

Abstract
Since late 2010, the outbreak of porcine epidemic diarrhea (PED) in China has resulted in the deaths of millions of suckling piglets. The main cause of the disease outbreak was unknown. In this study, partial spike (S), ORF3, and membrane (M) genes amplified from these variants were sequenced and analyzed. The results showed that the variants could be clustered into one to three subgroups and suggested that S genes were variable, while M genes were relatively conserved. Moreover, in comparison with the vaccine strain CV777, sequence alignment analyses revealed that the S genes of the newly isolated strains contained several mutations at the aa level. It is possible that these mutations have changed the hydrophobicity of the S protein and influenced the viral antigenicity and virulence. Interestingly, homology analyses based on ORF3 demonstrated that the isolates had an intact opening reading frame (ORF), which were different from the attenuated DR13 strain. In conclusion, the widespread PED virus (PEDV) isolates had virulent characteristics. Additionally, the high degree of variation in the genes, particularly S genes, might provide an explanation for the poor immunity and rapid spread of the disease.
1. Introduction
PED, caused by PEDV, is a highly contagious, enteric disease of swine. The main symptoms are vomiting, dehydration, and a high mortality in piglets [1]. PED is a serious health problem in suckling and nursing pigs that are typically 1-2 weeks of age. The mortality rate can be as high as 100% in 10-day-old or younger piglets. Pigs of all ages are susceptible to PEDV infection [2, 3]. PED occurrence has been reported in many countries, including Japan, Korea, France, Belgium, and Switzerland [4–6]; it was first reported in China in 1976 [7]. Since late 2010, millions of piglets have been killed in China during PED outbreaks, which significantly affected the pork meat supply and price [8]. PED becomes a severe disease and has led to a significant loss in the swine industry.
PEDV has a positive-sense single-stranded RNA that encodes major proteins, including spike (S), membrane (M), small membrane (sM), nucleocapsid (N), and envelope (E), translated from its subgenomic mRNA [9]. The S protein of PEDV is an important site of viral neutralization [10, 11]. The M protein, that is, structural M glycoprotein, plays an important role in the assembly process of the viral nucleocapsid and membrane [12]. Previous studies had revealed that the 624-nucleotide (nt) ORF3 in attenuated-type PEDVs contains 51 nt deletions compared to wild-type PEDVs, and this deletion region might be important for PEDV pathogenicity [13, 14].
The commercially produced vaccines, that is, inactivated transmissible gastroenteritis (TGEV H) and porcine epidemic diarrhea (CV777), were used to protect swine from the PED and TGE diseases in China. Despite the administration of the vaccine, PED still affected farms in many provinces in late 2010 [8]. The reason for this remains unknown. To understand the genetic variations of PEDV and explain the remaining instances of PEDV, PEDV RNA was extracted directly from nasal and rectal samples of naturally infected piglets. The partial S, ORF3, M genes were amplified, sequenced, and analyzed. The amino acid (aa) sequences were predicted, based on which the phylogenetic trees were constructed and the hydrophobicity was analyzed. The similarities and differences between current and reference PEDVs were elucidated. These analyses were also useful for further molecular biology studies of PEDV strains that were prevalent in China.
2. Materials and Methods
2.1. Samples
A total of 70 rectal swab samples were collected from 70 piglets infected with PEDV from October 2010 to March 2012. These infected piglets were chosen from 16 different swine farms located in 6 different provinces in China. Samples were subgrouped according to the geographic regions (Table 1). The rectal swabs were soaked in PBS buffer supplemented with penicillin G (10,000 IU/mL) and streptomycin (2 mg/mL) and centrifuged at 1,500 ×g for 15 min to collect the supernatant fluids. The supernatant fluids were stored at –80°C until used.
Table 1.
Detail information of present and reference sequences used in Figures 1(a), 2(a), and 3(a) including country/district and collection date.
| Country/District | Strain | Collection date |
|---|---|---|
| Nanning, Guangxi | GXNN04 | Nov-2010 |
| Heyuan, Guangdong | GDHY01 | Dec-2010 |
| Jiangmen, Guangdong | GDJM02 | Feb-2011 |
| Zhanjiang, Guangdong | GDZJ05 | Mar-2011 |
| Nanyang, Henan | HNNY03 | Apr-2011 |
| Zhengzhou, Henan | HNZZ01 | May-2011 |
| Cenzhou, Hunan | HNCZ02 | May-2011 |
| Haikou, Hainan | HNHK03 | Jun-2011 |
| Zhongluotan, Guangdong | GDZLT01 | Jul-2011 |
| Zhaoqing, Guangdong | GDZQ03 | Sep-2011 |
| Foshan, Guangdong | GDFS04 | Oct-2011 |
| Yangjiang, Guangdong | GDYJ02 | Dec-2011 |
| Heshan, Guangdong | GDHS02 | Mar-2012 |
| Changsha, Hunan | HNCS01 | Feb-2012 |
| Guangzhou, Guangdong | GDXMS01 | Mar-2012 |
| Guangzhou, Guangdong | GDXMS02 | Mar-2012 |
| Korea | Chinju99 | 1999 |
| Lanzhou (China) | LZC | Dec-2006 |
| Heilongjiang (China) | LJB/03 | 2003 |
| Belgium | CV777 | 1977 |
| Korea | DR13 | unknown |
| Korea | Attenuated DR13 | unknown |
| China | CH/S | 1986 |
2.2. RNA Extraction
RNA was extracted from the supernatant using TRIzol Reagent (Invitrogen Corp., Carlsbad, CA, USA) following the manufacturer's instructions. Briefly, the supernatant (200 μL) containing PEDV was mixed with 1 mL of TRIzol Reagent. Then, 200 μL of chloroform was added to the mixture, and the suspension was centrifuged for 15 min at 12,000 ×g. The RNA-containing aqueous phase was precipitated with the same volume isopropanol, maintained at –20°C for 1 h, and centrifuged for 10 min at 12,000 ×g. The resulted RNA pellet was washed with 1 mL of 75% ethanol, centrifuged for 10 min at 12,000 ×g and dried, and resuspended in 30 μL of diethyl-pyrocarbonate- (DEPC-) treated deionized water.
2.3. Primers
Pairs of sense and antisense primers were designed and aligned based on the nt sequence of the S, M, ORF3 genes from the GenBank database (National Center for Biotechnology Information, USA). These primers were used to amplify the PEDV gene of interest. The primer sequences and other details are shown in Table 2.
Table 2.
Primers used in RT-PCR for PEDV spike, ORF3, and membrane genes.
| Primer | Nucleotide sequence (5′-3′) | Size of product (bp) |
|---|---|---|
| S-F | TTCTGAGTCACGAACAGCCA | 651 |
| S-R | CATATGCAGCCTGCTCTGAA | |
| ORF3-F | TCCTAGACTTCAACCTTACG | 833 |
| ORF3-R | GGTGACAAGTGAAGCACAGA | |
| M-F | GTCTTACATGCGAATTGACC | 808 |
| M-R | GGCATAGAGAGATAATGGCA |
2.4. RT-PCR and Sequencing
RT-PCR was performed using PrimeScript One Step RT-PCR Kit Ver. 2 (TaKaRa Biotechnology Co., Ltd., Dalian, China). Primers S-F and S-R, ORF3-F and ORF3-R, and M-F and M-R, as shown in Table 2, were used to amplify the corresponding partial S, ORF3, and M genes from PEDV, respectively. For RT-PCR, 3 μL of extracted viral RNA was mixed with a reaction mixture containing 25 μL 2 × 1 Step Buffer, 2 μL each specific primer (10 pmol), 2 μL PrimeScript 1 Step Enzyme Mix, and 16 μL RNase Free water. The amplifications for the S, ORF3 genes were performed with reverse transcription at 50°C for 30 min and predenaturation at 95°C for 5 min, followed by 35 cycles of denaturation at 95°C for 1 min, annealing at 50°C for 1 min, and extension at 72°C for 1 min, and a final extension step of 72°C for 10 min. For the M gene, the amplification conditions were the same as above; however, the annealing temperature used was 56°C for 1 min. RT-PCR products were visualized via electrophoresis in a 1.5% agarose gel containing ethidium bromide. Bands of the correct size were excised and purified using TaKaRa MiniBEST Agarose Gel DNA Extraction Kit Ver. 3.0 (TaKaRa Biotechnology Co., Ltd., Dalian, China) following the manufacturer's instructions. The purified products were sequenced by BGI Biological Engineering Technology & Services Co., Ltd. (China, Shenzhen).
2.5. Sequence Analysis
Nucleotide and deduced aa sequences were analyzed using the CLUSTALX v1.83, Bioedit v7.0.5.2 programs, and MegAlign software for alignment and sequence analysis. S, M, ORF3 nt and deduced aa sequences were compared with the PEDV reference strains listed in Table 2. The resulting subsets were edited manually.
2.6. Phylogenetic Analysis
Using a neighbor-joining (NJ) method in MEGA version 5.1, phylogenetic trees were generated using an alignment of S, M, ORF3 gene nt and the aa sequences deduced from the reference PEDVs. To assess the relative support for each clade, bootstrap values were calculated from 1,000 analysis replicates, and the cut-off point for bootstrap replication was 50%.
3. Results
3.1. Amplification and Sequencing of S, ORF3, and M Genes
To obtain the nt sequence, the partial S, ORF3, M genes were amplified in PED positive samples using one-step RT-PCR. The predicted 651-bp, 833-bp, and 808-bp bands for S, ORF3, M genes, respectively, were detected in the resulted PCR products. Subsequently, these products were purified and sequenced.
3.2. S Gene Analysis
The sequence alignment results showed that the sequence homogeneity between the reference isolate and the newly isolated variants (88.7–97.3% for nt and 85.2–98.0% for aa) was lower than that observed within the newly isolated strains alone (96.7–99.8% for nt and 94.6–100% for aa). These newly isolated strains had nt homology of 94.6–97.3%, 88.6–91.1%, 92.8–95.5%, and aa homology of 94.4–98.0%, 85.2–88.8%, 91.4–95.4% when comparing to CV777, Chinju99, and CH/S sequences (data not shown).
A phylogenetic tree was generated based on the deduced aa sequences of the partial S gene. In all phylogenies, the reference aa sequences, observed in samples collected from previous outbreaks, tended to cluster near the root but had a poorly resolved branching structure. This was likely due to relatively low sequence divergence. In contrast, results analyzed using NJ method with bootstrap support indicated that viruses from more recent infections formed three well-supported lineages that tended to cluster far from the root. As expected. These lineages were denoted as groups I, II, and III in the S phylogenies (Figure 1(a)).
Figure 1.
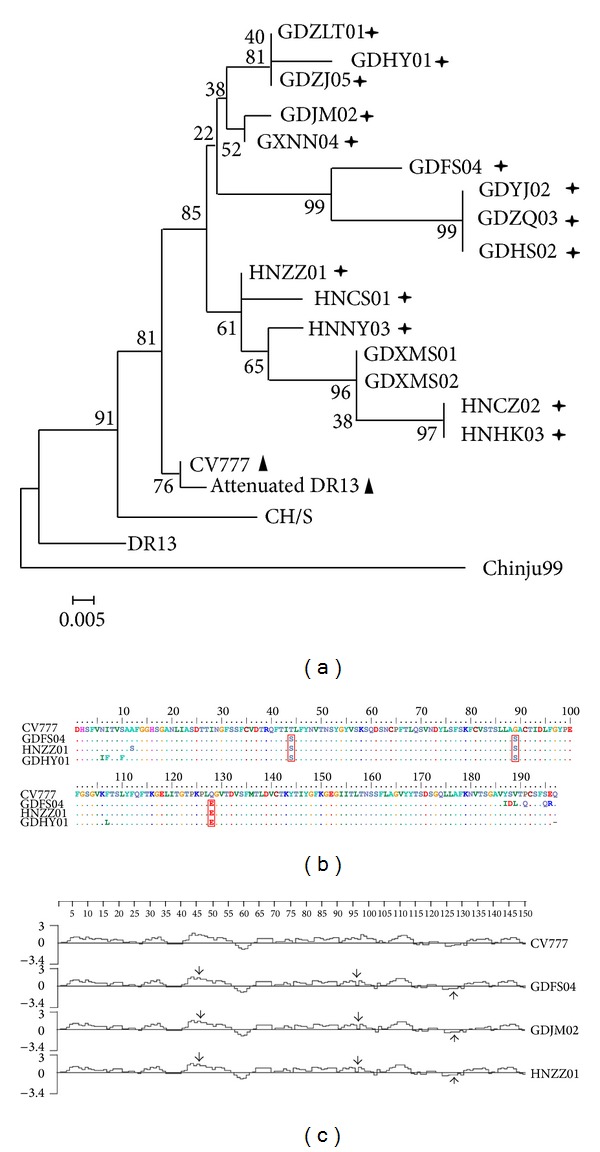
Comprehensive analysis of S genes from current PEDV with reference strains. (a) Phylogenetic analysis based on S aa; (b) point mutations analysis of S aa; (c) hydrophobic analysis of S aa. Phylogenetic tree is constructed by the neighbor-joining method with the MEGA 4.0 program. The percentage bootstrap support (per 1000 replicates) is indicated by the values at each node. Cross asterisk indicated present isolates; triangle indicated attenuated strains; box indicated point mutations.
To further investigate the nt and deduced aa substitutions, partial S sequences amplified from 16 isolates were sequenced and sorted according to the collection date and district. The fragments were aligned at position 1523–2115 of the complete S gene using Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi). In this study, three aa mutations were detected at positions 44 (T → S), 89 (G → S), and 128 (Q → E) (Figure 1(b)). Sun et al. [15, 16] suggested that this region might be considered an important immunodominant domain for PEDV. The hydrophobicity of the deduced aa for these newly isolated strains was then analyzed, and the mutation impact on the hydrophobicity of S protein was confirmed (Figure 1(c)).
3.3. ORF3 Gene Analysis
The nt sequence alignment results showed that the sequence homogeneity among the newly isolated strains (96.1–100%) was higher than that observed between the attenuated-type PEDVs and the newly isolated strains (89.6–98.8%). Likewise, the aa sequence alignment results showed a similar pattern (i.e., 96.7–100% among the current isolates and 91.1–100% between the attenuated-type PEDVs and the current isolates). More precisely, the newly isolated strain DR13 showed higher homology (97.2–98.7% for nt and 96.7–99.4% for aa) than both attenuated DR13 (89.6–90.7% for nt and 91.1–93.3% for aa) and CV777 (89.9–91% for nt and 91.1–93.3% for aa) (data not shown).
A phylogenic tree was constructed using the ORF3 aa sequences from the newly isolated strains and the references isolates. All of the sequences were divided into four phylogenetic groups: group I contained current isolates and reference strain CH/S, group II contained reference strains DR13 and Chinju99, group III contained attenuated DR13 and CV777, and group IV current isolated strains GXNN04, GDJM02, GDHY01, and GDZJ05. Further analysis showed that the current isolated groups (I and IV) were independent of the reference strain groups (II and III) (Figure 2(a)).
Figure 2.
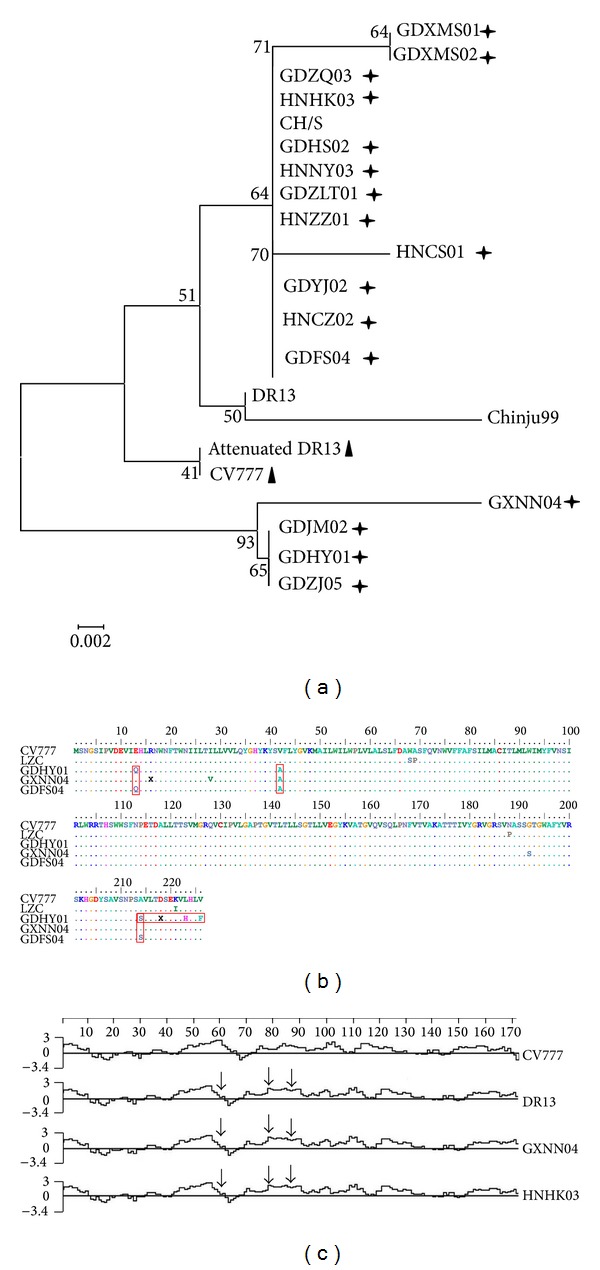
Comprehensive analysis of ORF3 genes from current PEDV with reference strains. (a) Phylogenetic analysis based on ORF3 aa; (b) point mutations analysis of ORF3 aa; (c) hydrophobic analysis of S aa. Phylogenetic tree is constructed by the neighbor-joining method with the MEGA 4.0 program. The percentage bootstrap support (per 1000 replicates) is indicated by the values at each node. Cross asterisk indicated present isolates; triangle indicated attenuated strains; box indicated point mutations.
Sequence analysis of the complete ORF3 genes showed that all of the newly isolated strains had intact ORFs that could regulate the translation of a 224 aa residues. Nevertheless, there was a large deletion region, approximately 51/49 nt (at positions 245–295/293), in the attenuated DR13 and CV777 PEDV, which accounted for the missing of 17/16 aa (YCPLLYYCGAFLDATI/I, at positions 83–98/97) (Figure 2(b)). The mutations observed at aa positions 7 (Q → X), 80 (V → F), and 182 (H → Q/Y) did not impact hydrophobicity (Figure 2(c)).
3.4. M Gene Analysis
Based on the complete M gene sequence, the newly isolated strains showed 98.5–100% nt sequence homogeneity within the group but 96.5–98.1% and 97.2–98.2% when compared to those stains previously isolated in China (LZC, CH/S, and LJB/03) and those foreign isolates (CV777 and Chinju99), respectively. Meanwhile, the newly isolated strains showed 97.8–100% aa sequence homogeneity within the group and 95.6–98.7% and 97.3–98.7% when compared to those previously isolated Chinese strains and foreign strains, respectively.
A phylogenetic tree was generated based on the deduced aa sequence of the M gene. With the exception of the GXNN04 strain, all of the newly isolated strains were placed into one subgroup. The other subgroup contained vaccine strains CV777 and early Chinese strains CH/S and LZC. It is noted that the GXNN04 isolate was closer to the Korean strains Chinju99 and LJB/03 than the other newly isolated strains (Figure 3(a)).
Figure 3.
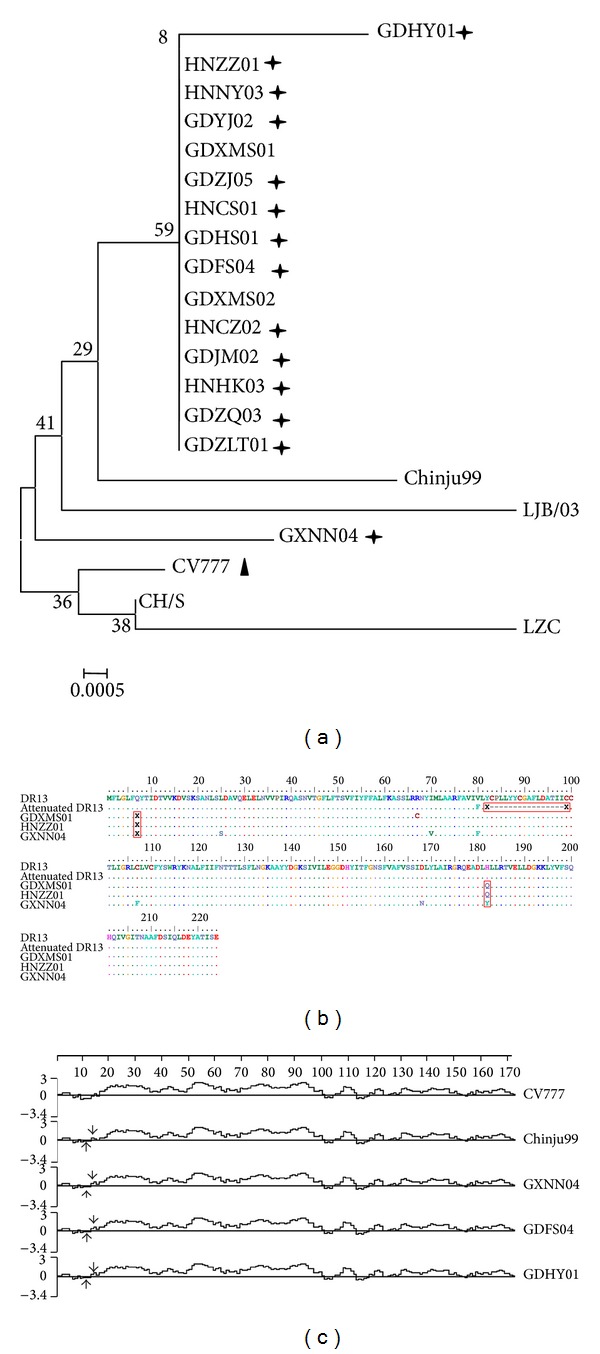
Comprehensive analysis of M genes from current PEDV with reference strains. (a) Phylogenetic analysis based on M aa; (b) point mutations analysis of M aa; (c) hydrophobic analysis of S aa. Phylogenetic tree is constructed by the neighbor-joining method with the MEGA 4.0 program. The percentage bootstrap support (per 1000 replicates) is indicated by the values at each node. Cross asterisk indicated present isolates; triangle indicated attenuated strains; box indicated point mutations.
All PEDV field strains isolated during the recent outbreaks had a single ORF of 681 nt, which encoded a protein of 227 aa. Except for several point mutations, there was no deletion or insertion within the ORFs of these isolates. The isolates had a conserved intergenic motif (ATAAAC) 5 nt upstream of the initiating ATG, which was previously recognized in Br1/87, CV777, LZC, and QH [17]. Compared to PEDV CV777, the new isolates had 11 nt mutations leading to 3 aa mutations. It is noted that the aa mutations at position 13 (E → Q) altered the hydrophobicity of the N terminus of the M protein (Figure 3(b)) and therefore had impact on its antigenic activity, whilst the other aa mutations, such as those at position 42 (V → A) and 214 (A → S), did not influence hydrophobicity of the M protein (Figure 3(c)).
4. Discussion
Gene sequencing analyses are useful tools to help us understand viral variation and pathogenicity. Several genes in PEDV have been analyzed previously, such as the N, S genes of PEDV Chinju99 isolated in 1999 in Korea, the M gene of PEDV CH/IMT/06 isolated in China, and the ORF3 gene of PEDV DR13 and attenuated DR13 [13, 17, 18]. More recently, some PEDV variants isolated in China (2010 to 2012) have also been reported [19]. However a comprehensive analysis to compare these three genes (i.e., partial S, ORF3, and M) has never been reported. In this study, partial S, ORF3, M genes from 16 PEDV strains isolated from the current Chinese PEDV outbreak were amplified and sequenced. The sequencing results were used to determine the genetic diversities and molecular characterizations of all of the PEDV strains.
The S protein of PEDV is exposed directly to the immune system of the host and therefore has always been used as a marker of viral variation. In this study, the data demonstrated that there was significant variation in S genes between the attenuated and virulent strains. Previous studies corroborated these results [8, 19]. Additionally, the newly isolated PEDV strains were highly homologous with each other but less similar to the vaccine strain CV777 or the virulent strain Chinju99 from Korea and CH/S from previous Chinese outbreaks. In the phylogenic trees, the current strains from different districts were genetically divided into different subgroups, but within each subgroup, the strains did not share the same geographical features. Also, these subgroups were genetically different from the groups of both attenuated strains (i.e., CV777 and attenuated DR13) and previous virulent strains (i.e., DR13, CH/S, and Chinju99). We can therefore draw a conclusion that the current strains in China represent a distinctive PEDV group and it is stable and virulent. The current strains displayed significant genetic variation from both the vaccine strains and previous strains. This may provide one possible answer for the nationwide outbreak and the poor efficacy of the vaccines. Previous studies showed that the S glycoprotein played an important role in viral attachment and penetration and was the neutralizing antibodies-inducing region due to the 2 B-cell linear epitopes [15]. In this study, the partial S proteins of current isolates, containing different aa to the previous strains due to mutations, were analyzed to determine the possible change on the protein hydrophobicity. When compared to the vaccine strain CV777, the current strains showed a significant hydrophobic alternation. This change was induced by a 4–9 aa mutations in the neutralizing epitope region of the current isolates and could provide one explanation for the poor immunity.
ORF3, located between the S and E genes, was recognized as a marker for the attenuated and virulent strains through an analysis of attenuated DR13 and wild-type DR13. These studies suggested that this area of the genome may be involved in cell tropism and the pathogenicity of the virus [13, 20, 21]. Park et al. [10] analyzed cell-culture-adapted PEDV (passage 100) with a smaller ORF3 gene and reduced virulence from the wild-type virus. In this study, the PEDV strains isolated from the current Chinese outbreaks had intact ORF3 genes that encoded 225 aa, which was in accordance with a previous study [13]. The phylogenic tree analysis demonstrated that the current strains should be clustered into the same group, despite of the different temporal and geographical origins (Figure 2(a)). This suggested that the ORF3 sequence was relatively conserved among stains from different temporal and geographical origins. A hydrophobicity analysis indicated that current isolates were different from the attenuated PEDV DR13 and vaccine strain CV777, which might be due to their adaptability and viral replication [14].
The 226 aa M protein of PEDV was proved to play an important role in viral package and budding. The current isolates shared 98.5–100% and 97.8–100% similarity in their nt and aa sequences, respectively. The phylogenetic tree suggested that all of the present PEDV strains, except for GXNN04, should be clustered into one subgroup, which demonstrated that the M gene was relatively conserved. This conclusion was consistent with the results from an earlier study [17]. A hydrophobicity analysis indicated there was only one mutation at aa 13 (E → Q) that would have affected the hydrophobicity of the M protein; however, the function of this mutation requires further investigation.
This study investigated the homology, phylogeny, and hydrophobicity of the partial S, full ORF3, and M genes from 16 currently prevalent PEDVs in China. The results suggested that most of the strains studied are highly homologous to each other, despite being clustered into different subgroups based on the S, M, and ORF3 genes. However, all of the current strains were distant from the vaccine strains genetically based on the S, M, and ORF3 genes analysis. The above analysis would help us to understand the origin of the virus and provided a reasonable explanation for both the outbreak and the poor efficacy of the commercial vaccine.
Acknowledgments
This work was supported by the Grants (no. 2012A020200014, no. 2010A040301010, and no. Yuenong2009-380) from Guangdong Provincial Department of Science and Technology and a Grant (no. 12C14071615) from Guangzhou Science and Technology and Information Bureau, respectively.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Ruiqin Sun, Zhangming Leng, and Shao-Lun Zhai contributed equally to this paper, they should be listed as co-first authors.
References
- 1.Have P, Moving V, Svansson V, Uttenthal A, Bloch B. Coronavirus infection in mink (Mustela vision). Serological evidence of infection with a coronavirus related to transmissible gastroenteritis virus and porcine epidemic diarrhea virus. Veterinary Microbiology. 1992;31(1):1–10. doi: 10.1016/0378-1135(92)90135-G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carvajal A, Lanza I, Diego R, Rubio P, Cármenes P. Seroprevalence of porcine epidemic diarrhea virus infection among different types of breeding swine farms in Spain. Preventive Veterinary Medicine. 1995;23(1-2):33–40. [Google Scholar]
- 3.Shibata I, Tsuda T, Mori M, Ono M, Sueyoshi M, Uruno K. Isolation of porcine epidemic diarrhea virus in porcine cell cultures and experimental infection of pigs of different ages. Veterinary Microbiology. 2000;72(3-4):173–182. doi: 10.1016/S0378-1135(99)00199-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kubota S, Sasaki O, Amimoto K, Okada N, Kitazima T, Yasuhara H. Detection of porcine epidemic diarrhea virus using polymerase chain reaction and comparison of the nucleocapsid protein genes among strains of the virus. Journal of Veterinary Medical Science. 1999;61(7):827–830. doi: 10.1292/jvms.61.827. [DOI] [PubMed] [Google Scholar]
- 5.Park SJ, Kim HK, Song DS, Moon H, Park B. Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) field isolates in Korea. Archives of Virology. 2011;156(4):577–585. doi: 10.1007/s00705-010-0892-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duarte M, Laude H. Sequence of the spike protein of the porcine epidemic diarrhoea virus. Journal of General Virology. 1994;75(5):1195–1200. doi: 10.1099/0022-1317-75-5-1195. [DOI] [PubMed] [Google Scholar]
- 7.Jinghui F, Yijing L. Cloning and sequence analysis of the M gene of porcine epidemic diarrhea virus LJB/03. Virus Genes. 2005;30(1):69–73. doi: 10.1007/s11262-004-4583-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun RQ, Cai RJ, Chen YQ, Liang P, Chen D, Song C. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerging Infectious Diseases. 2012;18(1):161–163. doi: 10.3201/eid1801.111259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofmann M, Wyler R. Quantitation, biological and physicochemical properties of cell culture-adapted porcine epidemic diarrhea coronavirus (PEDV) Veterinary Microbiology. 1989;20(2):131–142. doi: 10.1016/0378-1135(89)90036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park SJ, Song DS, Ha GW, Park B. Cloning and further sequence analysis of the spike gene of attenuated porcine epidemic diarrhea virus DR13. Virus Genes. 2007;35(1):55–64. doi: 10.1007/s11262-006-0036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sato T, Takeyama N, Katsumata A, Tuchiya K, Kodama T, Kusanagi K. Mutations in the spike gene of porcine epidemic diarrhea virus associated with growth adaptation in vitro and attenuation of virulence in vivo. Virus Genes. 2011;43(1):72–78. doi: 10.1007/s11262-011-0617-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Utiger A, Tobler K, Bridgen A, Ackermann M. Identification of the membrane protein of porcine epidemic diarrhea virus. Virus Genes. 1995;10(2):137–148. doi: 10.1007/BF01702594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park SJ, Moon HJ, Luo Y, et al. Cloning and further sequence analysis of the ORF3 gene of wild- and attenuated-type porcine epidemic diarrhea viruses. Virus Genes. 2008;36(1):95–104. doi: 10.1007/s11262-007-0164-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K, Lu W, Chen J, et al. PEDV ORF3 encodes an ion channel protein and regulates virus production. FEBS Letters. 2012;586(4):384–391. doi: 10.1016/j.febslet.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun D, Feng L, Shi H, et al. Identification of two novel B cell epitopes on porcine epidemic diarrhea virus spike protein. Veterinary Microbiology. 2008;131(1-2):73–81. doi: 10.1016/j.vetmic.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duarte M, Tobler K, Bridgen A, Rasschaert D, Ackermann M, Laude H. Sequence analysis of the porcine epidemic diarrhea virus genome between the nucleocapsid and spike protein genes reveals a polymorphic ORF. Virology. 1994;198(1):466–476. doi: 10.1006/viro.1994.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen JF, Sun DB, Wang CB, et al. Molecular characterization and phylogenetic analysis of membrane protein genes of porcine epidemic diarrhea virus isolates in China. Virus Genes. 2008;36(2):355–364. doi: 10.1007/s11262-007-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeo SG, Hernandez M, Krell PJ, Nagy E. Cloning and sequence analysis of the spike gene of porcine epidemic diarrhea virus Chinju99. Virus Genes. 2003;26(3):239–246. doi: 10.1023/A:1024443112717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J, Liu X, Shi D, Shi H, Zhang X, Feng L. Complete genome sequence of a porcine epidemic diarrhea virus variant. Journal of Virology. 2012;86(6):3408–3408. doi: 10.1128/JVI.07150-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwon H, Saif L, Jackwood D. Field isolates of transmissible gastroenteritis virus differ at the molecular level from the miller and purdue virulent and attenuated strains and from porcine respiratory coronaviruses. Journal of Veterinary Medical Science. 1998;60(5):589–597. doi: 10.1292/jvms.60.589. [DOI] [PubMed] [Google Scholar]
- 21.McGoldrick A, Lowings JP, Paton DJ. Characterisation of a recent virulent transmissible gastroenteritis virus from Britain with a deleted ORF 3a. Archives of Virology. 1999;144(4):763–770. doi: 10.1007/s007050050541. [DOI] [PMC free article] [PubMed] [Google Scholar]