LCLs for the study of statin pharmacogenomics and cholesterol metabolism, we noted that, even though the expression of several sterol metabolic genes such as HMGCR, LDLR and LSS was significantly different (q < 0.05) between LCLs and B-cells, overall the category of genes involved in cholesterol and lipid metabolism was not differentially expressed between B-cells and LCLs (q = 0.17).
Transcripts up-regulated in LCLs are potential EBNA2 targets
We hypothesized that genes involved in EBV transformation could be direct targets of transcriptional activators encoded within the EBV genome. Epstein–Barr Virus Nuclear Antigen 1 (EBNA1) and Epstein–Barr Virus Nuclear Antigen 2 (EBNA2) are known to be transcriptional activators encoded by EBV (13). Re-analysis of publicly available ChIP-seq data from Raji cells (14,21) identified EBNA1 peaks in the promoters (−1 to +1 kb from transcription start site) of 60 genes (Supplementary Material, Table S7), and EBNA2 peaks in the promoters of 2676 genes (Supplementary Material, Table S8). In total, 1081 genes with EBNA2 peaks were up-regulated (q < 0.05) in the LCLs compared with the B-cells, with a highly statistically significant (P < 1E−16) overlap identified between genes with EBNA2 peaks and those up-regulated in LCLs (Supplementary Material, Table S9, Fig. 2B). GO term analysis of these genes showed an enrichment in ‘cell cycle’ and ‘RNA processing’ (Supplementary Material, Tables S10–S12). Among the 51 up-regulated splicing factors only HNRNPA1 and SRFS3 (aka SRp20) have been previously shown to interact with an essential EBV protein, SM, that modulates splicing of both EBV and host cell pre-mRNAs (22, 23). Notably, expression levels of both SRFS3 and HNRNPA1 were higher in LCLs compared with B-cells (1.32-fold, q = 0.004 and 5.38-fold, q = 3.1E−14, respectively), and these genes had significant EBNA2 peaks in their promoters (Fig. 2C, Supplementary Material, Fig. S3, Table S2). We also identified a significant overlap (P = 5E−05) between genes down-regulated in LCLs versus B-cells and those with EBNA2 peaks; however there were substantially fewer genes (n = 591) compared with those that were differentially up-regulated in LCLs. Furthermore, there was no statistically significant enrichment of GO terms within this list (Supplementary Material, Fig. S4). Overall, these results strongly support the likelihood that EBV directly impacts host cell gene expression.
LCLs have reduced genome-wide methylation compared with B-cells
Next, we sought to determine whether differences in gene expression between LCLs and B-cells might be attributed to changes in DNA methylation. To test the effect of EBV transformation on methylation profiles, we compared the genome-wide methylation status of untreated LCL and B-cells obtained from four donors. We found 2002 regions that were significantly less methylated in LCLs (P < 0.05, paired t-test) and 756 regions that were significantly more methylated, indicating a net effect of global de-methylation (Fig. 2D, Supplementary Material, Tables S13 and S14). Specifically, analysis of promoter regions found evidence of reduced methylation in LCLs of 573 promoters, and increased methylation in only 104 promoters. No enrichment of GO terms was identified in the gene sets that demonstrated either increased or decreased methylation status between the two cell types (Supplementary Material, Table S15).
Statin-induced changes in gene expression are similar between B-cells and LCLs
Comparison of the statin versus sham-treated LCLs (n = 12) identified 43 genes whose expression was significantly changed by exposure to simvastatin (Supplementary Material, Table S16, q < 0.05). This is a much smaller number than that observed in LCLs derived from a larger study population (24), a result to be expected based on the limitation of statistical power of this data set. Nevertheless, consistent with the larger study, ‘sterol biosynthetic process’ was the top GO category (increased expression, q = 8.2E−10) identified from statin-responsive genes. With the lower threshold of P < 0.01, the expression of 699 genes was altered, of which 343 were up-regulated and 357 were down-regulated. GO analysis of up-regulated genes identified significant enrichment for ‘sterol biosynthetic process’ (q = 1.5E−11), ‘cholesterol biosynthetic process’ (q = 1.32E−08) and ‘lipid biosynthetic process’ (q = 0.005) (Supplementary Material, Table S17). Down-regulated GO categories included ‘RNA processing’ (q = 1.2E−05) and ‘mRNA splicing’ (q = 0.063) (Supplementary Material, Table S18).
Again, likely due to the limited statistical power of our data set, only eight genes demonstrated statistically significant changes in expression levels in the B-cells in response to statin treatment (n = 6, q < 0.05): ABCG1, ZC3HAV1, ABCA1, SQLE, HMGCR, RHOB, SCD, and CYP51A1. Of these genes only ZC3HAV1 was not previously reported to be statin responsive, while the remainder are known to be involved in sterol metabolism and transport. At a less stringent P-value threshold (P < 0.01), 218 genes changed in response to simvastatin (Supplementary Material, Table S19), of which 123 were up-regulated and 95 were down-regulated. These included many genes known to be sterol responsive such as LDLR and HMGCS1. GO term analysis of the 123 up-regulated genes showed enrichment in ‘sterol metabolic process’ (q = 6.5E−08) and ‘cholesterol metabolic process’ (q = 1.05E−05) (Supplementary Material, Table S20).
Of the genes with evidence of statin-responsiveness in both B-cells and LCLs (P < 0.01), 22 were up-regulated and one was down-regulated. (Fig. 3A, Table 1, Supplementary Material, Fig. S5). Importantly, the magnitude of statin-induced change in the expression of these genes was strongly correlated in LCLs versus. B-cells (R2 = 0.723, P = 4.0E−05) (Fig. 3B). Notably, these genes were primarily implicated in cholesterol metabolism with many known to be SREBF2 targets. Among the genes that were drug responsive in one cell type and not the other, we found enrichment of GO terms for only ‘mRNA processing’ and ‘mRNA splicing’ in genes that were down-regulated by statin treatment in LCLs but not B-cells (GO term, q = 4.23E−09 and q = 3.0E−05, respectively).
Figure 3.

Genes affected by statin treatment in B-cells and LCLs. (A) Overlap analysis of statin-responsive genes in B-cells and LCLs. Genes changing in response (P < 0.01) to statin treatment in B-cells (n = 7) and LCLs (n = 12). (B) Correlation of statin-induced changes in gene expression paired B-cells and LCLs. The average magnitude of change in genes identified to be statin responsive (P < 0.01) in both B-cells and LCLs (n = 23 genes) was plotted and linear regression used to assess their relationship.
Table 1.
Genes affected by statin treatment in B-cells and LCLs.
| Function | Symbol | B-cell logFC | LCL LogFC | B-cell P-value | LCL P-value |
|---|---|---|---|---|---|
| Cholesterol regulation | RHOB | 1.16 | 1.22 | 4.81E−06 | 1.65E−08 |
| INSIG1 | 0.52 | 0.50 | 2.67E−04 | 1.40E−05 | |
| Cholesterol biosynthesis | SQLE | 0.65 | 0.38 | 2.33E−06 | 9.46E−05 |
| CYP51A1 | 0.58 | 0.48 | 9.24E−06 | 2.97E−06 | |
| DHCR7 | 0.58 | 0.51 | 1.68E−03 | 4.14E−04 | |
| SC4MOL | 0.56 | 0.63 | 5.70E−05 | 1.04E−07 | |
| HMGCR | 0.56 | 0.52 | 3.46E−06 | 1.14E−07 | |
| HMGCS1 | 0.54 | 0.79 | 1.08E−04 | 1.73E−09 | |
| ANXA2P1 | 0.53 | 0.38 | 3.15E−03 | 4.83E−03 | |
| DHCR24 | 0.49 | 0.40 | 2.87E−03 | 1.91E−03 | |
| IDI1 | 0.44 | 0.39 | 5.64E−03 | 1.48E−03 | |
| FDFT1 | 0.42 | 0.45 | 4.20E−05 | 1.60E−07 | |
| Cholesterol biosynthesis NADPH source | IDH1 | 0.40 | 0.25 | 7.61E−04 | 5.58E−03 |
| Cholesterol uptake | ACAT2 | 0.40 | 0.54 | 1.86E−04 | 2.94E−08 |
| LDLR | 0.71 | 0.51 | 1.83E−04 | 3.78E−04 | |
| Cholesterol homeostasis and transport | ABCG1 | −1.18 | −0.32 | 2.41E−09 | 5.42E−03 |
| Adapter protein, function unknown | YWHAH | 0.38 | 0.30 | 3.84E−05 | 3.12E−05 |
| Lactosylceramide metabolism membrane related | B4GALT5 | 0.35 | 0.25 | 2.80E−03 | 4.73E−03 |
| Associated with microtubules | CLIP1 | 0.25 | 0.20 | 3.99E−03 | 2.94E−03 |
| Unknown | NRBP2 | 0.29 | 0.25 | 4.26E−03 | 1.62E−03 |
| FAM45A | 0.28 | 0.21 | 2.60E−03 | 3.29E−03 | |
| HCFC2 | 0.23 | 0.19 | 2.36E−03 | 1.70E−03 | |
| DEFB106A | 0.20 | 0.16 | 9.34E−03 | 7.02E−03 |
Identification of transcription factors involved in statin regulation
We found that among the genes up-regulated with statin treatment in LCLs, there was an enrichment in ChIP-seq identified-binding sites for transcription factors previously implicated in cholesterol metabolism including SREBF1 (q = 9.5E−11), SREBF2 (q = 1.8E−16) EGR1 (q = 2.0E−23) and HNF4A (q = 3.0E−19) (25,26). Similarly, binding sites for SREBF2 (q = 4.2E−10), SREBF1 (q = 2.0E−09), EGR1 (q = 1.2E−06) and HNF4A (q = 3.7E−06) were enriched in statin-induced up-regulated genes in B-cells. Analysis of enriched ChIP-seq peaks in down-regulated genes in LCLs identified 229 independent statistically enriched transcription factors (q < 0.01), of which MYC was the most significantly enriched (q = 1.5E−35). In contrast, statin-induced down-regulated genes in B-cells were not enriched for MYC-binding sites, but instead were enriched in EGR1 (q = 8.1E−06), RUNX1 (q = 7.2E−05) and HNF4A (7.2E−05) sites. These results are consistent with the increased levels of MYC transcripts in LCLs versus B-cells seen in our study (1.67-fold, q = 2.83E−10) and in other studies (14,27,28), This raises the possibility that MYC may play a role in statin-mediated reduction of gene expression in the LCLs but not the B-cells. For example, HNRNPA1, a well-documented splicing suppressor (29), had strong evidence of an MYC-binding peak in its promoter and was down-regulated by statin treatment in the LCLs, but not the B-cells (Supplementary Material, Fig. S3).
DISCUSSION
The main objective of this study was to assess the effects of EBV transformation of B-cells on transcriptional response to drug exposure, using simvastatin as an example of a widely prescribed class of drugs that is known to cause a robust transcriptional response through activation of the SREBF2 pathway. In matched B-cells and LCLs obtained from the same donor individuals, we documented that simvastatin exposure increased the expression of many known SREBF2 target genes involved in cholesterol metabolism in either B-cells or LCLs. Although relatively few genes were found to be statin responsive in both B-cells and LCLs, likely due to the limited sample size of this study, the directionality and magnitude of changes observed were highly correlated between B-cells and LCLs. This finding suggests that EBV transformation preserves the transcriptional response to statin treatment on either the genetic or epigenetic level. Hence, our results support the utility of LCLs as a model system for the study of statin pharmacogenetics.
Through hierarchical clustering analysis of the 8000 most highly expressed transcripts identified in our data set we found clustering of B-cells and LCLs by individual, irrespective of statin treatment. We further showed similar clustering using genes that were statin responsive in both B-cells and LCLs arrays. Caliskan et al. (15) also reported that LCLs derived from the same progenitor B-cell population clustered by gene expression. These findings suggest that EBV transformation does not disrupt the general inherent genetic or epigenetic regulation of transcript levels in at least a subset of genes, in contrast to the report of Fairfax et al. (20) that only 9.8% of eQTLs identified in LCLs were also observed in B-cells. However, it should be noted that our analysis was limited to a subset of transcripts and thus may not represent the transcriptome-wide effects of EBV transformation. Furthermore, we did not observe clustering by individual based on statin-induced differences in gene expression, either due to limited statistical power or to the possibility that genetic influences on the statin response are more modest than those contributing to variation in endogenous expression levels.
Notably, the expression of the class of genes involved in lipid metabolism was not statistically different between the two cell types, suggesting that LCLs can be a useful model system for the study of genetic and pharmacologic effects on cellular cholesterol homeostasis. The utility of LCLs in this regard is further supported by the recent evidence that ≥70% of the expressed genes in LCLs were also expressed in primary human hepatocytes and the human hepatoma cell line HepG2 (ET, personal communication). For the genes whose expression levels differed between B-cells and LCLs, the expression of cell cycle-related genes was higher and the expression of apoptosis-related genes was lower in LCLs compared with B-cells, consistent with previous reports and the transformed nature of the LCLs (15,30,31). Unexpectedly, we also identified a number of genes involved in RNA processing and splicing whose expression was altered by EBV transformation. These changes may be mediated at least in part by the EBV-produced transcriptional coactivator EBNA2, as we found a significant overlap between EBNA2 ChIP-seq identified target genes and genes up-regulated in LCLs compared with B-cells. Many of these target genes appear to be involved in mRNA splicing, suggesting that EBV may have direct effects on the structure and regulation of RNA generated by the host cell. Supporting this hypothesis is the observation that EBV utilizes individual endogenous B-cell splicing factors such as SFRS3 to splice its own transcriptome (22).
Among the genes that appear to be differentially regulated by statin treatment in the LCLs, we found that those involved in RNA processing and splicing were down-regulated by statin treatment in LCLs but not B-cells. While it is possible that the lack of identification of these effects in B-cells is due to the smaller number of B-cells evaluated (7 pairs) versus LCLs (12 pairs), or the increased heterogeneity of the B-cell isolates versus the LCLs, it is also possible that this may represent biologically meaningful differences. For example, we found enrichment for MYC transcription factor binding in the promoters of splicing factors and other genes down-regulated by statin treatment in LCLs but not in B-cells. MYC is known to be up-regulated by EBV and is essential for EBV-mediated B-cell to LCL transformation (14). Moreover, gene expression profiling studies by others have suggested that MYC can preferentially and substantially amplify the expression of specific targets including genes involved in alternative splicing (32,33). Furthermore, statin treatment has been shown to down-regulate and inactivate MYC (34,35). These findings are consistent with our observation of MYC up-regulation by EBV transformation, and down-regulation by statin exposure, and suggest a role for MYC in statin-induced down-regulation of genes involved in mRNA splicing.
An example of a mechanism by which MYC may modulate statin-induced down-regulation of genes involved in alternate splicing is provided by study of statin's effects on HNRNPA1. We have recently shown that this RNA-binding protein, which is known to interact with the EBV SM protein (23), modulates cholesterol response to statin by promoting alternative splicing of HMGCR, the target of statin inhibition (36). In the present study we have found that HNRNPA1 is up-regulated by EBV-induced transformation, is bound by both MYC and EBNA2, and is down-regulated by statin treatment (Supplementary Material, Fig. S6). Given our recent observations that statin treatment causes coordinated changes in alternative splicing of multiple genes involved in cholesterol metabolism (37), these findings highlight the complex interplay of statin and viral interaction within LCLs.
This study has several caveats. First, our results are only applicable to the utility of LCLs for the study of simvastatin-induced changes in gene expression, and may not be generalized for effects of other statins or drug classes. Secondly, while we demonstrated that gene expression ranking is generally preserved between LCLs and B-cells, we did not specifically monitor the effect of EBV transformation at the level of individual genes or gene variants. Thirdly, given our experimental design in which all LCLs were exposed to statin treatment in a single batch, while the native B-cells were statin treated individually, it is possible that some of the expression differences observed between cell types are due to batch effects. Finally, we did not assess the potential influence of EBV episome load on statin-induced differences in gene expression. However, while high (>10^6) EBV episomal load has been previously shown to be a confounder for statin-induced changes in gene expression, others have reported that low episomal load (thousands of copies) has little impact on gene expression in LCLs (15,38). In this regard, we note that the LCLs used for the present study are of a very low EBV copy number (tens to hundreds of copies). Moreover, our principal component analyses suggest that this and other potential confounders contribute <7% of the total variance of the gene expression levels.
Overall, this study supports the use of LCLs as a model system for studying differences in cellular regulation of cholesterol metabolism in response to statin. However, since we have identified differences between LCLs and native B-cells in the expression of genes involved in cell cycle, apoptosis and splicing, discoveries in those pathways should be validated in alternate model systems.
MATERIALS AND METHODS
B-cell isolation and EBV transformation
Sixty milliliters of blood was drawn from healthy unrelated adult donors. Complete details on ethnicity, gender and age of donors are shown in Supplementary Material, Table S21. The protocol was approved by the Children's Hospital Oakland Research Institute IRB and informed consent was obtained from all participants. Peripheral blood mononuclear cells (PBMCs) were isolated from 10 ml of blood using IsoPrep (Matrix Technologies) and transformed by EBV as described previously (39,40), n = 13. Viable cells remaining after 6 weeks were considered to be LCLs. PBMCs were isolated from the remaining 50 ml of blood using Lymphoprep (VWR), and B-lymphocytes were specifically isolated from PBMCs using the B-cell Negative Isolation kit (Invitrogen) according to the manufacturer's protocol. Aliquots of B-cells were incubated with FITC-conjugated CD19 fluorescent antibodies and the proportion of CD19-positive cells (B-cells only) was determined by FACS analysis (BD FACS Calibur) of 10 000-gated events. Only preparations with >90% B-lymphocyte purity were used for further analysis.
Statin incubation and RNA isolation
B-cells and LCLs were incubated with either 2 µm-activated simvastatin or a sham buffer for 24 h in RPMI media supplemented with 500 U/ml penicillin/streptomycin, 10% FBS and 2 nmol/L GlutaMAX as previously described (10), and all cultures were maintained at 37°C with 5% CO2. The B-cells were statin or sham exposed at the time of isolation (Supplementary Material, Table S21), while LCLs were frozen, thawed and exposed as a single batch. Simvastatin, obtained as a gift from Merck, was activated as previously described (10). RNA was isolated from all samples and stored at −80°C. All RNAs were converted to biotin-labeled cRNA using the Illumina TotalPrep-96 RNA amplification kit (Applied Biosystems, Foster City, CA) in a single batch. cRNA was randomized and hybridized in a single batch to Illumina HumanHT-12_V4 expression beadchips (Illumina, San Diego, CA) at Channing Laboratory (Division of Network Medicine, Brigham and Women's Hospital, Boston, MA) according to the manufacturer's protocols.
Gene expression analysis
Raw expression data were quantile normalized and log2 transformed using the Bioconductor package lumi (41) and P-values for expression of all annotated probes were derived using limma (42). Pairwise scatterplots were generated using R. Arrays with a significant number of outliers were discarded (Supplementary Material, Figs S7–S10). All discarded arrays were from B-cell samples, and likely reflect impure B-cell isolates. Thirty-eight arrays from 13 donors were used to assess B-cell versus LCL differences. Complete matched quartets of B-cell (statin and sham) and LCLs (statin and sham) were available for six donors (for a complete description see Supplementary Material, Table S21). Model-based comparisons between B-cells versus LCLs and statin versus sham treatment, controlled for individual, were performed using limma (42). All analyses were performed at the probe level. For results reported at the gene level, P-values shown reflect the most significant P-value for any probe within that gene. Q-values (q) were generated by using the ‘p-adjust’ function in R and refer to FDR-adjusted P-values (P). Biological process GO analysis was conducted using DAVID (43) using all annotated human genes as the background, and GO q-values indicate FDR-adjusted P-values. Venn diagrams were generated with Venny (44). Hypergeometric testing implemented in R (phyper function) was used to calculate overlap P-values in the Venn diagrams. Enrichment of ChIP-seq signal was identified using ChEA (45) and the Enrichr toolkit (http://amp.pharm.mssm.edu/Enrichr/) containing a database of publicly available ChIP-Seq results. All the analysis scripts are available upon request.
Clustering and principal component analysis
Global clustering analysis was performed using the hclust function in R on the complete expression data set from samples that passed quality control (15 statin and sham-treated B-cells, and 11 statin and sham-treated LCLs).
Expression arrays from the six individuals with a complete matched data set of all four treatment conditions (B-cell statin, B-cell sham, LCL statin and LCL sham) were filtered by subsetting on the 8000 most highly expressed genes and adjusted for EBV transformation and statin treatment status by linear modeling. The residuals were then clustered by the pvclust function in R, using ‘correlation distance’ and ‘average method’ parameters. Confidence in the branches was assessed by bootstrap resampling (n = 1000). Principal component analysis was conducted using the prcomp function in R, with default parameters.
Methylation assay
Genome-wide methylation status was assessed after sham-only treatment in four independent matched B-cell/LCL pairs. Strongly unmethylated islands (SUMI) or unmethylated CGs were assayed as previously described (46,47). Briefly, DNA was extracted by standard methods and digested overnight with the methylation-sensitive restriction enzyme HpaII (New England Biolabs). Indexed sequencing libraries were constructed from the HpaII digest using the standard Illumina kit, and sequenced using 50 bp paired end reads. Supplementary Material, Table S22 shows the number of reads collected for each sample that passed the quality filter. Alignments were analyzed with MetMap to assign to each HpaII site a probability of being unmethylated p(U) and a probability of being part of an unmethylated region p(I). MetMap annotates strongly unmethylated islands (SUMIs) as regions in which all HpaII sites have a p(I) >0.1 and where at least two HpaII fragments within the region are represented in the MethylSeq data. SUMIs shared between all eight samples were cross referenced using BedTools software package (48). Statistically significant differences in SUMI scores between LCLs and B-cells were identified using paired Student's t-test (additional details can be found in the Supplementary Material).
ChIP-seq analysis
EBNA2 and EBNA1 ChIP-seq raw sequence data were reanalyzed from previous studies (14,49). Sequences were aligned to the genome using Bowtie2 (50). Annotation, peak calling and quality control were conducted using the HOMER package (51) with default parameters. Although two EBNA2 data sets were available, one data set (GSM729851) did not pass quality control and was omitted from the analysis. Only the GSM729852 data set was used for EBNA2 analysis and both available ‘input’ data sets were used as background controls.
SUPPLEMENTARY MATERIAL
FUNDING
We thank Elizabeth Theusch for helpful discussions. This work was supported by National Institute of Health (1T32 HL 098057-01, U19 HL069757, R.M.K. and E.B.), (R01 HL 1041 33 M.W.M.), (U01 HL 065899, R01 HL 092197, R01 NR 013391 K.T.) and Dairy Research Institute (DMI#1052, R.M.K.).
Supplementary Material
ACKNOWLEGEMENTS
E.B. performed the array analysis and drafted the manuscript. A.A. isolated B-cells, and performed statin treatments and RNA-extractions. K.K. and E.B. developed and applied the PCA clustering algorithm. S.J.H. and D.B. performed the methylation assay and analysis. J.I.R. supervised transformation of B-cells. K.T. supervised the performance of expression array analysis. R.M.K. and M.W.M. conceived the project, guided analysis and drafted and revised the manuscript.
Conflict of Interest statement: The authors declare that they have no conflict of interest.
REFERENCES
- 1.Degner J.F., Pai A.A., Pique-Regi R., Veyrieras J.B., Gaffney D.J., Pickrell J.K., De Leon S., Michelini K., Lewellen N., Crawford G.E., et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482:390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixon A.L., Liang L., Moffatt M.F., Chen W., Heath S., Wong K.C., Taylor J., Burnett E., Gut I., Farrall M., et al. A genome-wide association study of global gene expression. Nat. Genet. 2007;39:1202–1207. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 3.Brown C.C., Havener T.M., Medina M.W., Krauss R.M., McLeod H.L., Motsinger-Reif A.A. Multivariate methods and software for association mapping in dose-response genome-wide association studies. BioData Min. 2012;5:21. doi: 10.1186/1756-0381-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwan T., Benovoy D., Dias C., Gurd S., Provencher C., Beaulieu P., Hudson T.J., Sladek R., Majewski J. Genome-wide analysis of transcript isoform variation in humans. Nat. Genet. 2008;40:225–231. doi: 10.1038/ng.2007.57. [DOI] [PubMed] [Google Scholar]
- 5.Monks S.A., Leonardson A., Zhu H., Cundiff P., Pietrusiak P., Edwards S., Phillips J.W., Sachs A., Schadt E.E. Genetic inheritance of gene expression in human cell lines. Am. J. Hum. Genet. 2004;75:1094–1105. doi: 10.1086/426461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stranger B.E., Forrest M.S., Dunning M., Ingle C.E., Beazley C., Thorne N., Redon R., Bird C.P., de Grassi A., Lee C., et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moffatt M.F., Kabesch M., Liang L., Dixon A.L., Strachan D., Heath S., Depner M., von Berg A., Bufe A., Rietschel E., et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 8.Welsh M., Mangravite L., Medina M.W., Tantisira K., Zhang W., Huang R.S., McLeod H., Dolan M.E. Pharmacogenomic discovery using cell-based models. Pharmacol. Rev. 2009;61:413–429. doi: 10.1124/pr.109.001461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mangravite L.M., Medina M.W., Cui J., Pressman S., Smith J.D., Rieder M.J., Guo X., Nickerson D.A., Rotter J.I., Krauss R.M. Combined influence of LDLR and HMGCR sequence variation on lipid-lowering response to simvastatin. Arterioscler Thromb Vasc Biol. 2010;30:1485–1492. doi: 10.1161/ATVBAHA.110.203273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Medina M.W., Gao F., Ruan W., Rotter J.I., Krauss R.M. Alternative splicing of 3-hydroxy-3-methylglutaryl coenzyme A reductase is associated with plasma low-density lipoprotein cholesterol response to simvastatin. Circulation. 2008;118:355–362. doi: 10.1161/CIRCULATIONAHA.108.773267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao F., Ihn H.E., Medina M.W., Krauss R.M. A common polymorphism in the LDL receptor gene has multiple effects on LDL receptor function. Hum Mol Genet. 2013;22:1424–1431. doi: 10.1093/hmg/dds559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Medina M.W., Theusch E., Naidoo D., Bauzon F., Stevens K., Mangravite L.M., Kuang Y.L., Krauss R.M. RHOA Is a modulator of the cholesterol-lowering effects of statin. PLoS Genetics. 2012;8:e1003058. doi: 10.1371/journal.pgen.1003058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Young L.S., Rickinson A.B. Epstein–Barr virus: 40 years on. Nat. Rev. Cancer. 2004;4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 14.Zhao B., Zou J., Wang H., Johannsen E., Peng C.W., Quackenbush J., Mar J.C., Morton C.C., Freedman M.L., Blacklow S.C., et al. Epstein–Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc. Natl Acad. Sci. USA. 2011;108:14902–14907. doi: 10.1073/pnas.1108892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caliskan M., Cusanovich D.A., Ober C., Gilad Y. The effects of EBV transformation on gene expression levels and methylation profiles. Hum. Mol. Genet. 2011;20:1643–1652. doi: 10.1093/hmg/ddr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Min J.L., Barrett A., Watts T., Pettersson F.H., Lockstone H.E., Lindgren C.M., Taylor J.M., Allen M., Zondervan K.T., McCarthy M.I. Variability of gene expression profiles in human blood and lymphoblastoid cell lines. BMC Genomics. 2010;11:96. doi: 10.1186/1471-2164-11-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Y.V., Turner S.T., Smith J.A., Hammond P.I., Lazarus A., Van De Rostyne J.L., Cunningham J.M., Kardia S.L. Comparison of the DNA methylation profiles of human peripheral blood cells and transformed B-lymphocytes. Hum Genet. 2010;127:651–658. doi: 10.1007/s00439-010-0810-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carter K.L., Cahir-McFarland E., Kieff E. Epstein–Barr virus-induced changes in B-lymphocyte gene expression. J. Virol. 2002;76:10427–10436. doi: 10.1128/JVI.76.20.10427-10436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding J., Gudjonsson J.E., Liang L., Stuart P.E., Li Y., Chen W., Weichenthal M., Ellinghaus E., Franke A., Cookson W., et al. Gene expression in skin and lymphoblastoid cells: refined statistical method reveals extensive overlap in cis-eQTL signals. Am. J. Hum. Genet. 2010;87:779–789. doi: 10.1016/j.ajhg.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fairfax B.P., Makino S., Radhakrishnan J., Plant K., Leslie S., Dilthey A., Ellis P., Langford C., Vannberg F.O., Knight J.C. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat. Genet. 2012;44:502–510. doi: 10.1038/ng.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu F., Wikramasinghe P., Norseen J., Tsai K., Wang P., Showe L., Davuluri R.V., Lieberman P.M. Genome-wide analysis of host-chromosome binding sites for Epstein–Barr Virus Nuclear Antigen 1 (EBNA1) Virol. J. 2010;7:262. doi: 10.1186/1743-422X-7-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verma D., Bais S., Gaillard M., Swaminathan S. Epstein–Barr virus SM protein utilizes cellular splicing factor SRp20 to mediate alternative splicing. J. Virol. 2010;84:11781–11789. doi: 10.1128/JVI.01359-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Key S.C., Yoshizaki T., Pagano J.S. The Epstein–Barr virus (EBV) SM protein enhances pre-mRNA processing of the EBV DNA polymerase transcript. J. Virol. 1998;72:8485–8492. doi: 10.1128/jvi.72.11.8485-8492.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mangravite L.M., Engelhardt B.E., Medina M.W., Smith J.D., Brown C.D., Chasman D.I., Mecham B.H., Howie B., Shim H., Naidoo D., et al. GATM Locus is associated with reduced incidence of statin-induced myopathy. Nature. 2013 doi: 10.1038/nature12508. doi:10.1038/nature12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gokey N.G., Lopez-Anido C., Gillian-Daniel A.L., Svaren J. Early growth response 1 (Egr1) regulates cholesterol biosynthetic gene expression. J. Biol. Chem. 2011;286:29501–29510. doi: 10.1074/jbc.M111.263509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin L., Ma H., Ge X., Edwards P.A., Zhang Y. Hepatic hepatocyte nuclear factor 4alpha is essential for maintaining triglyceride and cholesterol homeostasis. Arterioscler. Thromb. Vasc. Biol. 2011;31:328–336. doi: 10.1161/ATVBAHA.110.217828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pajic A., Polack A., Staege M.S., Spitkovsky D., Baier B., Bornkamm G.W., Laux G. Elevated expression of c-myc in lymphoblastoid cells does not support an Epstein–Barr virus latency III-to-I switch. J. Gen. Virol. 2001;82:3051–3055. doi: 10.1099/0022-1317-82-12-3051. [DOI] [PubMed] [Google Scholar]
- 28.Cherney B.W., Bhatia K., Tosato G. A role for deregulated c-Myc expression in apoptosis of Epstein–Barr virus-immortalized B cells. Proc. Natl Acad. Sci. USA. 1994;91:12967–12971. doi: 10.1073/pnas.91.26.12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clower C.V., Chatterjee D., Wang Z., Cantley L.C.L.C., Vander Heiden M.G., Krainer A.R. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc. Natl. Acad. Sci. USA. 2010;107:1894–1899. doi: 10.1073/pnas.0914845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maier S., Staffler G., Hartmann A., Hock J., Henning K., Grabusic K., Mailhammer R., Hoffmann R., Wilmanns M., Lang R., et al. Cellular target genes of Epstein–Barr virus nuclear antigen 2. J. Virol. 2006;80:9761–9771. doi: 10.1128/JVI.00665-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seto E., Moosmann A., Gromminger S., Walz N., Grundhoff A., Hammerschmidt W. Micro RNAs of Epstein–Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. 2010;6:e1001063. doi: 10.1371/journal.ppat.1001063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ji H., Wu G., Zhan X., Nolan A., Koh C., De Marzo A., Doan H.M., Fan J., Cheadle C., Fallahi M., et al. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PloS one. 2011;6 doi: 10.1371/journal.pone.0026057. e26057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seitz V., Butzhammer P., Hirsch B., Hecht J., Gutgemann I., Ehlers A., Lenze D., Oker E., Sommerfeld A., von der Wall E., et al. Deep sequencing of MYC DNA-binding sites in Burkitt lymphoma. PLoS One. 2011;6:e26837. doi: 10.1371/journal.pone.0026837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shachaf C.M., Perez O.D., Youssef S., Fan A.C., Elchuri S., Goldstein M.J., Shirer A.E., Sharpe O., Chen J., Mitchell D.J., et al. Inhibition of HMGcoA reductase by atorvastatin prevents and reverses MYC-induced lymphomagenesis. Blood. 2007;110:2674–2684. doi: 10.1182/blood-2006-09-048033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takwi A.A., Li Y., Becker Buscaglia L.E., Zhang J., Choudhury S., Park A.K., Liu M., Young K.H., Park W.Y., Martin R.C., et al. A statin-regulated microRNA represses human c-Myc expression and function. EMBO Mol. Med. 2012;4:896–909. doi: 10.1002/emmm.201101045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu C.Y., Theusch E., Lo K., Mangravite L.M., Naidoo D., Kutilova M., Medina M.W. HNRNPA1 regulates HMGCR alternative splicing and modulates cellular cholesterol metabolism. Hum. Mol. Genet., 2014;23:319–332. doi: 10.1093/hmg/ddt422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Medina M.W., Gao F., Naidoo D., Rudel L.L., Temel R.E., McDaniel A.L., Marshall S.M., Krauss R.M. Coordinately regulated alternative splicing of genes involved in cholesterol biosynthesis and uptake. PLoS One. 2011;6:e19420. doi: 10.1371/journal.pone.0019420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choy E., Yelensky R., Bonakdar S., Plenge R.M., Saxena R., De Jager P.L., Shaw S.Y., Wolfish C.S., Slavik J.M., Cotsapas C., et al. Genetic analysis of human traits in vitro: drug response and gene expression in lymphoblastoid cell lines. PLoS Genet. 2008;4:e1000287. doi: 10.1371/journal.pgen.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pressman S., Rotter J.I. Epstein–Barr virus transformation of cryopreserved lymphocytes: prolonged experience with technique. Am. J. Hum. Genet. 1991;49:467. [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson M.A., Gusella J.F. Use of cyclosporin A in establishing Epstein–Barr virus-transformed human lymphoblastoid cell lines. In Vitro. 1984;20:856–858. doi: 10.1007/BF02619631. [DOI] [PubMed] [Google Scholar]
- 41.Du P., Kibbe W.A., Lin S.M. Lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 42.Smyth G.K. Limma: Linear Models for Microarray Data. New York: Springer; 2005. [Google Scholar]
- 43.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 44.Oliveros J.C. VENNY. An interactive tool for comparing lists with Venn Diagrams. 2007 http://bioinfogp.cnb.csic.es/tools/venny/index.html . [Google Scholar]
- 45.Lachmann A., Xu H., Krishnan J., Berger S.I., Mazloom A.R., Ma'ayan A. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics. 2010;26:2438–2444. doi: 10.1093/bioinformatics/btq466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singer M., Boffelli D., Dhahbi J., Schonhuth A., Schroth G.P., Martin D.I., Pachter L. Metmap enables genome-scale Methyltyping for determining methylation states in populations. PLoS Comput. Biol. 2010;6:e1000888. doi: 10.1371/journal.pcbi.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vera J., Torres N.V. MetMAP: an integrated Matlab package for analysis and optimization of metabolic systems. In Silico Biol. 2004;4:97–108. [PubMed] [Google Scholar]
- 48.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dresang L.R., Vereide D.T., Sugden B. Identifying sites bound by Epstein-–Barr virus nuclear antigen 1 (EBNA1) in the human genome: defining a position-weighted matrix to predict sites bound by EBNA1 in viral genomes. J. Virol. 2009;83:2930–2940. doi: 10.1128/JVI.01974-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.