

Abstract
Identifying the early gene program induced by GnRH would help understand how GnRH-activated signaling pathways modulate gonadotrope secretory response. We previously analyzed GnRH-induced early genes in LβT2 cells, however these lack GnRH self-potentiation, a physiological attribute of gonadotropes. To minimize cellular heterogeneity, rat primary pituitary cultures were enriched for gonadotropes by 40–60% using a sedimentation gradient. Given the limited number of gonadotropes, RNA was amplified prior to microarray analysis. Thirty-three genes were up-regulated 40 minutes after GnRH stimulation. Real-time PCR confirmed regulation of several transcripts including fosB, c-fos, egr-2 and rap1b, a small GTPase and member of the Ras family. GnRH stimulated rap1b gene expression in gonadotropes, measured by a sensitive single cell assay. Immunocytochemistry revealed increased Rap1 protein in GnRH-stimulated gonadotropes. These data establish rap1b as a novel gene rapidly induced by GnRH and a candidate to modulate gonadotropin secretion in rat gonadotropes.
Keywords: GnRH, Rap1b, rat primary pituitary cultures, gonadotrope enrichment, early gene, single-cell gene expression
1. Introduction
GnRH plays a key role in the control of mammalian reproductive function. Upon its release from the hypothalamus, the neuropeptide interacts with high-affinity receptors (GnRH receptor) at the surface of the pituitary gonadotrope cells, thereby stimulating the synthesis and secretion of the gonadotropins LH and FSH, which in turn promote gametogenesis and sex steroid production in the ovaries and testes. The biosynthetic and secretory responses to GnRH stimulation are controlled via the modulation of cell signaling pathways and transcription factor activity. A number of genes are induced by GnRH, and their identification would provide insight into the molecular bases of the gonadotrope response to GnRH receptor activation.
Genomic studies using microarrays and high-throughput quantitative real-time PCR have allowed us and others to study the pattern of gene responses following GnRH receptor activation in the Lβ T2 gonadotrope cell line [1–5]. In particular, we were interested in the early gene program activated by GnRH in Lβ T2 cells. Hence, we previously identified 44 genes whose expression was increased within an hour of GnRH exposure, and another 31 up-regulated genes within 3–6 h of exposure ([3] and Ebersole, B. J., T. Yuen, E. Wurmbach, and S.C. Sealfon, unpublished data). Those genes were divided into three main categories: i) genes encoding inducible transcription factors, e.g. egr2, c-fos, klf4, which may be implicated in gonadotropin gene activation; ii) genes encoding modulators of signal transduction, such as ikb, rgs2, mkp1, which might contribute to feedback control of the GnRH-activated signaling cascade; iii) genes encoding cytoskeletal proteins. Furthermore, additional microarray analyses revealed that the amplitude of gene induction was dependent upon GnRH concentration. Most likely, the rates of transcription were influenced by the concentration of GnRH [5]. Another microarray study by Kakar et al. identified 149 up-regulated genes and 83 down-regulated genes following one-hour treatment with a GnRH agonist. Consonant with our previous results, the group reported c-fos, egr1, Egr2, klf4, and lrg21 among the up-regulated genes, and observed that most of the genes which exhibited an elevated expression returned approximately to basal levels after 3 h of treatment [1]. More recently, the effects of various GnRH pulse regimens on gene expression were examined in Lβ T2 cells. Distinct patterns of gene expression were associated with each pulse frequency, with the greatest changes occurring at an hour or less interpulse interval; Egr1 and Egr2, both of which may mediate GnRH induction of the LHβ gene promoter, were induced at high pulse frequency, whereas Egr corepressors Nab1 and Nab2 were induced at low pulse frequency [2]. Importantly, Egr-1, in synergy with SF-1, was demonstrated to mediate GnRH induction of the LHβ gene promoter both in vitro and in vivo [6–9].
Advances in our understanding of gene responses elicited by GnRH interaction with its receptor have been obtained using Lβ T2 cells. Consistent with the results obtained in in vivo studies, GnRH-stimulated Lβ T2 cells show a significant induction of LH protein secretion, LHβ mRNA, as well as of LHβ gene promoter activity [10–14]. However, these immortalized cells do not exhibit self-priming of LH secretory response to GnRH [13], in contrast with primary cultures of female rodent anterior pituitary cells [14–17]. GnRH self-priming or self-potentiation is a signal amplification device that manifests as enhanced gonadotropin secretion in response to a second, identical GnRH stimulation which is not dependent on changes in LH synthesis or the GnRH receptor [18]. GnRH priming is a critical event in the triggering of ovulation [19] and has been demonstrated in vivo in humans and in vivo and in vitro in rodent models [16,18–22]. Therefore, studying GnRH-activated gene program in primary cultures of anterior pituitary cells is important from a physiological standpoint. Such studies have been impeded due to the heterogeneity of the anterior pituitary cell population, with gonadotropes representing only 5 to 15% of the five secretory cell types contained in the pituitary gland (somatotropes, lactotropes, corticotropes, thyrotropes).
In the present study, we examined early genomic events following GnRH receptor activation in rat primary pituitary cell cultures enriched for gonadotropes. A duration of 40 min post-GnRH stimulation was chosen based on our previous studies establishing a window for the transcription-dependent action of GnRH in self-potentiation [15]. Because the number of gonadotropes in the anterior pituitary is very restricted, we opted to pre-amplify the RNA prior to conducting high-density oligonucleotide microarray analysis. Expression data were validated by quantitative real-time PCR.
2. Materials and Methods
2.1. Materials
Adult female Sprague-Dawley rats (Charles River Laboratories, Hollister, CA) were maintained in controlled light conditions (12-h light, 12-h dark) for 2 weeks before use. Pituitary glands were removed after CO2 narcosis and decapitation. The protocols employed in these experiments were reviewed and approved by the University of California Davis Institutional Animal Care and Use Committee. Media and sera for cell culture were purchased from Invitrogen-GIBCO (Carlsbad, CA). Trypsin, kanamycin sulfate, BSA fraction V, 17β-estradiol (E2), 8-bromo-cAMP, and GnRH were purchased from Sigma-Aldrich (St. Louis, MO). For immunofluorescence, normal goat serum and mounting medium were obtained from Vector Laboratories (Burlingame, CA), and tetramethyl rhodamine isothiocyanate (TRITC)-conjugated goat anti-mouse IgG and fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit IgG were from Sigma-Aldrich; TOTO-3 iodide was from Invitrogen-GIBCO (Carlsbad, CA). Affinity purified rabbit polyclonal antibody against Rap1 was from Santa Cruz Biotechnology (Santa Cruz, CA); mouse monoclonal antibody against Rap1 was from BD Biosciences (San Jose, CA). Monoclonal antibody 518B7 against bovine LHβ was provided by Dr. Jan Roser (University California, Davis) [23]. Fura-2/AM and pluronic F-127 were from Invitrogen-Molecular Probes (Carlsbad, CA). Pipettes were pulled from Corning pyrex 7740 glass (Garner Glass, Claremont, CA) and coated with R6010 (K. R. Anderson, Santa Clara, CA).
2.2. Pituitary cell culture
Gonadotrope-enriched cultures
Anterior pituitaries (13–15/experiment) from rats ovariectomized for two weeks were enzymatically dispersed with trypsin and resuspended in Ca2+-free Minimum Essential Medium (MEM) containing 0.5% BSA and loaded into a unit gravity sedimentation chamber containing a gradient of 1.0–3.0% BSA in Ca2+-free MEM [24]. After equilibration, cells were collected from the bottom of the gradient in 20 × 50 ml fractions. Fractions 17–20 contained large cells and were combined; this pool represented ~6% of the total cells recovered from the sedimentation chamber. As determined by subsequent immunocytochemistry (Fig. 1), LH-positive cells in this pool ranged from 40–60% compared to < 7% for unfractionated cells. The gonadotrope-enriched pool was pelleted and resuspended in supplemented MEM culture medium [MEM plus 200 μM kanamycin sulfate, 0.2 nM E2, and 10% fetal bovine serum (FBS) that had been charcoal-treated to remove endogenous steroids]. For RNA studies, cells from the gonadotrope-enriched pool were plated at 22.5 × 104 cells per 22-mm well. For immmunocytochemical quantification of LH-positive cells, aliquots from the gonadotrope-enriched pool and a pre-fractionation sample were plated at 0.5 × 104 cells on 12-mm glass coverslips inserted into 22-mm wells. All cells were maintained in a humidified atmosphere (37ºC in 5% CO2). On day 2, medium was replenished (day of plating = day 0).
Fig. 1. Localization of LH (red) in gonadotrope-enriched primary pituitary cultures using confocal microscopy.
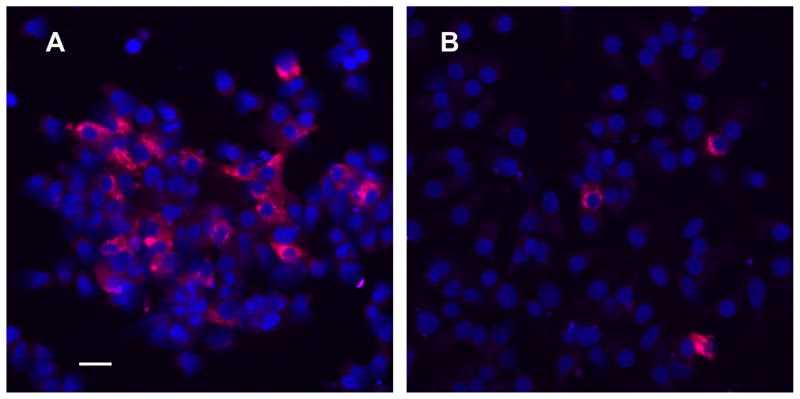
Gonadotrope-enriched (panel A) and unfractionated cells (panel B) from the same pituitary dispersion were cultured for 3 days and fixed and immunostained for confocal microscopy. Red identifies LH-positive cells (gonadotropes). Dark blue (TOTO-3) identifies cell nuclei. Scale bar (20 μm) in panel A applies to both panels.
Heterogeneous pituitary cultures for Rap1 immunocytochemistry and single-cell studies
Each dispersion consisted of one anterior pituitary enzymatically dispersed with trypsin and prepared for culture [25]. For immunocytochemistry, cells were plated at 1.8 × 104 cells on 12-mm glass coverslips inserted into 22-mm wells. For single cell studies, plating was in 35-mm dishes incorporating a 22-mm glass-bottomed well (Ted Pella, Inc., Redding, CA) at 1.5 × 104 cells per well. Flooding with supplemented MEM culture medium and maintenance were as above.
2.3. Experimental protocols
RNA studies in gonadotrope-enriched cultures
On day 3 of culture, GnRH was added to experimental wells to achieve a final concentration of 10 nM; control wells received an equivalent volume of vehicle medium. After 40 min, cells were rinsed with ice-cold PBS, lysis buffer containing guanidinium thiocyanate was added, cells were scraped, triturated, and stored at −70ºC. Cell lysates were processed for total RNA using a Qiagen RNeasy mini kit (Qiagen, Valencia, CA) and included an on-column DNase I digestion step.
Heterogeneous pituitary cell immunocytochemistry studies
On day 3 of culture, medium was replaced with control or 1 nM GnRH-containing medium. After 15 min, all media were replaced with control medium and incubation continued for an additional 45 min, following which cells were rinsed with PBS and fixed for immunocytochemistry. Additional GnRH-treated and control wells were rinsed and fixed at that end of the initial 15 min pulse.
Single gonadotrope studies
Cells were loaded with the Ca2+ indicator Fura-2 (fura-2/AM, 30 min, 37° C), and culture medium was replaced with an extracellular (EC) medium [26]. Dishes were transferred to an inverted microscope with a temperature regulated stage maintained at 22ºC and perfused with EC medium using a peristaltic pump (at 1 ml/min). Once a putative gonadotrope was identified, a tight seal was obtained with a patch pipette; solution flow was switched from control EC medium to challenge EC medium using a valve placed close to the microscope stage. Challenge EC medium contained either 10 nM GnRH, 1 mM 8-bromo-cAMP, or control medium. The rise in [Ca2+]i, which occurs within a few seconds of GnRH contact, was recorded and used to confirm the cell as a gonadotrope; [Ca2+]i was determined by Fura-2 radiometric fluorescence spectroscopy [26] using hardware and software from Photon Technology International (Birmingham, NJ). Groups: a GnRH pulse was administered for 15 min followed by control medium for 25 min, after which cell contents were harvested. In other groups, a pulse of either 8-bromo-cAMP or control medium was administered for 15 min, followed by control medium for 25 min, after which GnRH was administered for ~1 min and [Ca2+]i increase recorded for gonadotrope confirmation before harvest. Only one cell per dish was harvested. For short term control group a 1 min pulse of GnRH was administered to a putative gonadotrope ~ 5 min after selection and [Ca2+]i increase was recorded before harvest. For all groups, cell contents were harvested by aspiration into the patch pipette containing 10μl reverse transcriptase (AccuRT) cocktail and expelled into a PCR tube. The reaction mixture was immediately incubated for 1 h at 60° C to generate cDNA, and stored at −70ºC until determination of Rap1b expression level by quantitative real-time PCR.
2.4. Immunocytochemistry
Single label (LH)
On day 3, cultured cells from the gonadotrope-enriched studies were rinsed with PBS, fixed with pre-cooled methanol:acetone (1:1) for 7 min at −20ºC, rinsed in PBS, and blocked in 5% normal goat serum/PBS. Cells were exposed to 1:500 anti-LH for 2 h at room temperature and stained with TRITC-conjugated goat antimouse IgG and 2.4 nM TOTO-3 nuclear DNA stain for 1 h.
Dual label (Rap1, LH)
Following control or GnRH stimulation, pituitary cells were rinsed in PBS, fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, treated with −20ºC methanol, and blocked in 5% normal goat serum/PBS. Cells were exposed to 1:100 anti-Rap1 and 1:500 anti-LH overnight at 4ºC. Two different Rap1 antibodies, a rabbit polyclonal and a mouse monoclonal antibody, were tested with similar results; the Rap1 polyclonal antibody was used for the results reported here. Both antibodies recognize Rap1b and Rap1a, which are 95% homologous proteins (for review, see [27]. Immunofluorescence staining with FITC- and TRITC-conjugated second antibodies was accomplished in successive incubations. As negative controls, either or both primary antibodies were omitted. To establish absence of overlap between the detection of FITC and TRITC, single-labeled cells were imaged under similar conditions as dual-labeled cells to confirm proper signal isolation.
2.5. Confocal microscopy and image analysis
A Zeiss LSM 510 confocal microscope (Carl Zeiss MicroImaging, Thornwood, NY) with 40x (gonadotrope-enriched cultures) or 63x (Rap1 immunofluorescence) oil-immersion lenses was used as previously described [28]. For Rap1 protein studies, 2 μm overlapping sections through the entire cell volume of identified gonadotropes were analyzed using the Object Counter3D plugin in ImageJ (http://rsb.info.nih.gov/ij). To quantify Rap1-associated punctate areas, threshold criteria for brightness and size for the analysis were established based on preliminary screening of the data: the software identified qualifying number of objects and determined their extent throughout the cell via optical slicing. Object volume and total volume/cell were determined and data expressed as the fold-change (GnRH-treated cells/vehicle-treated cells) for groups terminated at 15 min of treatment or at 60 min. Analysis was carried out on cells from 4 separate pituitary dispersions (n = 4); for each group, data represent 1–2 coverslips/dispersion with 2–5 cells analyzed/coverslip. Differences between two groups were analyzed with the t test using SigmaStat (Systat Software, San Jose, CA).
2.6. RNA pre-amplification and cDNA microarray analysis
Comparison of Amplification Protocols
The relative performance of different amplification approaches was first determined using RNA from the Lβ T2 gonadotrope cells in order to select the most reliable protocol to use for amplification of the rat gonadotrope cells.
A) RNA amplification using terminal continuation
Lβ T2 cells obtained from the Mellon laboratory (University of California, San Diego) were maintained at 37ºC in 5% CO2 in humidified air in DMEM (Mediatech, Herndon, VA) containing 10% fetal bovine serum (Gemini, Calabasas, CA) [13]. Approximately 2 x 107 cells were seeded in triplicate 100-mm dishes and medium was replaced 24 h later with DMEM containing 25 mM HEPES (Mediatech) and glutamine. On the next day, cells were treated with either 100 nM GnRH or vehicle and were re-incubatated for 1 h. Total RNA was isolated using an RNeasy Mini kit (Qiagen, Valencia, CA). The terminal continuation method [29] relies on the use of a terminal continuation (TC) primer containing a SP6 bacteriophage promoter sequence and a span of CTPs. It has been estimated that over 60% of all human genes are located near CpG islands [30]. While the mechanism of annealing of the TC primer to the 5’ regions of mRNA was still under investigation, Che and Ginsberg (2004) proposed that the TC primers preferentially anneal within CpG islands [29]. Briefly, 100 ng RNA from each sample was reverse transcribed in a 20-μl volume containing 10 ng/μl Oligo(dT) primer, 10 ng/μl TC primer, 1X first strand buffer (Invitrogen, Carlsbad, CA), 1 mM dNTPs, 5 mM DTT, 20 U RNase inhibitor (Roche), and 5U Superscript II reverse transcriptase (Invitrogen). Reverse transcription was allowed to proceed for 2 h at 42°C. Second strand cDNA synthesis was performed in a 100-μl volume containing 10 mM Tris (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.5 U RNase H (Invitrogen), 5 U Platinum Taq DNA polymerase (Invitrogen), and 20 μl first-strand reaction mix. The reaction was allowed to proceed as follows: 10 min at 37°C, 3 min at 95°C, 3 min at 50°C, and 30 min at 72°C. Double-stranded cDNA products were purified using a QIAquick PCR purification kit (Qiagen). Sense RNA was synthesized by in vitro transcription using a MegaScript SP6 kit (Ambion, Austin, TX), according to the manufacturer’s protocol. The quality of amplified products was assessed by quantitative real-time PCR after reverse transcription of sense RNA using Oligo(dT) primers.
B. RNA isothermal amplification using hybrid DNA-RNA primer
Isothermal amplification of RNA samples for microarray probe labeling was prepared using an Ovation Biotin RNA Amplification and Labeling System (NuGen, San Carlos, CA), following the manufacturer’s recommendations. Briefly, 100 ng total RNA was incubated for 60 min at 48ºC with 1 μl First Strand Primer, 1.75 μl First Strand Buffer, and 0.25 μl First Strand Enzyme in a 5-μl volume. Second strand cDNA synthesis was performed by incubating the 5 μl first strand cDNA products with 4.5 μl Second Strand Buffer and 0.5 μl Second Strand Enzyme Mix at 37ºC for 30 min. RNA amplification and labeling were carried out by incubating the 10 μl second strand cDNA products with 18 μl SPIA Buffer Mix, 1 μl SPIA Primer Mix, 1 μl water, and 10 μl SPIA Enzyme Mix at 50ºC for 60 min. Labeled probes were used to hybridize with 4 Murine Genome U74Av2 GeneChip microarrays (Affymetrix, Santa Clara, CA; two vehicle-treated and two GnRH-treated samples). Data analysis was performed using Microarray Suite 5.0 (Affymetrix).
C. RNA amplification using two-cycle T7 RNA polymerase
Two-cycle target labeling was performed according to the manufacturer’s protocol (Affymetrix). Briefly, 100 ng total RNA was incubated in a 10-μl volume containing 2 μl T7-Oligo(dT) primer, 2 μl 5X first strand buffer, 1 μl 0.1 M DTT, 0.5 μl RNase inhibitor, 0.5 μl 10 mM dNTP, and 1 μl SuperScript II (Invitrogen) at 42 ºC for 1 h. Second strand cDNA synthesis was carried out by incubating 10 μl first strand cDNA products with 4.8 μl water, 4 μl 17.5 mM MgCl2, 0.4 μl 10 mM dNTP, 0.6 μl E. coli DNA Polymerase I, and 0.2 μl RNase H at 16ºC for 2 h. In vitro transcription was performed using the MegaScript T7 Kit (Ambion). Second strand cDNA products (20 μl) were incubated with 5 μl 10X Reaction buffer, 5 μl each of ATP, CTP, UTP and GTP solutions, and 5 μl Enzyme Mix at 37ºC for 16 h. The cRNA generated was purified using an RNeasy kit (Qiagen) and the manufacturer’s RNA cleanup protocol. cRNA was subsequently subjected to a second cycle of T7 amplification. First strand cDNA synthesis was performed by incubating 9 μl cRNA with 2 μl 0.2 μg/μl Random Primer, 4 μl 5X first strand buffer, 2 μl 0.1M DTT, 1 μl RNase inhibitor, 1 μl 10 mM dNTP and 1 μl SuperScript II (Invitrogen) at 42ºC for 1 h. First strand cDNA products were treated with 1 μl RNase H at 37ºC for 20 min. Reaction was stopped by heating at 95ºC for 5 min. Second strand cDNA synthesis was carried out by incubating the 21 μl first strand cDNA with 4 μl 5 μM T7-Oligo(dT) Primer, 88 μl RNase-free water, 30 μl 5X second strand buffer, 3 μl 10 mM dNTP, and 4 μl E. coli DNA Polymerase I at 16ºC for 2 h. The reaction was allowed to proceed for another 10 min at 16ºC after the addition of 2 μl T4 DNA Polymerase. Double-stranded cDNA products were then purified using a QIAquick PCR purification kit (Qiagen) according to the manufacturer’s recommendations. cRNA generation and labeling were performed using a BioArray HighYield RNA transcript labeling kit (Enzo Diagnostics). Labeled probes were used to hybridize with 4 Murine Genome U74Av2 GeneChip microarrays (Affymetrix; two vehicle-treated and two GnRH-treated samples). Data analysis was performed using Microarray Suite 5.0 (Affymetrix). Based on these results, the two-cycle amplification was used for the rat gonadotrope RNA samples.
Microarray experiments
A total of 12 Affymetrix Rat Expression Array 230 v2.0 (6 GnRH-treated and 6 vehicle-treated samples), each containing 31,000 gene clusters, were used. Data were analyzed using Affymetrix GeneChip Operating System (GCOS), with candidate regulated genes identified by data concordance across pairwise comparisons.
2.7. Quantitative real-time PCR
One hundred nanograms of total RNA were converted into cDNA and 1/100 utilized in a 40-cycle three-step PCR (95ºC/15 sec, 55ºC/20 sec, 72ºC/30 sec) in 20 mM Tris pH 8.4, 50 mM KCl, 3 mM MgCl2, 200 μM dNTPs, 0.5X SYBR Green I (Molecular Probes, Eugene, OR), 200 nM each primer, and 0.5 U Platinum Taq DNA polymerase (Invitrogen) using an ABI Prism 7900 thermal cycler (Applied Biosystems, Foster City, CA). Amplicon size (typically between 100 and 200 bp) and reaction specificity were confirmed by agarose gel electrophoresis. Threshold cycle (Ct) values were determined using SDS software v2.3 (Applied Biosystems). Primer sequences were as follows: r-cjun/S, TGTCTGTATGCTGGGGTGAG; r-cjun/A, TTGCAACACCCTCTTCTTCC; r-cfos/F, CAGCTGCACTACCTATACGTC; r-cfos/R, GTAATGCACCAGCTCAGTCAG; r-fosB/S, CAGGGTCAACATCCGCTAAG; r-fosB/A, GGTGAGGACAAACGAGGAAG; r-egr1/S2, TGTTTCAGGGGAGGCTTTAG; r-egr1/A2, CAGTAGGTAACCGCAGCATTC; r-egr2/S, CAGTGGTGTTGCAAGTGTCC; r-egr2/A, CAGCTGCAGTGACGTTAAGC; r-egr3/S, CAGCGACCACCTCACTACTC; r-egr3/A, CCCCTTTCTCCGACTTCTTC; r-Nr4a3/S, CGTGGTGACTTGGATACACC; r-Nr4a3/A, GCTGCCTTAAAGGTCACTGC; r-Rap1b/S, GTGGATGTGAATGTGGGAAG; r-Rap1b/A, ATACTGTGGCTCCCTGTTGG; r-GAPDH/S1, CATGGCCTCCAAGGAGTAAG; r-GAPDH/A1, AGATGGTGAAGGTCGGTGTG; r-actin/S1, AGGTGACAGCATTGCTTCTG; r-actin/A1, GCCTCAACACCTCAAACCAC.
2.8. Single-cell quantitative real-time PCR protocol
We adapted a hemi-nested PCR assay for single gonadotrope gene quantitation of Rap1b and Ppia (a reference house-keeping gene), respectively. The assay used two consecutive PCRs, the first with an outer primer set and the second with an inner primer set. Amplification linearity was maintained over a large number of cycles with low non-specific amplification of PCR by-products. Spike oligonucleotides were used to reduce nonspecific amplification by competing with abundant, low affinity DNA sequences for primer binding (random background amplifications), thus improving assay accuracy and detection limit. For the first PCR, single-cell cDNA was split in two aliquots for each gene: sample plus Master Mix 1 (PCR buffer, MgCl2, dNTP, Platinum Taq DNA polymerase, gene-specific spike oligonucleotide and outer primer set) in a 10-μl final volume were incubated in a thermocycler (95ºC/2 min followed by 8 cycles of 95ºC/15 sec, 55ºC/20 sec, 72ºC/30 sec). The second PCR was performed in a 7900 Prism RT- PCR machine (Applied Biosystems) using 5 μl of a 1:10 dilution of the first PCR product plus Master Mix 2 (PCR buffer, MgCl2, dNTP, Platinum Taq DNA polymerase, appropriate TaqMan probe and inner primer set) in a 10-μl final volume (95ºC/2 min and 40 cycles of 95ºC/15 sec, 60ºC/1 min). Copy numbers were estimated by intrapolating Ct values against a standard curve generated using standard oligonucleotides. Assay control wells included non-template controls from PCR1 and 2 Master Mixes. Differences between groups were analyzed by one-way ANOVA followed by Student-Newman-Keuls for multiple comparisons, using SigmaStat. Primer sequences used in the first PCR were as follows: r- Rap1b/S2, TGCTTGAAATCCTGGATACTG; r-Rap1b/A, AGCCTTGTCCGTTCTTCATGTAC; r- Ppia/S2, TCTTGTCCATGGCAAATGCT; r-Ppia/A, 5’-GCCATCCAGCCACTCAGTCT; r-Rap1b-spike, TGCTTGAAATCCTGGATACTGTCTAGAGAGTACCGACATTTGACGAGGCAAGGACGTACATGA AGAACGGACAAGGCT; r-Ppia-spike, TCTTGTCCATGGCAAATGCTACCGTCACGTCTATTTTTTGACCCTTGGTAAACACAAACCAGG AGACTGAGTGGCTGGATGGC. Primers used in the second PCR were: r-Rap1b/S, TGCAGGAACGGAGCAGTTT; r-Rap1b/A; r-Ppia/S, GCTGGACCAAACACAAATGGT; r-Ppia/A; r-Rap1b/probe, FAM-CAGCCATGAGAGATC-MGB; r-Ppia/probe, VIC-CCAGTTTTTTATCTGCACTGC-MGB. Standard oligonucleotide sequences were as follows: r-Rap1b/std, TGCTTGAAATCCTGGATACTGCAGGAACGGAGCAGTTTACAGCCATGAGAGATCTGTACATGA AGAACGGACAAGGCT; r-Ppia/std, TCTTGTCCATGGCAAATGCTGGACCAAACACAAATGGTTCCCAGTTTTTTATCTGCACTGCCA AGACTGAGTGGCTGGATGGC.
3. Results
3.1. Microarray analysis using gonadotrope-enriched primary pituitary cultures requires RNA amplification due to a limited number of cells
Because gonadotropes represent only 5–15% of anterior pituitary cells, a relatively small number of gonadotrope cells were purified from rat primary pituitary cultures. Therefore, it became crucial to reliably amplify the RNA in order to carry out high-throughput gene expression profiling. To this aim, the following three amplification methods were evaluated. While the goal of the present study is the regulation in primary rat gonadotropes, studies to test the protocols used RNA samples from Lβ T2 gonadotrope cells because they are an abundant and well-characterized source. The rat gonadotrope RNA is limited and therefore is not suitable for protocol development. Using 100 ng of total RNA isolated from the Lβ T2 gonadotrope cell line, we evaluated: terminal continuation (TC) RNA amplification [29], isothermal amplification using a hybrid DNA-RNA primer, and two-cycle T7 RNA polymerase amplification; these alternative amplification protocols were compared to a standard Affymetrix one-cycle T7 RNA polymerase amplification using 40 μg of total RNA. The quality of amplified products was assessed by quantitative real-time PCR, by targeting both the 5’ and 3’ ends of a 1.2 kb mouse gapdh transcript and a 1.9 kb β-actin transcript. The comparison of 5’- and 3’- PCR results was used to determine the degree of 3’ biasing of the amplification (the absence of biasing would give a ratio of unity). We compared the amplification approaches to identify which approach gave the maximal amplification yield with minimal 3’ biasing. The TC RNA amplification method generated heavily 3’ biased products, with 3’/5’ ratios of 30 and 50 for the gapdh and β-actin transcripts, respectively (data not shown); therefore it was eliminated. The isothermal amplification method resulted in uneven amplification, as revealed by a skewed scatter plot (Fig. 2B), suggesting sequence dependency.
Fig. 2. Comparison of three RNA amplification protocols for microarray analysis.
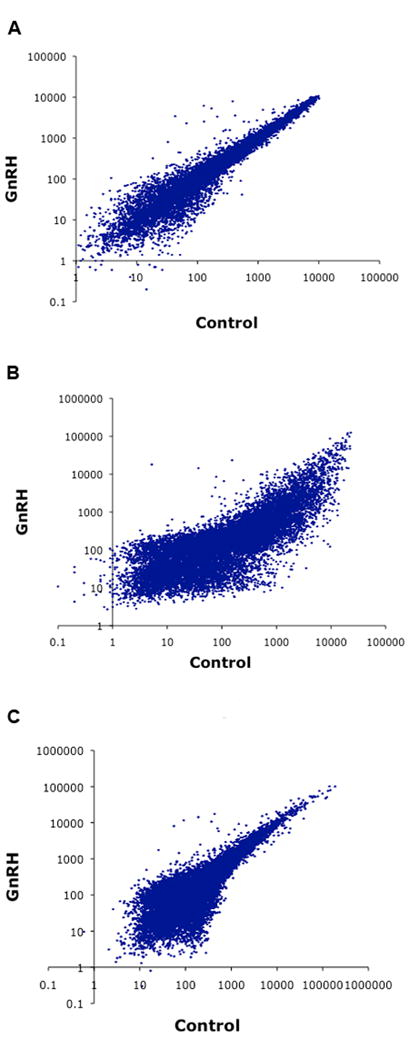
Scatter plots of microarray data obtained from three different RNA amplification methods. Lβ T2 cells were treated with 100 nM GnRH or vehicle (Control) for 1 h. RNA was isolated and aliquots were used for amplification procedures. Labeled probes were hybridized to Affymetrix GeneChip U74A microarrays. A, Amplification using a standard Affymetrix one-cycle T7 protocol with 40 μg total RNA. B, Hybrid DNA-RNA primer isothermal amplification with 100 ng total RNA. C, Amplification using a modified Affymetrix two-cycle T7 protocol with 100 ng total RNA. Copy numbers were estimated by intrapolation of CT values against a serial dilution of known concentrations of plasmid DNA containing either mouse GAPDH or mouse β-actin cDNA. Ideally, copy numbers reported for the 3’ and 5’ assays should yield a ratio of 1.
In the two-cycle amplification scatter plot (Fig. 2C), more low-intensity signals were observed in the lower-left quadrant, as compared to the one-cycle amplification scatter plot (Fig. 2A), and fewer high-intensity signals were spotted in the upper-right quadrant, overall denoting some loss of sensitivity. While the two-cycle T7 amplification method tended to generate 3’-biased amplified products, with a 3’/5’ ratio of 7 for β-actin (Fig. 2C) versus <2 using a standard Affymetrix one-cycle T7 RNA polymerase amplification (Fig. 2A), the bias was relatively low, and the expression profile was overall very similar to that of the standard procedure. Thus, the two-cycle T7 amplification was selected as the RNA amplification method for subsequent high-density microarray experiments with rat gonadotropes.
3.2. Microarray analysis of early gene expression in GnRH-stimulated primary gonadotropes identifies 33 up-regulated genes
To investigate the pattern of early gene responses following activation of the GnRH receptor, primary rat pituitary cultures enriched for gonadotropes were treated with 10 nM GnRH or vehicle for 40 min, the RNA amplified by two-cycle T7 amplification and subjected to high-density microarray analysis. Six independent replicate experiments were performed to identify differentially expressed genes. Data revealed the increased expression of 33 genes following GnRH exposure, by at least a 1.5 fold factor (Table 1; the raw data were deposited in the NCBI GEO-Database Accession no. GSE31399). In contrast, no genes were found to be down-regulated. Transcripts induced by GnRH receptor activation included the immediate early genes c-fos, fosB, and egr2, which encode transcription factors. Other GnRH-induced transcripts most notably encoded signal transduction proteins and cell signaling modulators (e.g. Rap1b, and Rgs4), protein or electron transporters (e.g. Bet1, Rab1, Slc38a2), cell adhesion proteins (e.g. Cldn4, Itga6), as well as proteins whose molecular functions are still poorly understood (e.g. Rtn4, Dnajb4).
Table 1.
Fold-changes from the early gene microarray experiments
| Probe Set | Fold | Gene Symbol | Gene Title |
|---|---|---|---|
| 1373759_at | 4.17 | fosB | FBJ osteosarcoma oncogene B |
| 1387306_a_at | 2.85 | Egr2 | early growth response 2 |
| 1398940_at | 2.14 | Srrm2 | serine/arginine repetitive matrix 2 FBJ murine osteosarcoma viral oncogene |
| 1375043_at | 2.05 | Fos | homolog |
| 1368888_a_at | 1.86 | Rtn4 | reticulon 4 |
| 1373093_at | 1.85 | Errfi1 | ERBB receptor feedback inhibitor 1 blocked early in transport 1 homolog |
| 1372710_at | 1.80 | Bet1 | (S.cerevisiae) |
| 1372131_at | 1.79 | Ubqln2 | Ubiquilin 2 |
| 1368151_at | 1.78 | Matr3 | matrin 3 |
| 1372347_at | 1.78 | Skil | SKI-like, transcript variant 2 |
| 1368044_at | 1.76 | Scg2 | secretogranin 2 |
| 1371583_at | 1.73 | Rbm3 | RNA binding motif (RNP1, RRM) protein 3 |
| 1387076_at | 1.72 | Hif1a | hypoxia inducible factor 1, alpha subunit |
| 1398841_at | 1.71 | Rab1 | RAB1, member RAS oncogene family |
| 1371755_at | 1.71 | EST | Similar to RIKEN cDNA B230219D22 |
| 1388547_at | 1.69 | Cldn4 | claudin 4 |
| 1369939_at | 1.66 | Cycs | cytochrome c, somatic |
| 1398797_at | 1.65 | Hnrpk | heterogeneous nuclear ribonucleoprotein K |
| 1371388_at | 1.64 | Pdhb | pyruvate dehydrogenase (lipoamide) beta |
| 1387806_at | 1.63 | Rap1b | RAS related protein 1b |
| 1383401_at | 1.61 | Tes | similar to Testis derived transcript |
| 1368505_at | 1.60 | Rgs4 | regulator of G-protein signaling 4 |
| 1379255_at | 1.60 | Atp6ap2 | ATPase, H+ transporting, lysosomal accessory protein 2 |
| 1370286_at | 1.60 | Slc38a2 | solute carrier family 38, member 2 |
| 1373156_at | 1.59 | Alex2 | armadillo repeat protein ALEX2 |
| 1371642_at | 1.59 | Eif4a2 | eukaryotic translation initiation factor 4A2 |
| 1367971_at | 1.55 | Ptp4a2 | protein tyrosine phosphatase 4a2 |
| 1376762_at | 1.54 | Plekha1 | pleckstrin homology domain containing, family A (phosphoinositide binding specific) member 1 |
| 1367512_at | 1.54 | Chmp5 | chromatin modifying protein 5 |
| 1372722_at | 1.53 | Dnajb4 | DnaJ (Hsp40) homolog, subfamily B, member 4 |
| 1383240_at | 1.53 | Itga6 | Integrin, alpha 6 |
| 1370238_at | 1.51 | Usmg5 | upregulated during skeletal muscle growth 5 |
| 1374529_at | 1.50 | TSP-2 | Thrombospondin 2 |
Values meeting the criteria for up-regulation are listed in descending order. Primary rat gonadotrope cells were exposed to 10 nM GnRH for 40 min, then harvested and processed for RNA extraction, as described in Materials and Methods. Data analysis was performed by Affymetrix GeneChip Operating System (GCOS). A gene was considered to be up-regulated by GnRH if there is at least 50% concordance across multiple pairwise comparisons of GnRH- vs. vehicle-treated microarrays, and if the fold-change was at least 1.50. Note: using the same selection criteria, no gene was found to be down-regulated after a 40-min GnRH treatment.
3.3. Rap1b is confirmed as a novel early gene induced by GnRH in primary gonadotropes
In order to validate the microarray data, we conducted a real-time PCR assay on a subset of up-regulated and mechanistically interesting genes regulated in the primary gonadotropes. With the exception of rap1b and nr4a3, the genes selected for real-time PCR analysis were previously found to be activated in Lβ T2 cells exposed to GnRH for one hour [[1,3,5,6] and Ebersole, B. J., T. Yuen, E. Wurmbach, and S.C. Sealfon, unpublished data]. Our quantitative assay confirmed the up-regulation of c-fos, fosB, egr2, and rap1b, while others that did not meet our criteria on the microarray (below 1.5-fold change, see Table 1) were found to be up-regulated, e.g. c-jun, egr1, egr3, nr4a3 (Fig. 3). Intriguingly, two novel genes, nr4a3 and rap1b, were established as GnRH-induced. Because previous studies reported a regulatory role for Rap1 in secretion in several exocrine and endocrine cells [31–34], rap1b was further investigated in this work.
Fig. 3. Validation of the early gene microarray data by real-time PCR.
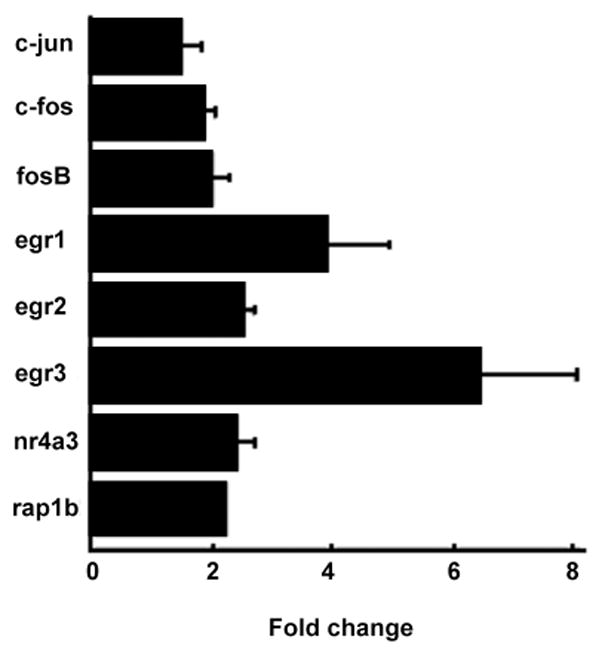
The expression levels of eight candidate genes were evaluated by real-time PCR in vehicle- vs. GnRH-treated primary rat gonadotropes. Represented are fold changes relative to the control conditions (i.e. with vehicle). Data from three independent experiments were calculated and plotted. Bars show standard deviations. All genes shown were significant at p<.05.
3.4. Single-cell studies confirm that GnRH induces rap1b gene expression in primary gonadotropes
Despite a significant enrichment for gonadotrope cells in purified gonadotropes (40–60%) as compared to pituitary cultures, the presence of other pituitary cell populations cannot be completely discounted. Hence, the potential cellular heterogeneity may be responsible for some degree of heterogeneous gene expression. To discriminate the contribution of gonadotropes to the increase in rap1b mRNA expression under GnRH stimulation, we established a single-cell quantitative real-time PCR assay in individual rat gonadotrope cells. As shown in Fig. 4, GnRH induced a 6-fold (P<0.001) increase in the level of rap1b transcripts in single gonadotrope cells, as compared to the unstimulated control. These results confirmed the microarray and real-time PCR data obtained in enriched gonadotrope cultures. As GnRH is known to stimulate an early increase in cAMP in rat pituitary cells [35], we examined the effect of 8-bromo-cAMP on rap1b gene expression: there was a 2-fold increase (P<0.02) in the level of rap1b transcripts. This result suggested that GnRH-induced increase in rap1b mRNA may be partially mediated by cAMP.
Fig. 4. GnRH activates Rap1b gene expression in single rat gonadotropes.

Individual rat gonadotrope cells were perfused with a 15-min pulse of extracellular medium only (Ctrl, control) or with medium containing either 10 nM GnRH or 1 mM 8-bromo-cAMP. Perfusion with control medium only was continued, and 25 min later cells were harvested and immediately processed for reverse transcription, as described in Materials and Methods. Rap1b data were normalized to Ppia transcripts determined in the same gonadotrope. Single cell data for all groups were obtained from two separate pituitary dispersions. The number of individual gonadotropes is in parentheses. Compared to control: ** P < 0.001, *P < 0.02.
3.5. GnRH-induced increase in rap1b gene expression has a similar effect on Rap1b protein levels in primary gonadotropes
Rap1b protein production following GnRH stimulation was evaluated by dual immunofluorescence and confocal microscopy. Pituitary cells were co-exposed to anti-LH and anti-Rap1 antibodies to examine signal co-localization to gonadotropes. As shown in Fig. 5C, at 1 h after initiation of a 15-min GnRH pulse, Rap1 complexes appeared as focal points of intense immunofluorescence staining in LH-positive cells with a distribution that suggested positioning of the complexes at the periphery, in areas compact with LH secretory granules. Although these immunofluorescent objects had a diameter of 500 nm or greater, their roundness was only apparent because they usually extended into multiple optical slices. As the diameter of rat gonadotrope secretory granules is typically 150–250 nm (reviewed in [36]), we could not determine whether the Rap1 complexes were associated with LH secretory granules. Moreover, only a subpopulation of objects was yellow suggesting subcellular co-localization. In contrast, in control cells (Fig. 5A) or in cells fixed after only 15 min of GnRH stimulation (Fig. 5B), Rap1 focal points appeared in smaller aggregates and were more diffusely distributed in the cytoplasm. Object volume quantification (expressed as fold-change, GnRH-treated/vehicle-treated) revealed that Rap1 immunofluorescence was significantly greater 1 h after GnRH stimulation as compared to cells fixed 15 min after GnRH treatment (6-fold; P<0.03; Fig. 5D). Thus, the results on protein expression were consistent with the mRNA data, indicating that the effects observed were specific and mediated by the GnRH receptor.
Fig. 5. GnRH activates Rap1 protein expression in primary gonadotropes.

Rat pituitary cells were either incubated in the presence of vehicle for 60 min (A), challenged with a 15-min pulse of 1nM GnRH and fixed immediately (B) or 45 min later (C). Fixed cells were subjected to dual immunofluorescence staining for LH (red) and Rap1 (green) and optical sectioning. Panels A–C: Single 2 μm optical sections at maximum cell diameter (A and B) or at intermediate diameters maximum for the field (C). Inset of panel C, an expanded view of immunofluorescent punctuate Rap1-associated objects. Scale bars = 5 μm; the dimension of the expanded view (C) is approximately 9 x 3 μm. D, Rap1-associated object volume determined from 2 μm overlapping optical sections through each cell: Fold-change in Rap1 immunofluorescence in gonadotropes (GnRH-treated/vehicle-treated) fixed at either 15 min (open bar) or 60 min (hatched bar). * P < 0.03 compared to cells fixed at 15 min.
4. Discussion
Pituitary gonadotropes must decipher instructions received from GnRH receptor activation to respond with the appropriate amount and pattern of gonadotropin secretion. We have investigated the gene network that is activated shortly after GnRH stimulation of gonadotrope-enriched primary cultures of rat pituitary cells, as these early induced genes may encode proteins that notably modulate cellular signaling. We identified 33 up-regulated genes, among which rap1b, a widely expressed small GTPase implicated in a number of cellular processes, was novel. We provided evidence for GnRH-induced increase in both Rap1b transcript and protein, and confirmed the induction of rap1b gene expression at the single gonadotrope level.
Among the up-regulated genes identified in this work, there are genes encoding transcription factors (e.g.c-Fos, FosB, and Egr2), proteins involved in signal transduction or in the regulation of signal transduction (e.g. Rap1b, and Rgs4), proteins involved in transport (e.g. Bet1, Rab1, Slc38a2), cell adhesion (e.g. Cldn4, Itga6), and various other processes. By contrast, more than half of the 28 genes whose expression has been found to increase in Lβ T2 cells in response to GnRH are transcription factors (e.g. Egr2, c-Fos, Klf4); the remainder are modulators of signal transduction (e.g. Rgs2) and cytoskeletal proteins (e.g. γ-actin and transgelin) [3]. Importantly, our microarray data for primary pituitary cells were validated by an independent determination of the expression levels of a subset of genes by real-time PCR. In the present study, most GnRH-regulated genes showed less than a 2-fold induction as determined by microarray analysis (see Table 1). The greatest change detected for primary gonadotropes is 4-fold, as compared to ~50-fold in Lβ T2 cells [3]. Species differences between rat and mouse, primary cells vs. transformed cells, and duration of GnRH treatment − 40 minutes in the primary gonadotropes vs. 1 hour in the Lβ T2 cell line - may account for these discrepancies. Additionally, despite a marked enrichment in gonadotrope cells, our primary pituitary cultures may still be contaminated by other secretory cell types, thereby resulting in some degree of cell heterogeneity. We conjecture that high basal expression of a given gene in the contaminating cells could potentially mask the detection of gene induction in the gonadotropes. Because differences in the nature of contaminating cells are a potential source of variability between replicated experiments, we performed a total of six replicate experiments in order to minimize the effect of sample variance. More generally, the degree of variability that subsists within any cell population, not to mention noise or random fluctuations among identical cell types, contribute to cell-to-cell variation in gene expression; this can be eliminated by monitoring gene expression at the single-cell level (for review, see [37]). We therefore optimized and implemented a single-cell quantitative real-time PCR assay, which unambiguously substantiated the up-regulation of rap1b in single rat gonadotropes. Despite the cellular heterogeneity of primary pituitary cultures, employing them to study the actions of GnRH is physiologically more pertinent than using the Lβ T2 cell line, as primary cultures mimic in vivo conditions more closely than transformed cells. Supporting this concept, we previously demonstrated that rat or mouse pituitary cultures responded to estradiol stimulation with an increase in progesterone receptor expression, while Lβ T2 cells did not. Furthermore, GnRH-stimulated LH secretion was augmented by progesterone in rat or mouse pituitary cells, but not in Lβ T2 gonadotropes [14]. This finding is consistent with the absence of GnRH self-priming in the immortalized cell line [13] and the notion that the progesterone receptor plays a role in GnRH self-priming [24]. Pituitary adenylate cyclase-activating polypeptide (PACAP) was previously shown to stimulate the CGA and LHβ genes and reduce FSHβ mRNA levels in primary pituitary cells [38], but not in Lβ T2 cells [39,40]; thus, based on the data obtained in primary pituitary cells, PACAP was thought to enhance responsiveness to GnRH and regulate the differential secretion of LH and FSH (for review, see [41]). Although PACAP’s role was not examined in our study, this peptide has been shown to signal through PKA-independent and as well as PKA-dependent pathways. For example, select neurotropic actions of PACAP have been shown to operate through cAMP activation of EPAC, a guanine nucleotide exchange factor (GEF) and the GTPase, Rap1 (reviewed in [53]).
In the present work, all of the rat gonadotrope early genes found to be up-regulated by GnRH as determined by real-time PCR were previously shown to be modulated by GnRH in Lβ T2 cells following a one-hour GnRH treatment [1,3,5,6], except for nr4a3 and rap1b. Nr4a3, also known as neuron-derived orphan nuclear receptor 1 (or NOR-1), is a member of the nuclear hormone receptor superfamily; it is constitutively active, operates in a ligand-independent manner, and is expressed in a variety of energy-dependent tissues such as skeletal muscle, brain, adipose tissue, brain, liver, therefore suggesting a potential role in energy metabolism (for review, see [42]). Rap1, a member of the Ras superfamily of small GTP-binding proteins, is involved in a diverse range of functions, e.g., cell adhesion, polarity, and proliferation [43–45]. Activation of Rap1 involves conversion of the GDP-bound to the GTP-bound form by GEFs, such as Epac1 and Epac2, which are regulated by cAMP, or CalDAG-GEF, which is activated by Ca2+ and diacylglycerol [43]. Rap1 has two isoforms, Rap1a and Rap1b, which seem to mediate similar but cell type-specific actions (for review, see [43,46,47]). Rap1b is the predominant isoform and the most abundant Ras family member in platelets [48]. In fact, rap1b knockout mice exhibit 85% lethality, and in surviving rap1b-null mice Rap1b deficiency results in a bleeding defect due to abnormal platelet function [49]. Rap1b was also shown to stimulate mitogenesis in the thyroid both in vitro and in vivo in a cAMP-dependent manner [44,45].
Of more relevance to gonadotrope function, Rap1 also has a regulatory role in protein secretion, specifically amylase release from parotid and pancreatic acinar cells, cAMP-stimulated neurotensin secretion in an endocrine cell line, and insulin secretion from pancreatic beta cells [31–34]. A parallel potentially exists between the role of Rap1b in insulin secretion and in the priming activity for LH secretion in gonadotropes. Glucagon-like peptide-1 (GLP-1), an intestinally derived incretin, potentiates or primes glucose-stimulated insulin secretion from pancreatic beta cells, and Rap1b has been implicated in this process [34,50]. GLP-1 receptor agonists are important tools in the treatment of type 2 diabetes mellitus, and studies to delineate the signaling pathway have focused on regulation of Rap1b activation and its downstream targets [50,51]. Epac2, the cAMP-regulated activator of Rap1b, is reported to have a primary role in GLP-1 action on insulin secretion [34,50,52]. Unresolved is whether the potentiating action of GLP-1-induced cAMP elevation in pancreatic beta cells operates solely through the Epac2 and Rap1b mechanisms or additionally through a PKA pathway [50,53].
To our knowledge, the effect of GLP-1 or insulin secretagogues on upregulation of rap1b transcripts has not been examined in pancreatic beta cells. In our studies, we observed that cAMP stimulates rap1b gene expression in single gonadotropes. GnRH previously has been shown to stimulate an early increase in cAMP in rat pituitary cells [35], and elevated cAMP, which by itself does not stimulate LH secretion, can substitute for the initial GnRH pulse to potentiate subsequent responses to secretagogues [24,54]. Although our results are consistent with cAMP involvement in GnRH priming, they do not address whether cAMP directly affects Epac/Rap1 activation in addition to increasing Rap1b.
Subcellular compartmentalization is a spatial regulator in signaling pathways, and Epac2 has been found associated with pancreatic beta cell exocytotic machinery (reviewed in [52]) and Rap1 localizes with secretory granule membranes in parotid and pancreatic acini as well as pancreatic beta cells [31,33,34]. Consistent with this, for gonadotropes we observed a distribution of Rap1 immunofluorescent complexes in areas compact with LH secretory granules that was prominent 1 h after GnRH stimulation. Further studies are required to elucidate its precise role in GnRH-stimulated gonadotropes, but we hypothesize that the small GTPase, Rap1b, is a candidate contributor to GnRH priming.
5. Conclusion
This study demonstrates that GnRH up-regulates Rap1b shortly after stimulation of rat primary gonadotropes. Owing to an optimized amplification of the RNA in gonadotrope-enriched primary pituitary cells, we were able to carry out high-throughput gene expression analysis and identify the early genes that are regulated by GnRH in a highly physiological context. Results were independently validated by real-time PCR. Further, we showed that GnRH induced a significant increase in rap1b transcript levels in single gonadotrope cells, thereby corroborating our findings in cell populations that may exhibit cell-to-cell variation, and to some extent cell heterogeneity. This early increase in rap1b transcript was followed by an increase in the protein detected in the vicinity of LH secretory granules. Rap1b was previously proposed to potentiate protein secretion in other cell types and may be directly implicated in gonadotropin secretion. Further studies are required to elucidate its precise role in GnRH-stimulated gonadotropes, but we hypothesize that the small GTPase, Rap1b, is a candidate contributor to GnRH priming.
Highlights.
Optimized RNA amplification allowed microarray analysis of GnRH-stimulated gonadotrope-enriched primary pituitary cultures.
rap1b is a novel early gene induced by GnRH in primary gonadotropes.
Single-cell real-time PCR demonstrated the rap1b is induced by GnRH in the gonadotropes.
Rap1b protein was markedly increased in GnRH-stimulated primary gonadotropes.
Acknowledgments
Our work was supported by the National Institutes of Health Grants DK46943 (SCS), DK66606 (JLT, SCS), and HD12137 (JLT, DWW). We are grateful to Dr. Pamela L. Mellon for providing the Lβ T2 cells. We are indebted to Dr. Tearina T. Chu for kindly providing access to microarray instrumentation. Use of the ABI Prism 7900 at UC Davis was supported by the Nutrition Research Unit, NIH DK35747. We thank Dr. Lidija Ivic for her help in figure preparation.
References
- 1.Kakar SS, Winters SJ, Zacharias W, Miller DM, Flynn S. Identification of distinct gene expression profiles associated with treatment of LbetaT2 cells with gonadotropin-releasing hormone agonist using microarray analysis. Gene. 2003;308:67–77. doi: 10.1016/s0378-1119(03)00446-3. [DOI] [PubMed] [Google Scholar]
- 2.Lawson MA, Tsutsumi R, Zhang H, Talukdar I, Butler BK, Santos SJ, Mellon PL, Webster NJ. Pulse sensitivity of the luteinizing hormone beta promoter is determined by a negative feedback loop Involving early growth response-1 and Ngfi-A binding protein 1 and 2. Mol Endocrinol. 2007;21:1175–91. doi: 10.1210/me.2006-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wurmbach E, Yuen T, Ebersole BJ, Sealfon SC. Gonadotropin-releasing hormone receptor-coupled gene network organization. J Biol Chem. 2001;276:47195–201. doi: 10.1074/jbc.M108716200. [DOI] [PubMed] [Google Scholar]
- 4.Wurmbach E, Yuen T, Sealfon SC. Focused microarray analysis. Methods. 2003;31:306–16. doi: 10.1016/s1046-2023(03)00161-0. [DOI] [PubMed] [Google Scholar]
- 5.Yuen T, Wurmbach E, Ebersole BJ, Ruf F, Pfeffer RL, Sealfon SC. Coupling of GnRH concentration and the GnRH receptor-activated gene program. Mol Endocrinol. 2002;16:1145–53. doi: 10.1210/mend.16.6.0853. [DOI] [PubMed] [Google Scholar]
- 6.Dorn C, Ou Q, Svaren J, Crawford PA, Sadovsky Y. Activation of luteinizing hormone beta gene by gonadotropin-releasing hormone requires the synergy of early growth response-1 and steroidogenic factor-1. J Biol Chem. 1999;274:13870–6. doi: 10.1074/jbc.274.20.13870. [DOI] [PubMed] [Google Scholar]
- 7.Kaiser UB, Halvorson LM, Chen MT. Sp1, steroidogenic factor 1 (SF-1), and early growth response protein 1 (egr-1) binding sites form a tripartite gonadotropin-releasing hormone response element in the rat luteinizing hormone-beta gene promoter: an integral role for SF-1. Mol Endocrinol. 2000;14:1235–45. doi: 10.1210/mend.14.8.0507. [DOI] [PubMed] [Google Scholar]
- 8.Lee SL, Sadovsky Y, Swirnoff AH, Polish JA, Goda P, Gavrilina G, Milbrandt J. Luteinizing hormone deficiency and female infertility in mice lacking the transcription factor NGFI-A (Egr-1) Science. 1996;273:1219–21. doi: 10.1126/science.273.5279.1219. [DOI] [PubMed] [Google Scholar]
- 9.Mouillet JF, Sonnenberg-Hirche C, Yan X, Sadovsky Y. p300 regulates the synergy of steroidogenic factor-1 and early growth response-1 in activating luteinizing hormone-beta subunit gene. J Biol Chem. 2004;279:7832–9. doi: 10.1074/jbc.M312574200. [DOI] [PubMed] [Google Scholar]
- 10.Haisenleder DJ, Ferris HA, Shupnik MA. The calcium component of gonadotropin-releasing hormone-stimulated luteinizing hormone subunit gene transcription is mediated by calcium/calmodulin-dependent protein kinase type II. Endocrinology. 2003;144:2409–16. doi: 10.1210/en.2002-0013. [DOI] [PubMed] [Google Scholar]
- 11.Haisenleder DJ, Workman LJ, Burger LL, Aylor KW, Dalkin AC, Marshall JC. Gonadotropin subunit transcriptional responses to calcium signals in the rat: evidence for regulation by pulse frequency. Biol Reprod. 2001;65:1789–93. doi: 10.1095/biolreprod65.6.1789. [DOI] [PubMed] [Google Scholar]
- 12.Liu F, Austin DA, Mellon PL, Olefsky JM, Webster NJ. GnRH activates ERK1/2 leading to the induction of c-fos and LHbeta protein expression in LbetaT2 cells. Mol Endocrinol. 2002;16:419–34. doi: 10.1210/mend.16.3.0791. [DOI] [PubMed] [Google Scholar]
- 13.Turgeon JL, Kimura Y, Waring DW, Mellon PL. Steroid and pulsatile gonadotropin-releasing hormone (GnRH) regulation of luteinizing hormone and GnRH receptor in a novel gonadotrope cell line. Mol Endocrinol. 1996;10:439–50. doi: 10.1210/mend.10.4.8721988. [DOI] [PubMed] [Google Scholar]
- 14.Turgeon JL, Waring DW. Differential expression and regulation of progesterone receptor isoforms in rat and mouse pituitary cells and LbetaT2 gonadotropes. J Endocrinol. 2006;190:837–46. doi: 10.1677/joe.1.06923. [DOI] [PubMed] [Google Scholar]
- 15.Turgeon JL, Waring DW. The timing of progesterone-induced ribonucleic acid and protein synthesis for augmentation of luteinizing hormone secretion. Endocrinology. 1991;129:3234–9. doi: 10.1210/endo-129-6-3234. [DOI] [PubMed] [Google Scholar]
- 16.Turgeon JL, Waring DW. Luteinizing hormone secretion from wild-type and progesterone receptor knockout mouse anterior pituitary cells. Endocrinology. 2001;142:3108–15. doi: 10.1210/endo.142.7.8282. [DOI] [PubMed] [Google Scholar]
- 17.Waring DW, Turgeon JL. LHRH self priming of gonadotrophin secretion: time course of development. Am J Physiol. 1983;244:C410–8. doi: 10.1152/ajpcell.1983.244.5.C410. [DOI] [PubMed] [Google Scholar]
- 18.Turgeon JL, Waring DW. Functional cross-talk between receptors for peptide and steroid hormones. Trends Endocrinol Metab. 1992;3:360–5. doi: 10.1016/1043-2760(92)90002-i. [DOI] [PubMed] [Google Scholar]
- 19.Fink G. The self-priming effect of LHRH: a unique servomechanism and possible cellular model for memory. Front Neuroendocrinol. 1995;16:183–90. doi: 10.1006/frne.1995.1006. [DOI] [PubMed] [Google Scholar]
- 20.Chappell PE, Schneider JS, Kim P, Xu M, Lydon JP, O'Malley BW, Levine JE. Absence of gonadotropin surges and gonadotropin-releasing hormone self-priming in ovariectomized (OVX), estrogen (E2)-treated, progesterone receptor knockout (PRKO) mice. Endocrinology. 1999;140:3653–8. doi: 10.1210/endo.140.8.6895. [DOI] [PubMed] [Google Scholar]
- 21.Hoff JD, Lasley BL, Yen SS. The functional relationship between priming and releasing actions of luteinizing hormone-releasing hormone. J Clin Endocrinol Metab. 1979;49:8–11. doi: 10.1210/jcem-49-1-8. [DOI] [PubMed] [Google Scholar]
- 22.Sollenberger MJ, Carlsen EC, Booth RA, Jr, Johnson ML, Veldhuis JD, Evans WS. Nature of gonadotropin-releasing hormone self-priming of luteinizing hormone secretion during the normal menstrual cycle. Am J Obstet Gynecol. 1990;163:1529–34. doi: 10.1016/0002-9378(90)90620-m. [DOI] [PubMed] [Google Scholar]
- 23.Matteri RL, Roser JF, Baldwin DM, Lipovetsky V, Papkoff H. Characterization of a monoclonal antibody which detects luteinizing hormone from diverse mammalian species. Domest Anim Endocrinol. 1987;4:157–65. doi: 10.1016/0739-7240(87)90011-7. [DOI] [PubMed] [Google Scholar]
- 24.Turgeon JL, Waring DW. Activation of the progesterone receptor by the gonadotropin-releasing hormone self-priming signaling pathway. Mol Endocrinol. 1994;8:860–9. doi: 10.1210/mend.8.7.7984148. [DOI] [PubMed] [Google Scholar]
- 25.Turgeon JL, Waring DW. Rapid augmentation by progesterone of agonist-stimulated luteinizing hormone secretion by cultured pituitary cells. Endocrinology. 1990;127:773–80. doi: 10.1210/endo-127-2-773. [DOI] [PubMed] [Google Scholar]
- 26.Waring DW, Turgeon JL. Ca2+-activated K+ channels in gonadotropin-releasing hormone-stimulated mouse gonadotrophs. Endocrinology. 2009;150:2264–72. doi: 10.1210/en.2008-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitra RS, Zhang Z, Henson BS, Kurnit DM, Carey TE, D'Silva NJ. Rap1A and rap1B ras-family proteins are prominently expressed in the nucleus of squamous carcinomas: nuclear translocation of GTP-bound active form. Oncogene. 2003;22:6243–56. doi: 10.1038/sj.onc.1206534. [DOI] [PubMed] [Google Scholar]
- 28.Turgeon JL, Shyamala G, Waring DW. PR localization and anterior pituitary cell populations in vitro in ovariectomized wild-type and PR-knockout mice. Endocrinology. 2001;142:4479–85. doi: 10.1210/endo.142.10.8425. [DOI] [PubMed] [Google Scholar]
- 29.Che S, Ginsberg SD. Amplification of RNA transcripts using terminal continuation. Lab Invest. 2004;84:131–7. doi: 10.1038/labinvest.3700005. [DOI] [PubMed] [Google Scholar]
- 30.Antequera F, Bird A. Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci U S A. 1993;90:11995–9. doi: 10.1073/pnas.90.24.11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.D'Silva NJ, Jacobson KL, Ott SM, Watson EL. Beta-adrenergic-induced cytosolic redistribution of Rap1 in rat parotid acini: role in secretion. Am J Physiol. 1998;274:C1667–73. doi: 10.1152/ajpcell.1998.274.6.C1667. [DOI] [PubMed] [Google Scholar]
- 32.Li J, O'Connor KL, Cheng X, Mei FC, Uchida T, Townsend CM, Jr, Evers BM. Cyclic adenosine 5'-monophosphate-stimulated neurotensin secretion is mediated through Rap1 downstream of both Epac and protein kinase A signaling pathways. Mol Endocrinol. 2007;21:159–71. doi: 10.1210/me.2006-0340. [DOI] [PubMed] [Google Scholar]
- 33.Sabbatini ME, Chen X, Ernst SA, Williams JA. Rap1 activation plays a regulatory role in pancreatic amylase secretion. J Biol Chem. 2008;283:23884–94. doi: 10.1074/jbc.M800754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M, Zhang C, Tamamoto A, Satoh T, Miyazaki J, Seino S. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci U S A. 2007;104:19333–8. doi: 10.1073/pnas.0707054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waring DW, Turgeon JL. A pathway for luteinizing hormone releasing-hormone self-potentiation: cross-talk with the progesterone receptor. Endocrinology. 1992;130:3275–82. doi: 10.1210/endo.130.6.1317780. [DOI] [PubMed] [Google Scholar]
- 36.Thomas P, Waring DW. Modulation of stimulus-secretion coupling in single rat gonadotrophs. J Physiol. 1997;504 ( Pt 3):705–19. doi: 10.1111/j.1469-7793.1997.705bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Longo D, Hasty J. Dynamics of single-cell gene expression. Mol Syst Biol. 2006;2:64. doi: 10.1038/msb4100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsujii T, Ishizaka K, Winters SJ. Effects of pituitary adenylate cyclase-activating polypeptide on gonadotropin secretion and subunit messenger ribonucleic acids in perifused rat pituitary cells. Endocrinology. 1994;135:826–33. doi: 10.1210/endo.135.3.7915230. [DOI] [PubMed] [Google Scholar]
- 39.Fujii Y, Okada Y, Moore JP, Jr, Dalkin AC, Winters SJ. Evidence that PACAP and GnRH down-regulate follicle-stimulating hormone-beta mRNA levels by stimulating follistatin gene expression: effects on folliculostellate cells, gonadotrophs and LbetaT2 gonadotroph cells. Mol Cell Endocrinol. 2002;192:55–64. doi: 10.1016/s0303-7207(02)00109-0. [DOI] [PubMed] [Google Scholar]
- 40.Kanasaki H, Mutiara S, Oride A, Purwana IN, Miyazaki K. Pulse frequency-dependent gonadotropin gene expression by adenylate cyclase-activating polypeptide 1 in perifused mouse pituitary gonadotroph LbetaT2 cells. Biol Reprod. 2009;81:465–72. doi: 10.1095/biolreprod.108.074765. [DOI] [PubMed] [Google Scholar]
- 41.Winters SJ, Moore JP., Jr PACAP, an autocrine/paracrine regulator of gonadotrophs. Biol Reprod. 2011;84:844–50. doi: 10.1095/biolreprod.110.087593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pearen MA, Muscat GE. Minireview: Nuclear hormone receptor 4A signaling: implications for metabolic disease. Mol Endocrinol. 2010;24:1891–903. doi: 10.1210/me.2010-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bos JL, de Rooij J, Reedquist KA. Rap1 signalling: adhering to new models. Nat Rev Mol Cell Biol. 2001;2:369–77. doi: 10.1038/35073073. [DOI] [PubMed] [Google Scholar]
- 44.Ribeiro-Neto F, Leon A, Urbani-Brocard J, Lou L, Nyska A, Altschuler DL. cAMP-dependent oncogenic action of Rap1b in the thyroid gland. J Biol Chem. 2004;279:46868–75. doi: 10.1074/jbc.M406858200. [DOI] [PubMed] [Google Scholar]
- 45.Ribeiro-Neto F, Urbani J, Lemee N, Lou L, Altschuler DL. On the mitogenic properties of Rap1b: cAMP-induced G(1)/S entry requires activated and phosphorylated Rap1b. Proc Natl Acad Sci U S A. 2002;99:5418–23. doi: 10.1073/pnas.082122499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raaijmakers JH, Bos JL. Specificity in Ras and Rap signaling. J Biol Chem. 2009;284:10995–9. doi: 10.1074/jbc.R800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stork PJ. Does Rap1 deserve a bad Rap? Trends Biochem Sci. 2003;28:267–75. doi: 10.1016/S0968-0004(03)00087-2. [DOI] [PubMed] [Google Scholar]
- 48.Klinz FJ, Seifert R, Schwaner I, Gausepohl H, Frank R, Schultz G. Generation of specific antibodies against the rap1A, rap1B and rap2 small GTP-binding proteins. Analysis of rap and ras proteins in membranes from mammalian cells. Eur J Biochem. 1992;207:207–13. doi: 10.1111/j.1432-1033.1992.tb17039.x. [DOI] [PubMed] [Google Scholar]
- 49.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH, White GC., 2nd Rap1b is required for normal platelet function and hemostasis in mice. J Clin Invest. 2005;115:680–7. doi: 10.1172/JCI22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leech CA, Chepurny OG, Holz GG. Epac2-dependent rap1 activation and the control of islet insulin secretion by glucagon-like peptide-1. Vitam Horm. 2010;84:279–302. doi: 10.1016/B978-0-12-381517-0.00010-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dzhura I, Chepurny OG, Kelley GG, Leech CA, Roe MW, Dzhura E, Afshari P, Malik S, Rindler MJ, Xu X, Lu Y, Smrcka AV, Holz GG. Epac2-dependent mobilization of intracellular Ca(2)+ by glucagon-like peptide-1 receptor agonist exendin-4 is disrupted in beta-cells of phospholipase C-epsilon knockout mice. J Physiol. 2010;588:4871–89. doi: 10.1113/jphysiol.2010.198424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol. 2010;50:355–75. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- 53.Hatakeyama H, Takahashi N, Kishimoto T, Nemoto T, Kasai H. Two cAMP-dependent pathways differentially regulate exocytosis of large dense-core and small vesicles in mouse beta-cells. J Physiol. 2007;582:1087–98. doi: 10.1113/jphysiol.2007.135228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Turgeon JL, Waring DW. cAMP augmentation of secretagogue-induced luteinizing hormone secretion. Am J Physiol. 1986;250:E62–8. doi: 10.1152/ajpendo.1986.250.1.E62. [DOI] [PubMed] [Google Scholar]