

Abstract
We describe a boy presenting with intellectual disability and dysmorphic features in whom a cryptic microdeletion in chromosome band 2q12.1 was identified with array CGH. The deletion results in a loss of the POU3F3 and MRPS9 genes. In this paper, we discuss the possible role of POU3F3 haploinsufficiency in relation to the boy's phenotype.
Key Words : Array CGH, Intellectual disability, MRPS9, POU3F3, 2q12.1
Introduction
Intellectual disability (ID) has a prevalence of about 1-3% in the general population and is a major health care problem. There is a strong genetic etiology for this condition since in around 28% of patients with ID [Stevenson et al., 2003], a genetic aberration can be identified. Until now, approximately 450 genes have been described to be implicated in ID, but it is estimated that many more need to be discovered [Regan and Willatt, 2010].
The advent of genomic microarrays has led to a strong improvement in diagnostic yield in ID patients [Menten et al., 2006], and as such microarrays have become the first tier diagnostic tool to investigate the genetic basis of ID and/or learning disabilities. The introduction of molecular karyotyping has also led to the identification of several new microdeletion and -duplication syndromes and also of new genes implicated in ID [Raymond, 2010; Vissers and Stankiewicz, 2012; Weise et al., 2012]. Besides recurrent deletion and duplication events, rarer copy number variations (CNVs) have also been described in ID [Koolen et al., 2008; Mikhail et al., 2011].
In this paper, we present a child with ID and dysmorphic features with a de novo deletion in chromosome band 2q12.1 affecting only 2 genes: POU3F3 (OMIM 602480) and MRPS9 (OMIM 611975).
We propose the POU3F3 gene as a candidate gene for ID.
Case Report
The proband is the third child from healthy, nonconsanguineous parents. The family history is not contributory. The boy was born at full term with a birth weight of 3,140 g. No feeding problems were observed.
Upon physical examination, mild dysmorphic features were noted, with a flat nose, prominent ears and eyebrows, mild hypertelorism, low anterior hairline, and ogival palate. Blood analysis with metabolic screening and MRI of the brain showed no abnormalities, and EEG revealed no epileptiform alterations.
The boy presented at the age of 20 months with a mild motor delay and ID. He was not able to walk independently nor to produce any words. The boy showed hypotonia with excessive drooling, normal deep tendon reflexes and joint hyperlaxity. He had an average weight and length and a relative small head circumference (10th percentile). At the age of 22 months, he had a motor developmental age of 13 months and a cognitive level of 18 months. When he was 2 years old, he could walk but with some rigidity, and he lacked good postural responses. Multidisciplinary reassessment at the age of 3 years showed a cognitive level of 27 months, a developmental coordination disorder and a language impairment with mainly weak semantics. There were no behavioral problems nor social disturbances noted.
Materials and Methods
Informed consent was obtained from the parents, and paternity was evaluated.
Conventional karyotyping was performed with G-banding by standard procedures on short-term lymphocyte cultures. For array CGH analysis, DNA was isolated from total blood using the QIAamp DNA Blood Mini Kit (Qiagen, Venlo, The Netherlands) according to the manufacturer's instructions. Copy number profiling was performed on 180K oligonucleotide arrays (Agilent Technologies, Diegem, Belgium) according to the manufacturer's instructions with minor modifications as described [Buysse et al., 2009]. CNV evaluation was performed using the in-house developed software tool arrayCGHbase [Menten et al., 2005].
The deletion was confirmed by FISH with probes RP11-762O15 and RP11-574D15 using standard procedures.
Results
Chromosome analysis revealed a normal male karyotype: 46,XY.
Subsequently, array CGH analysis revealed the presence of 3 CNVs: a de novo deletion in chromosome band 2q12.1, a maternally inherited triplication in 2q24.2 and a maternally inherited deletion in 7q21.11. The inherited triplication and deletion were considered as benign events, since they contained no known genes and were inherited from a healthy parent.
The karyotype is thus 46,XY.arr[hg19] 2q21.1 (105,337,224-105,696,461)×1 dn.
The de novo deletion in 2q12.1 spans 360 kb and contains 2 known genes, POU3F3 and MRPS9 (fig. 1). Consultation of the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home) did not reveal this region as a benign copy variable region.
Fig. 1.

Array CGH detail of the proband's chromosome band 2q12.1, showing the deletion of POU3F3 and MRPS9.
FISH analysis confirmed the deletion in the patient (fig. 2).
Fig. 2.
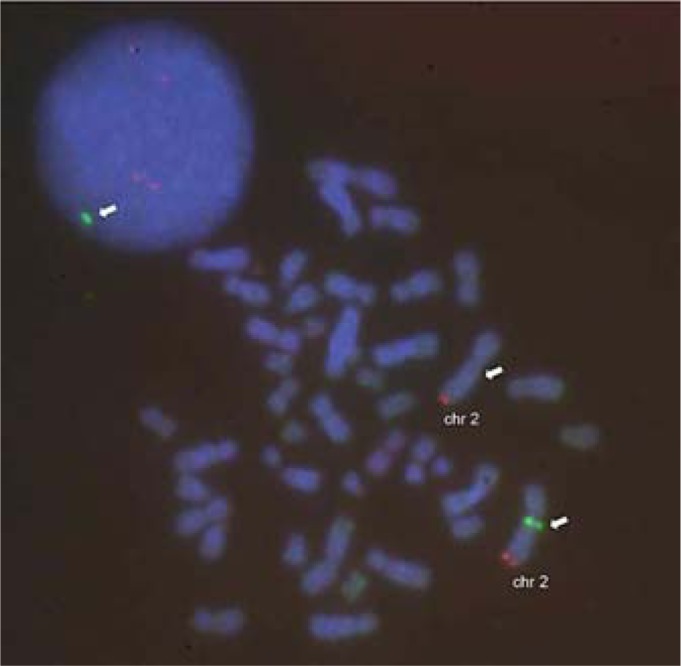
Confirmation of the deletion by FISH in an interphase nucleus (upper left) and a metaphase nucleus: the RP11-574D15 probe (spectrum green, white arrow) shows only one signal in the normal chromosome 2 (2q12.1 locus). The 2q telomeric probe (spectrum orange, Abbott Molecular, Ottignies, Belgium) shows 2 normal signals (2 split signals in interphase nucleus).
Discussion
This report describes a patient with ID and dysmorphic features carrying a de novo 360-kb deletion in chromosome band 2q12.1. The deletion results in a loss of both the POU3F3 and MRPS9 gene.
POU3F3 is a member of the class III POU family of transcription factors which are expressed in the central nervous system. These proteins are characterized by the presence of a POU domain which recognizes and binds an octamer sequence in promoter regions by which they can regulate the transcription of target genes [Hara et al., 1992; Schonemann et al., 1998]. The POU domain proteins play essential roles, not only in the development of highly specialized tissues, such as complex neuronal systems, but also in more general cellular processes [Philips and Luisi, 2000; Andersen and Rosenfeld, 2001].
POU3F3 is a proneural factor that, together with ASCL1, plays an essential role in the activation of DLL1 and other neuronal genes, and as such it is very important in neurogenesis [Castro et al., 2006].
Besides this, POU3F3 is also a transactivator of SOX11, meaning that these 2 transcription factors work together to exert their function [Kuhlbrodt et al., 1998; Kim et al., 2008]. It has been established that SOX11 is an essential actor in normal development of the nervous system [Jankowski et al., 2009; Penzo-Méndez, 2010; Mu et al., 2012]. Interestingly, a patient with ID, dysmorphic features and autism carrying a deletion of the SOX11 gene has been reported [Lo-Castro et al., 2009].
Pou3f3 is important in neuronal development as shown in Pou3f3 deficient mice, which present with severe brain deformities [McEvilly et al., 2002; Sugitani et al., 2002]. Furthermore, Pou3f3 and Pou3f2 are essential for the normal migration of neurons to the cortex [Wolf et al., 2009].
Besides POU3F3, the deletion in our patient also spans MRPS9, a gene that encodes a mitochondrial ribosomal protein. MRPS9 is one of more than 70 protein components of mitochondrial ribosomes that are encoded by the nuclear genome [Rötig, 2011]. Although these genes play an important role in mitochondrial functioning, and mutations in some of the family members have been described in relation to human diseases, these disorders all show an autosomal recessive inheritance pattern and are indicative for mitochondrial diseases.
Deletions in chromosome band 2q12.1 and of POU3F3 in particular appear to be rare. This is the first report describing a patient with a cryptic deletion in chromosome band 2q12.1. A large deletion of 2q12q14 in a neonate with severe multiple malformations has been reported [Antich et al., 1983], but comparison of our case with this one is not useful due to the large difference in the deletion size.
The deletion of POU3F3 in a patient with developmental delay further emphasizes the role of this gene in neurodevelopment and ID. Additional patients and further phenotype-genotype studies are however warranted to support this hypothesis.
Acknowledgements
The authors are indebted to the patients. S.V. is supported by a PhD fellowship of the Research Foundation – Flanders (FWO). This article presents research results of the Belgian program of Interuniversity Poles of attraction initiated by the Belgian State, Prime Minister's Office, Science Policy Programming (IUAP).
References
- 1.Andersen B, Rosenfeld MG. POU domain factors in the neuroendocrine system: lessons from developmental biology provide insights into human disease. Endocr Rev. 2001;22:2–35. doi: 10.1210/edrv.22.1.0421. [DOI] [PubMed] [Google Scholar]
- 2.Antich J, Carbonell X, Mas J, Clusellas N. De novo interstitial deletion of the long arm of chromosome 2 in a malformed newborn with a karyotype: 46, XY, del(2)(q12q14) Acta Paediatr Scand. 1983;72:631–633. doi: 10.1111/j.1651-2227.1983.tb09785.x. [DOI] [PubMed] [Google Scholar]
- 3.Buysse K, Delle Chiaie B, Van Coster R, Loeys B, De Paepe A, et al. Challenges for CNV interpretation in clinical molecular karyotyping: lessons learned from a 1001 sample experience. Eur J Med Genet. 2009;52:398–403. doi: 10.1016/j.ejmg.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Castro DS, Skowronska-Krawczyk D, Armant O, Donaldson IJ, Parras C, et al. Proneural bHLH and Brn proteins coregulate a neurogenic program through cooperative binding to a conserved DNA motif. Dev Cell. 2006;11:831–844. doi: 10.1016/j.devcel.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 5.Hara Y, Rovescalli AC, Kim Y, Nirenberg M. Structure and evolution of four POU domain genes expressed in mouse brain. Proc Natl Acad Sci USA. 1992;89:3280–3284. doi: 10.1073/pnas.89.8.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jankowski MP, McIlwrath SL, Jing X, Cornuet PK, Salerno KM, et al. Sox11 transcription factor modulates peripheral nerve regeneration in adult mice. Brain Res. 2009;1256:43–54. doi: 10.1016/j.brainres.2008.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim DK, Han SB, Hong ST, Choi YJ, Sun W, et al. Expression of Sox11 and Brn transcription factors during development and following transient forebrain ischemia in the rat. Neurosci Lett. 2008;433:259–264. doi: 10.1016/j.neulet.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 8.Koolen DA, Sistermans EA, Nilessen W, Knight SJ, Regan R, et al. Identification of non-recurrent submicroscopic genome imbalances: the advantage of genome-wide microarrays over targeted approaches. Eur J Hum Genet. 2008;16:395–400. doi: 10.1038/sj.ejhg.5201975. [DOI] [PubMed] [Google Scholar]
- 9.Kuhlbrodt K, Herbarth B, Sock E, Enderich J, Hermans-Borgmeyer I, Wegner M. Cooperative function of POU proteins and SOX proteins in glial cells. J Biol Chem. 1998;273:16050–16057. doi: 10.1074/jbc.273.26.16050. [DOI] [PubMed] [Google Scholar]
- 10.Lo-Castro A, Giana G, Fichera M, Castiglia L, Grillo L, et al. Deletion 2p25.2: a cryptic chromosome abnormality in a patient with autism and mental retardation detected using aCGH. Eur J Med Genet. 2009;52:67–70. doi: 10.1016/j.ejmg.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 11.McEvilly RJ, de Diaz MO, Schonemann MD, Hooshmand F, Rosenfeld MG. Transcriptional regulation of cortical neuron migration by POU domain factors. Science. 2002;295:1528–1532. doi: 10.1126/science.1067132. [DOI] [PubMed] [Google Scholar]
- 12.Menten B, Pattyn F, De Preter K, Robbrecht P, Michels E, et al. arrayCGHbase: an analysis platform for comparative genomic hybridization microarrays. BMC Bioinformatics. 2005;6:124. doi: 10.1186/1471-2105-6-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menten B, Maas N, Thienpont B, Buysse K, Vandesompele J, et al. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: a new series of 140 patients and review of published reports. J Med Genet. 2006;43:625–633. doi: 10.1136/jmg.2005.039453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mikhail FM, Lose EJ, Robin NH, Descartes MD, Rutledge KD, et al. Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am J Med Genet A. 2011;155A:2386–2396. doi: 10.1002/ajmg.a.34177. [DOI] [PubMed] [Google Scholar]
- 15.Mu L, Berti L, Masserdotti G, Covic M, Michaelidis TM, et al. SoxC transcription factors are required for neuronal differentiation in adult hippocampal neurogenesis. J Neurosci. 2012;32:3067–3080. doi: 10.1523/JNEUROSCI.4679-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Penzo-Méndez AI. Critical roles for SoxC transcription factors in development and cancer. Int J Biochem Cell Biol. 2010;42:425–428. doi: 10.1016/j.biocel.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillips K, Luisi B. The virtuoso of versatility: POU proteins that flex to fit. J Mol Biol. 2000;302:1023–1039. doi: 10.1006/jmbi.2000.4107. [DOI] [PubMed] [Google Scholar]
- 18.Raymond FL. Monogenic causes of mental retardation. In: Knight SJL, editor. Genetics of Mental Retardation. Monogr Hum Genet. Vol. 18. Basel: Karger; 2010. pp. 89–100. [Google Scholar]
- 19.Regan R, Willatt L. Knight SJL. Genetics of Mental Retardation. Monogr Hum Genet. Vol. 18. Basel: Karger; 2010. Mental retardation: definition, classification and etiology; pp. 16–30. [Google Scholar]
- 20.Rötig A. Human diseases with impaired mitochondrial protein synthesis. Biochim Biophys Acta. 2011;1807:1198–1205. doi: 10.1016/j.bbabio.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 21.Schonemann MD, Ryan AK, Erkman L, McEvilly RJ, Bermingham J, Rosenfeld MG. POU domain factors in neural development. Adv Exp Med Biol. 1998;449:39–53. doi: 10.1007/978-1-4615-4871-3_4. [DOI] [PubMed] [Google Scholar]
- 22.Stevenson RE, Procopio-Allen AM, Schroer RJ, Collins JS. Genetic syndromes among individuals with mental retardation. Am J Med Genet A. 2003;123A:29–32. doi: 10.1002/ajmg.a.20492. [DOI] [PubMed] [Google Scholar]
- 23.Sugitani Y, Nakai S, Minowa O, Nishi M, Jishage K, et al. Brn-1 and Brn-2 share crucial roles in the production and positioning of mouse neocortical neurons. Genes Dev. 2002;16:1760–1765. doi: 10.1101/gad.978002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vissers LE, Stankiewicz P. Microdeletion and microduplication syndromes. Methods Mol Biol. 2012;838:29–75. doi: 10.1007/978-1-61779-507-7_2. [DOI] [PubMed] [Google Scholar]
- 25.Weise A, Mrasek K, Klein E, Mulatinho M, Llerena JC, Jr, et al. Microdeletion and microduplication syndromes. J Histochem Cytochem. 2012;60:346–358. doi: 10.1369/0022155412440001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolf M, Lommes P, Sock E, Reiprich S, Friedrich RP, et al. Replacement of related POU transcription factors leads to severe defects in mouse forebrain development. Dev Biol. 2009;332:418–428. doi: 10.1016/j.ydbio.2009.06.011. [DOI] [PubMed] [Google Scholar]