

Abstract
Background
Pediatric sudden cardiac arrest (CA) is an unfortunate and devastating condition, often leading to poor neurologic outcomes. However, little experimental data on the pathophysiology of pediatric CA is currently available due to the scarcity of animal models.
New Method
We developed a novel experimental model of pediatric cardiac arrest and cardiopulmonary resuscitation (CA/CPR) using postnatal day 20–25 mice. Adult (8–12 weeks) and pediatric (P20–25) mice were subjected to 6 min CA/CPR. Hippocampal CA1 and striatal neuronal injury were quantified 3 days after resuscitation by hematoxylin and eosin (H&E) and Fluoro-Jade B staining, respectively.
Results
Pediatric mice exhibited less neuronal injury in both CA1 hippocampal and striatal neurons compared to adult mice. Increasing ischemia time to 8 min CA/CPR resulted in an increase in hippocampal injury in pediatric mice, resulting in similar damage in adult and pediatric brains. In contrast, striatal injury in the pediatric brain following 6 or 8 min CA/CPR remained extremely low. As observed in adult mice, cardiac arrest causes delayed neuronal death in pediatric mice, with hippocampal CA1 neuronal damage maturing at 72 hours after insult. Finally, mild therapeutic hypothermia reduced hippocampal CA1 neuronal injury after pediatric CA/CPR.
Comparison with Existing Method
This is the first report of a cardiac arrest and CPR model of global cerebral ischemia in mice
Conclusions
Therefore, the mouse pediatric CA/CPR model we developed is unique and will provide an important new tool to the research community for the study of pediatric brain injury.
Keywords: Pediatric, Juvenile, Cardiac Arrest, Global cerebral ischemia, hypothermia
1. Introduction
Cardiac arrest (CA) is an important cause of morbidity and mortality in both the adult and juvenile populations (Knudson et al., 2012; Roger et al., 2012). It is estimated that approximately 600,000 adults, primarily due to ventricular fibrillation or tachycardia and 16,000 children, primarily resulting from asystolic arrest, suffer sudden cardiac arrest each year in the United States. A significant amount of research has focused on improving rates of return of spontaneous circulation (ROSC), leading to increased survival rates. However, therapeutic options to improve neurologic outcome after ROSC remain limited. To date, mild therapeutic hypothermia is the only therapy shown to be effective in increasing survival and decreasing morbidity in adult cardiac arrest patients (The Hypotherrmia after Cardiac Arrest Study Group 2002; Arrich et al., 2012; Bernard et al., 2002; Choi et al., 2012). There are no therapies that have been proven effective in improving outcome after pediatric CA, although a clinical study evaluating the efficacy of therapeutic hypothermia after pediatric CA is ongoing (THAPCA, NCT00878644). Emerging clinical evidence is emphasizing the importance of considering the juvenile cardiac arrest population independently, not treating them as small adults (Hickey and Painter, 2006; Rice and Barone, 2000). However, little experimental data exists to directly assess differences in brain injury following injury in the adult versus juvenile. This is due in large part to scarcity of models of this important juvenile population; therefore we modified our adult mouse CA/CPR model (Kofler et al., 2004) to examine injury at this developmental stage.
Improved resuscitation techniques and the widespread use of therapeutic hypothermia has reduced mortality, however the unfortunate consequence of these improvements is the increased numbers of people suffering from long-term neurological deficits. The majority of survivors exhibit significant neurological sequelae, including impaired memory and executive cognitive function (Bunch et al., 2004; Lim et al., 2004; Mateen et al., 2011; O'Reilly et al., 2003; Peskine et al., 2010). These deficits are thought to be a result of neuronal cell death in ischemia-sensitive brain regions such as the hippocampus and basal ganglia (striatum) (Bottiger et al., 1998; Garcia and Anderson, 1989; Kofler et al., 2004). Injury to these sensitive neuron populations have been extensively studied in adult animal models, revealing a multitude of mechanisms, including excitotoxicity, oxidative stress, apoptosis, neuroinflammation, among others (Iadecola and Anrather, 2011; Moskowitz et al., 2010; Szydlowska and Tymianski, 2010). In contrast, relatively little is known about pediatric brain responses to global cerebral ischemia. Neonatal hypoxia-ischemia has been relatively well studied and indicates a possible window of high vulnerability at this developmental stage (Ikonomidou et al., 1989; Muramatsu et al., 1997; Towfighi et al., 1997; Yager et al., 1996). However, differences between the experimental models used in the neonates and adults makes direct comparisons complicated. In contrast, little is known regarding the relative vulnerability of the juvenile brain to ischemia. A model of asphyxia juvenile cardiac arrest has recently been developed using rats (Manole et al., 2012; Shoykhet et al., 2012; Tang et al., 2010; Walson et al., 2011), however to our knowledge there are no reports directly comparing ischemic vulnerabilities of juvenile and adult animals following global cerebral ischemia.
This manuscript describes a new model of cardiac arrest in juvenile mice (postnatal day 20–25) that is directly amenable for age-dependent comparisons. We utilized the new mouse model of juvenile cardiac arrest and CPR (CA/CPR) to test the hypothesis that young (pediatric) animals are more resistant to global cerebral ischemia than adult animals. Further, the efficacy of mild therapeutic hypothermia was assessed in this new model. Data presented here demonstrate the development of a reproducible mouse model of juvenile cardiac arrest that will provide an important new tool to the neuroscience research community, allowing the use of genetically engineered mice to begin to unravel the complex molecular interactions contributing to injury in the juvenile brain.
2. Materials and Methods
2.1 Experimental animals
All experimental protocols were approved by the Institutional Animal Care and Use Committee and conformed to the National Institutes of Health guidelines for the care and use of animals in research. Male C57Bl/6 20–25 day (P20–25) pediatric and 8–12 week old adult mice were used for this study. Pediatric mice were weaned and not with dam at time of experiment. Mice housed in a standard 12 hr light and 12 hr dark cycle and had free access to food and water.
2.2 Cardiac arrest and cardiopulmonary resuscitation model
Anesthesia was induced with 3% isoflurane and maintained with 1.5–2% isoflurane in oxygen enriched air (Fraction of Inspired Oxygen (FiO2) 30%) via face mask. Temperature probes were placed into the left ear canal and rectum. Rectal temperature was controlled at near 37 °C during surgery with a heating lamp and heating pad. For drug administration, a PE-10 catheter was inserted into the right internal jugular vein and flushed with heparinized 0.9% saline solution. Animals were endotracheally intubated using an intravenous catheter (22G for adult mouse, 24G for pediatric mouse), connected to a mouse ventilator (Minivent, Hugo Sachs Elektronik, March-Hugstetten, Germany) set to a respiratory rate of 150 breaths/min in adult mice and 160 breaths/min in pediatric mice. Needle electrodes were placed subcutaneously on the chest for electrocardiogram (EKG) monitoring throughout the experimental procedures. Cardiac arrest was induced by injection of 50 µL (30 µL for juvenile mice) 0.5M KCl via the jugular catheter, and confirmed by the appearance of asystole on the electrocardiography monitor and no spontaneous breathing. Animals gasped for about 30 seconds after injection of KCl, but the EKG remained a flat line. Spontaneous breathing was not observed during cardiac arrest and a paralytic agent was not required. The endotracheal tube was disconnected from the ventilator and anesthesia was stopped. Body warming was ceased one minute before induction of cardiac arrest. During cardiac arrest, the pericranial temperature was maintained at 37.5 ± 0.2 °C by using a water-filled coil, which was placed around the animal’s head and heated to around 40.0 °C by running through a warm water bath. Body temperature was allowed to fall spontaneously during the arrest to 35 °C. CPR was begun 6 or 8 min after induction of cardiac arrest, by slow injection of 0.5–1.0 ml (0.2–0.5 ml for juvenile mice) of epinephrine (16 µg epinephrine/ml 0.9% saline), chest compressions at a rate of approximately 300 min−1, and ventilation with 100% oxygen at a respiratory rate of 200 breaths/min for adult and 210 breaths/min for pediatric mice. As soon as ROSC was achieved, defined as electrocardiographic activity with visible cardiac contractions, chest compressions were stopped. If ROSC could not be achieved within 2 min of CPR, resuscitation was stopped and the animal was excluded from the study.
After ROSC was achieved, pediatric mice which underwent 8 min cardiac arrest were randomized into 2 groups: normothermic and hypothermic-30 minutes. In animals of the hypothermic groups, rapid cooling was started by using a water-filled pad placed underneath the animal which was chilled by running through an ice-water bath. The body temperature dropped to 32°C quickly and was maintained at 32 ±0.5°C for 30 minutes. Then the animal was rewarmed by the heating lamp and pad for about 10–15 min until body temperature reached 36°C. In animals of the normothermic group, the body was rewarmed to reach 36 °C by using the heating lamp and pad at a rate of 0.3–0.5 °C/min, and maintained around 36 °C for 30 minutes. Body and ear temperature were monitored closely during the experiment.
Five minutes after resuscitation, FiO2 was decreased to 50%. When spontaneous breathing rate reached 30 breaths/min, respiratory rate was adjusted to reach 150 breaths/min in adults and 160 breaths/min in pediatrics. In case of insufficient spontaneous breathing, mechanical ventilation was continued until the animals breathed at least 60 times/min and then the endotracheal tube was removed. Temperature probes and catheters were removed, and the skin wounds were closed. The animal was then placed into its home cage for complete recovery.
2.3 Health Assessment Score
Mice were weighed daily, and a health assessment score was calculated for each mouse daily for three days after CA/CPR. The graded scoring systems ranged from 0 to 2, 0 to 3, or 0 to 5 depending on the behavior assessed, with 0 indicating no deficit and the upper limit indicating the most impaired. The behaviors assessed included consciousness (0–3), interaction (0–2), ability to grab wire top (0–2), motor function (0–5), and activity (0–2) (Allen et al., 2011; Kosaka et al., 2012). Scores in each category were summated to generate an overall health assessment score.
2.4 Hematoxylin & Eosin and Fluoro-Jade B Staining
Twenty-four hours, three days or seven days after CA/ CPR, animals were anesthetized with 3% isoflurane and transcardially perfused with 0.9% saline followed by 4% paraformaldehyde. Brains were removed, post-fixed with paraformaldehyde and embedded in paraffin. Coronal sections 6 µm thick were serially cut and stained with hematoxylin and eosin (H&E). The hippocampal CA1 region was analyzed, three levels (100 µm apart), beginning from −1.5 mm bregma. Nonviable neurons were determined by the presence of hypereosinophilic cytoplasm and pyknotic nuclei. The percentage of nonviable neurons was calculated for each brain region (average of 3 levels per region). The investigator was blinded to treatment before analyzing neuronal damage. For cell density analysis, images were captured at 400× from each section and number of dead (dark pyknotic nuclei and pink cytoplasm) and live cells/area were analyzed using ImageJ software.
Fluoro-Jade B staining was performed to specifically analyze the ischemic neurons in the rostral and caudal caudoputamen. Briefly, 6µ paraffin sections on Superfrost Plus Microscope Slides were deparaffinized by immersing in three 5 minute changes of xylene and rehydrated in absolute and 95% alcohol. The slides were then immersed in a solution containing 1% Sodium hydroxide in 80% alcohol for 5 minutes. Following a 2 minute immersion in 70% alcohol and a 2 minute rinse in running distilled water, the slides were placed in a 0.06% solution of Potassium permanganate for 20 minutes. The staining step followed using a 0.0004% working solution of Fluoro-Jade B (Millipore, USA) in 0.1% acetic acid. Following 20 minutes of staining the slides were rinsed in running distilled water for 2 minutes, drained of water and vertically dried on paper towels while being protected from light. To complete the drying process, the slides were put on a 50°C slide warmer for 10 minutes. The dried slides were briefly dipped in xylene and coverslipped with DPX mountant (Electron Microscopy Sciences, Inc., USA). All staining steps were conducted using agitation and both the preparation of the Fluoro Jade B staining solution and the staining steps were carried out in dim light to minimize photo bleaching. Fluorescence images were obtained by a fluorescence microscopy (Leica DM 2000, USA). The Fluor-Jade B positive cells which indicated ischemic neurons appeared green fluorescence. Two random high field areas (317 µM × 237 µM at 400× magnification) were picked in each caudoputamen (average of 2 levels per region) and the number of Fluoro-Jade B positive cells per millimeter was determined. The investigator was blinded to treatment before analyzing neuronal damage.
2.5 Statistical Analysis
All data are presented as mean±SEM. Histological damage and health assessment scores were compared using 1-way ANOVA followed by Newman–Keuls multiple comparison test. Survival comparisons were made using a χ2 test. Probability values <0.05 were considered statistically significant.
3. Results
3.1 Pediatric mice less sensitive to CA/CPR than adult mice
To compare the vulnerability of the adult and pediatric brain to ischemic insult, hippocampal and striatal damage was analyzed in both age groups. Adult (8–12 weeks) and pediatric (P20–25) male mice were subjected to 6 min cardiac arrest followed by CPR and neuronal injury was analyzed 3 days after resuscitation. Immediate asystolic arrest was observed in all mice after injection of KCl. Body weight was significantly different between adult and pediatric mice, CPR duration was not different between groups (except when ischemic duration was altered), epinephrine dose per body weight was higher in pediatric mice compared to adults and survival was not different between groups (Table 1 & 2). Health assessment scores were not different among experimental groups, with the exception of the adult 8 min CA/CPR group which exhibited significantly greater impairment, consistent with our histology analyses (see below). These experiments revealed an age-dependent effect on CA1 injury, such that pediatric mice had less injury than adult mice, exhibiting hippocampal CA1 injury of 21.7 ± 6.8% (n=6) in the pediatric group and 55.4 ± 4.0% (n=8, p < 0.01) in the adult group (Figure 1). Similarly, striatal injury was greater in adult (191 ± 62 Fluoro-Jade B positive cells/mm2 (n=9) compared to 0.5 ± 0.2 Fluoro-Jade B positive cells/mm2 (n=6) in pediatric mice (Figure 2).
Table 1. Body weight, cardiac arrest parameters, survival rate, and general health assessment.
Total ischemia represents time from induction of cardiac arrest to successful resuscitation. Postoperative day (POD) refers to day following CA/CPR.
| CA 6 minutes | CA 8 minutes | ||||
|---|---|---|---|---|---|
| Adult | Pediatric | Adult | Pediatric | ||
| Nomothermia | Hypothermia | ||||
| N | 8 | 6 | 7 | 8 | 6 |
| Body weight (BW, g) | 23.2 ± 0.6 | 9.7 ± 0.2 | 23.6 ± 0.6 | 9.9 ± 0.4 | 10.3 ± 0.2 |
| Total ischemia time (sec) | 461 ± 7 | 436 ± 5 | 590 ± 6 | 594 ± 7 | 590 ± 9 |
| Epinephrine (µg/g BW) | 0.26 ± 0.03 | 0.42 ± 0.02* | 0.33 ± 0.04 | 0.52 ± 0.05* | 0.52 ± 0.07 |
| Surviving animals (%) | 89(8/9) | 100(6/6) | 70(7/10) | 89(8/9) | 100(6/6) |
| Health Assessment Score | |||||
| POD 1 | 1.6 ± 0.3 | 1.0 ± 0.5 | 4.9 ± 0.8* | 2.1 ± 0.3 | 2.2 ± 0.2 |
| POD 2 | 1.1 ± 0.2 | 0.2 ± 0.2 | 2.0 ± 0.8 | 1.4 ± 0.3 | 1.5 ± 0.2 |
| POD 3 | 0.5 ± 0.2 | 0.0 ± 0.0 | 2.1 ± 1.2* | 0.5 ± 0.2 | 0.7 ± 0.2 |
* in epinephrine indicates P < 0.05 compared to corresponding adult group and * in health assessment score indicates P < 0.05 compared to pediatric normothermia 8 minute group.
CA/CPR, cardiac arrest and cardiopulmonary resuscitation. BW, body weight.
Table 2. Body weight, cardiac arrest parameters, survival rate, and general health assessment among pediatric 24 hours, 3 days and 7 days groups.
Postoperative day (POD) refers to day following CA/CPR.
| 24 hours | 3 days | 7 days | |
|---|---|---|---|
| N | 4 | 8 | 5 |
| Body weight (BW, g) | 9.9±0.2 | 9.9 ± 0.4 | 9.8±0.6 |
| Total ischemia time (sec) | 597±21 | 594 ± 7 | 586±6 |
| Epinephrine (µg/g BW) | 0.62±0.02 | 0.52 ± 0.05 | 0.56±0.04 |
| Surviving animals (%) | 80(4/5) | 89(8/9) | 100(8/8) |
| Health Assessment Score | |||
| POD 1 | 2.0±0.0 | 2.1 ± 0.3 | 3.0±0.5 |
| POD 2 | 1.4 ± 0.3 | 1.6±0.3 | |
| POD 3 | 0.5 ± 0.2 | 0.9±0.2 |
BW, body weight.
Figure 1. Adult hippocampal neuronal damage greater than pediatric.

Representative photomicrographs of hippocampal CA1 neurons in adult mice following 6 min cardiac arrest (A) and 8 min cardiac arrest (B) and pediatric mice following 6 min cardiac arrest (C) and 8 min cardiac arrest (D) and stained with H & E 3 days later. E) Quantification of ischemic neurons in the CA1 region of the hippocampus 3 days after CA/CPR, indicating significantly less injured neurons in pediatric mice that underwent 6 min cardiac arrest. * P < 0.05
Figure 2. Adult striatal neuronal damage greater than pediatric.

Representative fluorescent photomicrographs of striatal neurons in adult mice following 6 min cardiac arrest (A) and 8 min cardiac arrest (B) and pediatric mice following 6 min cardiac arrest (C) and 8 min cardiac arrest (D) and stained with FluoroJade B 3 days later. E) Quantification of ischemic neurons in the striatum 3 days after CA/CPR, indicating significantly less injured neurons in pediatric mice that underwent 6 min cardiac arrest. * P < 0.05
To further characterize the age-dependent effect of ischemia, we increased ischemic time to 8 min in both age groups and assessed hippocampal CA1 and striatal neuronal injury 3 days after resuscitation. In contrast to the observation made using 6 min CA/CPR, adult and pediatric mice exhibited similar ischemic damage, with hippocampal CA1 injury of 44.0 ± 10.1% (n=7) and 47.5 ± 6.6 (n=8,), respectively (Figure 1). Surprisingly, injury in the striatum was greater in the adult brain (107 ± 50 Fluoro-Jade B positive cells/mm2 (n=6) compared to 3.6 ± 1.2 Fluoro-Jade B positive cells/mm2 (n=7) in pediatric mice (Figure 2). The injury of hippocampus and striatum between 6 min and 8 min cardiac arrest in adults are not different.
3.2 Cardiac arrest causes delayed neuronal death in pediatric mice
To assess the time course of injury in the pediatric mouse brain (P20–25), 8 min CA/CPR was performed in P20–25 male mice and hippocampal CA1 neuronal injury analyzed 1, 3 and 7 days after resuscitation. As previously observed in adult mice (Kofler et al., 2004; Kosaka et al., 2012), global cerebral ischemia in this model exhibited selective, delayed cell death of hippocampal CA1 neurons. Quantification of CA1 neuronal injury at each time-point following resuscitation from 8 min cardiac arrest was performed using H&E staining. The results indicate that ischemic damage of hippocampal CA1 neurons increased with reperfusion time, with a peak at 3 days after CPR (Figure 3). Specifically, hippocampal neuronal injury increased from 5.6 ± 1.6% (n=4) at 24 hours to 47.5 ± 6.6% (n=8, p<0.01) at 3 days and appears to decline at 7 days after CPR (22.1 ±4.5%; n=8; P <0.05 compared to 3 day injury). To address the apparent decrease in CA1 injury 7 days after resuscitation, density of live and dead cells were analyzed at 3 and 7 days, revealing a decrease in the number of dead cells at 7 days; 1575 ± 292 dead cells/mm2 at three days (n=8) compared to 686 ± 180 dead cells/mm2 at seven days (n=7; P < 0.05). No further reduction in the number of viable neurons was observed at 7 compared to 3 days; 2367 ± 411 live cells/mm2 at three days (n=8) compared to 2893 ± 267 live cells/mm2 at seven days. These data demonstrate the removal of dead neurons between 3 and 7 days, indicating that analysis of neuronal injury at 3 days is optimal.
Figure 3. Delayed neuronal death of hippocampal CA1 neurons following 8 min cardiac arrest in pediatric mice.

Representative photomicrographs of hippocampal CA1 neurons from 24 hour (A), 3 days (B) and 7 days (C) after resuscitation and stained with H & E. D) Quantification of ischemic neurons in CA1 region of hippocampus at different points after CA/CPR. E) Quantification of density of ischemic and live neurons in the CA1 region of the hippocampus at 3 and 7 days after resuscitation. * P < 0.01 compared with 3 days.
3.3 Hypothermia reduces neuronal injury in pediatric mice
Hypothermic therapy is a promising treatment to improve neurological outcome after ventricular arrhythmia-induced cardiac arrest in adults and neonatal asphyxia. However, the effect of hypothermia therapy in infants and children with cardiac arrest remains understudied. In order to test whether therapeutic hypothermia is neuroprotective in our novel pediatric CA/CPR model, mild hypothermia (32 ± 0.5°C) was administered for 30 minutes after resuscitation and then rewarmed. Three days after CPR, histological analysis revealed that hippocampal CA1 neuronal damage was reduced following hypothermia compared to the nomothermic group (Figure 4). Hypothermic group mice (n=6) had 17.7± 2.0% ischemic neuronal injury compared to 47.5 ± 6.6% in the normothemic group (n=8, p<0.05).
Figure 4. Mild therapeutic hypothermia significantly reduces CA1 neuronal injury in pediatric mice.

Representative photomicrophraphs of hippocampal CA1 neurons from pediatric mice exposed to 8 min normothermic cardiac arrest (A) and 8 min cardiac arrest followed by 30 min mild hypothermia (B). C) Quantification of ischemic CA1 neurons 3 days after CA/CPR, indicating significant reduction in neuronal injury in mice treated with post-arrest hypothermia. P < 0.05
4. Discussion
Our results demonstrated that hippocampal CA1 pyramidal cells are selectively injured in the novel mouse model of pediatric global cerebral ischemia induced by cardiac arrest, and time course analysis showed that the neuronal death is matured at 72 hours after insult. Mild therapeutic hypothermia provided neuroprotection after pediatric CA/CPR. Most importantly, the hippocampus and striatum of pediatric mice are less susceptible to ischemia compared to adult mice, however, pediatric hippocampal neuronal damage proportionately increased with ischemic time. Therefore, our novel pediatric global cerebral ischemic model is unique and a valuable new tool to investigate mechanisms of injury and repair following cardiac arrest in the pediatric population.
Delayed neuronal death was first demonstrated by Ito et al in 1975, and further confirmed in the gerbil hippocampus following ischemia in the rat four-vessel occlusion model (Ito et al., 1975; Kirino, 1982; Pulsinelli, 1985; Pulsinelli et al., 1982). It is used to describe a phenomenon that certain neuronal populations are selectively injured and undergo delayed cell death at 2–3 days after transient cerebral ischemia. The hippocampus, particularly the CA1 pyramidal neurons, is well known to be susceptible to ischemia. Consistent with the literature using adult animals, we also observed that hippocampal neuronal death is a slow process and showed a mature morphologic outcome at 3 days in our pediatric CA/CPR model. The mechanisms of delayed neuronal death are not well understood, although there are several proposed mechanisms. For example, glutamate excitotoxicity, prolonged inhibition of protein synthesis, lipid metabolism and free radicals, apoptosis and mitochondrial hypothesis (Abe et al., 1995). Little research has focused on cell death mechanisms in the pediatric population and our new model provides a new tool to begin to understand the etiology of brain injury in this age group. Histological studies in adults often demonstrate that most of the CA1 neurons are still present at the 2nd day after insult, begin to fade at the 4th day, and are completely gone by the 7th day (Abe et al., 1995; Kirino, 1982). However, data presented here demonstrates that injured neurons in the pediatric mouse brain begin to fade by the 7th day, but that many are still present. The reason for the apparent discrepancies among previous findings may be due to different ischemic models, species or age. One commonly used global cerebral ischemia model is bilateral common carotid artery occlusion or four-vessel occlusion (Kirino, 1982; Pulsinelli and Buchan, 1988). In contrast, we utilize cardiac arrest to stimulate global cerebral ischemia and indeed previous data from our laboratory demonstrates that 7–10 days after CA/CPR in adult mice, a portion of injured CA1 neurons remain (Allen et al., 2011). Regardless, our analysis of the novel pediatric mouse model of CA/CPR demonstrates delayed neuronal cell death and indicates that analysis of injury at 3 days post-resuscitation is optimal for future studies into mechanisms of injury and protection.
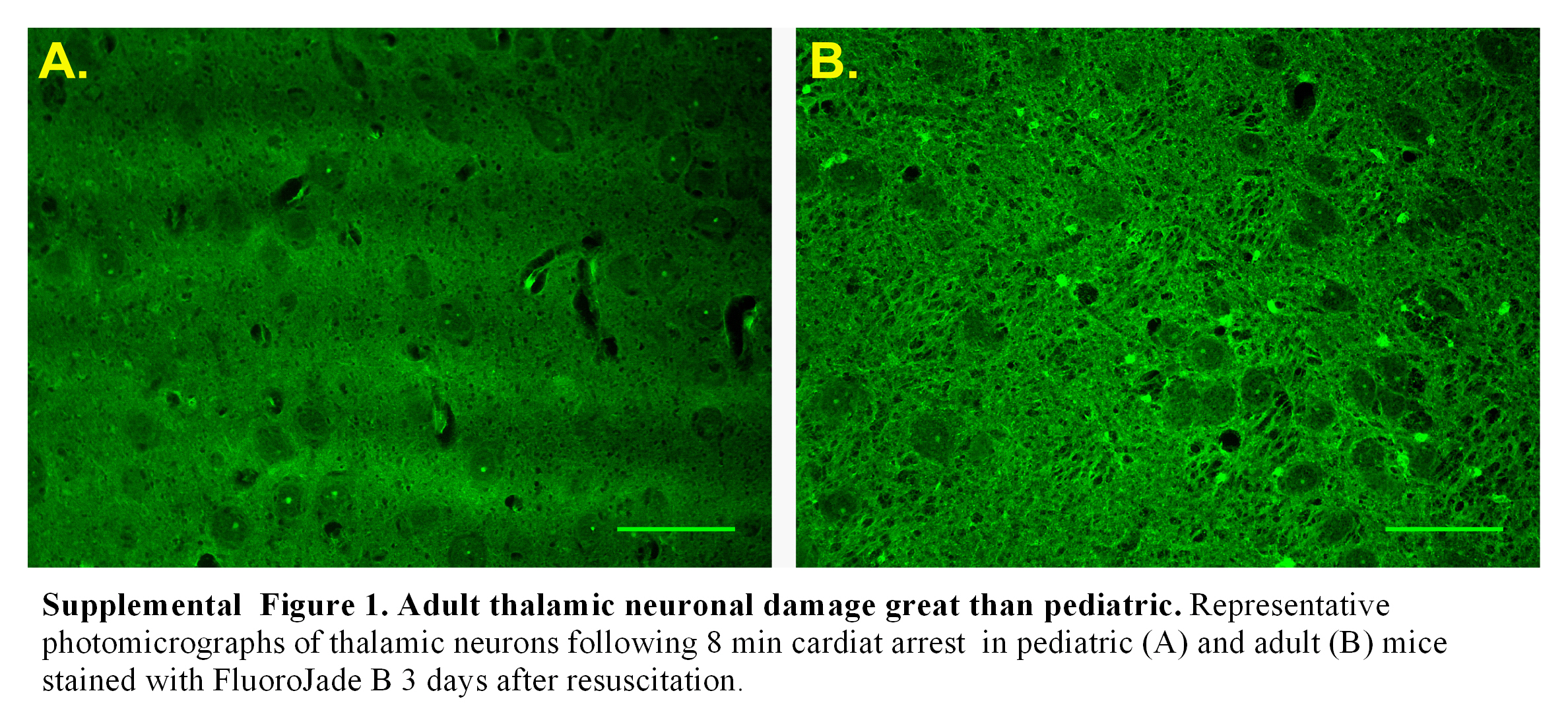
Neuronal vulnerability does not only relate to strain and species differences, but also to the stage of brain development. Brain development begins during early fetal stages and continues for at least the first decade of human childhood, during which time there is increased brain plasticity or capacity to be shaped or molded by experience (Johnston, 2004; Johnston et al., 2001). Similarly, the rodent brain develops during early embryonic stages and continues for the first few weeks postnatally. It is believed that developing brain recovers better from ischemia than mature brain, but this remains controversial (Anderson et al., 2011; Rice and Barone, 2000). Our findings suggest that pediatric mice have better histological neuronal outcome compared to adult mice. Interestingly, we observe that the relative ischemic tolerance in the hippocampus is lost when exposed to more severe ischemia. In contrast, striatal vulnerability to ischemia remains extremely low in the pediatric brain, even when exposed to severe ischemia. Similarly, pediatric mice exhibited undetectable injury in the thalamus following 8 min CA, in contrast to small but detectable injury in the adult (Supplemental Figure 1). The mechanism underlying this unexpected regional difference is unclear and warrants further study. Glutamate excitotoxicity is considered an early and important initiator of ischemic neuronal damage (Moskowitz et al., 2010; Prass and Dirnagl, 1998) and consistent with this the hippocampal CA1 region has extremely dense excitatory input, but few inhibitory neurons (Monaghan et al., 1983; Onodera et al., 1987). Brain development studies in mice have observed that hippocampal neurons and excitatory synapses are rapidly forming during the second and third postnatal week, continuously maturing until relatively full maturity is reached by around P28 (Mody et al., 2001). Furthermore, increasing vulnerability has shown to be correlated with maturation of excitatory synapses and maximal synaptogenesis (Hickey and Painter, 2006). Hence, it is likely that the reduced ischemic injury observed in our pediatric mice may in part be explained by the developmental stage of the brain resulting in relatively immature excitatory synapses. Further, increased ischemic injury observed in our 8 min CA/CPR group is likely due to excitotoxicity that overwhelms this relative advantage. In addition to differences in synaptic development, it is possible that age-dependent differences in sensitivity to high oxygen during resuscitation or brain oxidative stress mechanisms underlie differential injury observed across the ages studied. Regardless of mechanism, development of a mouse model of pediatric CA/CPR has allowed direct comparison of ischemic sensitivity between pediatric and adult mice and provides an important new research tool to dissect mechanisms of injury and possible new interventions specifically for the pediatric population.
Extensive basic and clinical research has shown that mild hypothermia improves neurologic outcome and long-term survival following cardiac arrest (Choi et al., 2012; Safar and Kochanek, 2002). Moreover, hypothermia therapy is recommended as an effective treatment for ventricular arrhythmia-induced cardiac arrest in adults and for hypoxic ischemic encephalopathy in newborns, and also has been added in the guideline by the American Heart Association for children who remain comatose after resuscitation (Doherty et al., 2009; Nolan et al., 2003). However, emerging data from this study and others clearly demonstrates that unique brain features exist during childhood development that likely alter the etiology of brain injury in the young compared to adult brain. Hence, supportive data for therapeutic hypothermia treatment after pediatric CA/CPR is still needed. Here, we demonstrated that inducing hypothermia treatment after resuscitation in our pediatric CA/CPR model significantly reduced hippocampal neuronal damage, consistent with recent studies in the rat (Fink et al., 2005). Although the mechanisms of neuroprotection following hypothermia remain poorly understood, several studies have hypothesized that reduced brain temperature may minimize cerebral metabolic demand (Eisenburger et al., 2001). Additionally, recent studies have demonstrated that hypothermia can mitigate various other pathological processes after ischemia, i.e. excitotoxicity, apoptosis, free radical production and neuroinflammation (Bayir et al., 2009; Ceulemans et al., 2010; Polderman, 2009). Our finding provides pre-clinical data supporting the use of therapeutic hypothermia in children who suffer cardiac arrest, although further research is necessary to elucidate the mechanism and long-term effect of hypothermia after pediatric resuscitation.
5. Conclusions
We have developed a novel experimental model of pediatric CA/CPR in mice which mimics the clinical situation. This new mouse model differs from the reported rat model in the method of inducing cardiac arrest. The use of KCl to stop the heart in our mouse model has the advantage of being extremely reproducible regarding ischemic duration, compared to the age variability inherent in asphyxial cardiac arrest, which introduced the possibility that juvenile and adult rats would require different durations of anoxia to cause the heart to stop. Indeed, reports indicate that asphyxial arrest requires 3–4 minutes to cause loss of circulation in adult rats (Katz et al., 1995) and 60–90 seconds in pediatric rats (Fink et al., 2004; Shoykhet et al., 2012). This new model will provide an important and clinically relevant platform for the study of cardiac arrest in the pediatric brain. Our data demonstrates that cardiac arrest produces global cerebral ischemia and neuronal injury in selectively vulnerable brain regions in juvenile mice. However, the injured area and degree of injury is different from the adult brain. Hypothermia as an effective therapy after adult cardiac arrest, and our data shows that hypothermic neuroprotection is evident in our pediatric CA/CPR model.
Supplementary Material
{kind=link}
Highlights.
New mouse model of pediatric cardiac arrest & CPR to study global cerebral ischemia
Pediatric brain less sensitive to global ischemia than adult brain
Pediatric striatum relatively resistant to ischemia
Mild hypothermia protects pediatric mouse brain from ischemic damage
Acknowledgments
Project funded by NIH grant NS046072, Walter S. and Lucienne Driskill Foundation grant
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- The Hypotherrmia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. The New England journal of medicine. 2002;346:549–556. doi: 10.1056/NEJMoa012689. [DOI] [PubMed] [Google Scholar]
- Abe K, Aoki M, Kawagoe J, Yoshida T, Hattori A, Kogure K, Itoyama Y. Ischemic delayed neuronal death. A mitochondrial hypothesis. Stroke. 1995;26:1478–1489. doi: 10.1161/01.str.26.8.1478. [DOI] [PubMed] [Google Scholar]
- Allen D, Nakayama S, Kuroiwa M, Nakano T, Palmateer J, Kosaka Y, Ballesteros C, Watanabe M, Bond CT, Lujan R, Maylie J, Adelman JP, Herson PS. SK2 channels are neuroprotective for ischemia-induced neuronal cell death. J Cereb Blood Flow Metab. 2011;31:2302–2312. doi: 10.1038/jcbfm.2011.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson V, Spencer-Smith M, Wood A. Do children really recover better? Neurobehavioural plasticity after early brain insult. Brain. 2011;134:2197–2221. doi: 10.1093/brain/awr103. [DOI] [PubMed] [Google Scholar]
- Arrich J, Holzer M, Havel C, Mullner M, Herkner H. Hypothermia for neuroprotection in adults after cardiopulmonary resuscitation. Cochrane Database Syst Rev. 2012;9:CD004128. doi: 10.1002/14651858.CD004128.pub3. [DOI] [PubMed] [Google Scholar]
- Bayir H, Adelson PD, Wisniewski SR, Shore P, Lai Y, Brown D, Janesko-Feldman KL, Kagan VE, Kochanek PM. Therapeutic hypothermia preserves antioxidant defenses after severe traumatic brain injury in infants and children. Critical care medicine. 2009;37:689–695. doi: 10.1097/CCM.0b013e318194abf2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N. Engl. J. Med. 2002;346:557–563. doi: 10.1056/NEJMoa003289. [DOI] [PubMed] [Google Scholar]
- Bottiger BW, Schmitz B, Wiessner C, Vogel P, Hossmann KA. Neuronal stress response and neuronal cell damage after cardiocirculatory arrest in rats. J Cereb Blood Flow Metab. 1998;18:1077–1087. doi: 10.1097/00004647-199810000-00004. [DOI] [PubMed] [Google Scholar]
- Bunch TJ, White RD, Smith GE, Hodge DO, Gersh BJ, Hammill SC, Shen WK, Packer DL. Long-term subjective memory function in ventricular fibrillation out-of-hospital cardiac arrest survivors resuscitated by early defibrillation. Resuscitation. 2004;60:189–195. doi: 10.1016/j.resuscitation.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Ceulemans AG, Zgavc T, Kooijman R, Hachimi-Idrissi S, Sarre S, Michotte Y. The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. Journal of neuroinflammation. 2010;7:74. doi: 10.1186/1742-2094-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HA, Badjatia N, Mayer SA. Hypothermia for acute brain injury--mechanisms and practical aspects. Nature reviews. Neurology. 2012;8:214–222. doi: 10.1038/nrneurol.2012.21. [DOI] [PubMed] [Google Scholar]
- Doherty DR, Parshuram CS, Gaboury I, Hoskote A, Lacroix J, Tucci M, Joffe A, Choong K, Farrell R, Bohn DJ, Hutchison JS. Hypothermia therapy after pediatric cardiac arrest. Circulation. 2009;119:1492–1500. doi: 10.1161/CIRCULATIONAHA.108.791384. [DOI] [PubMed] [Google Scholar]
- Eisenburger P, Sterz F, Holzer M, Zeiner A, Scheinecker W, Havel C, Losert H. Therapeutic hypothermia after cardiac arrest. Current opinion in critical care. 2001;7:184–188. doi: 10.1097/00075198-200106000-00007. [DOI] [PubMed] [Google Scholar]
- Fink EL, Alexander H, Marco CD, Dixon CE, Kochanek PM, Jenkins LW, Lai Y, Donovan HA, Hickey RW, Clark RS. Experimental model of pediatric asphyxial cardiopulmonary arrest in rats. Pediatric critical care medicine : a journal of the Society of Critical Care Medicine and the World Federation of Pediatric Intensive and Critical Care Societies. 2004;5:139–144. doi: 10.1097/01.pcc.0000112376.29903.8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink EL, Marco CD, Donovan HA, Alexander H, Dixon CE, Jenkins LW, Stange CJ, Kochanek PM, Clark RS. Brief induced hypothermia improves outcome after asphyxial cardiopulmonary arrest in juvenile rats. Developmental neuroscience. 2005;27:191–199. doi: 10.1159/000085992. [DOI] [PubMed] [Google Scholar]
- Garcia JH, Anderson ML. Physiopathology of cerebral ischemia. Critical reviews in neurobiology. 1989;4:303–324. [PubMed] [Google Scholar]
- Hickey RW, Painter MJ. Brain injury from cardiac arrest in children. Neurologic clinics. 2006;24:147–158. doi: 10.1016/j.ncl.2005.10.002. viii. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Mosinger JL, Salles KS, Labruyere J, Olney JW. Sensitivity of the developing rat brain to hypobaric/ischemic damage parallels sensitivity to N-methyl-aspartate neurotoxicity. J Neurosci. 1989;9:2809–2818. doi: 10.1523/JNEUROSCI.09-08-02809.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito U, Spatz M, Walker JT, Jr, Klatzo I. Experimental cerebral ischemia in mongolian gerbils. I. Light microscopic observations. Acta neuropathologica. 1975;32:209–223. doi: 10.1007/BF00696570. [DOI] [PubMed] [Google Scholar]
- Johnston MV. Clinical disorders of brain plasticity. Brain & development. 2004;26:73–80. doi: 10.1016/S0387-7604(03)00102-5. [DOI] [PubMed] [Google Scholar]
- Johnston MV, Nishimura A, Harum K, Pekar J, Blue ME. Sculpting the developing brain. Advances in pediatrics. 2001;48:1–38. [PubMed] [Google Scholar]
- Katz L, Ebmeyer U, Safar P, Radovsky A, Neumar R. Outcome model of asphyxial cardiac arrest in rats. J Cereb Blood Flow Metab. 1995;15:1032–1039. doi: 10.1038/jcbfm.1995.129. [DOI] [PubMed] [Google Scholar]
- Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain research. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- Knudson JD, Neish SR, Cabrera AG, Lowry AW, Shamszad P, Morales DL, Graves DE, Williams EA, Rossano JW. Prevalence and outcomes of pediatric in-hospital cardiopulmonary resuscitation in the United States: an analysis of the Kids' Inpatient Database*. Critical care medicine. 2012;40:2940–2944. doi: 10.1097/CCM.0b013e31825feb3f. [DOI] [PubMed] [Google Scholar]
- Kofler J, Hattori K, Sawada M, DeVries AC, Martin LJ, Hurn PD, Traystman RJ. Histopathological and behavioral characterization of a novel model of cardiac arrest and cardiopulmonary resuscitation in mice. J. Neurosci. Methods. 2004;136:33–44. doi: 10.1016/j.jneumeth.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Kosaka Y, Quillinan N, Bond C, Traystman R, Hurn P, Herson P. GPER1/GPR30 activation improves neuronal survival following global cerebral ischemia induced by cardiac arrest in mice. Translational stroke research. 2012;3:500–507. doi: 10.1007/s12975-012-0211-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C, Alexander MP, LaFleche G, Schnyer DM, Verfaellie M. The neurological and cognitive sequelae of cardiac arrest. Neurology. 2004;63:1774–1778. doi: 10.1212/01.wnl.0000144189.83077.8e. [DOI] [PubMed] [Google Scholar]
- Manole MD, Kochanek PM, Foley LM, Hitchens TK, Bayir H, Alexander H, Garman R, Ma L, Hsia CJ, Ho C, Clark RS. Polynitroxyl albumin and albumin therapy after pediatric asphyxial cardiac arrest: effects on cerebral blood flow and neurologic outcome. J Cereb Blood Flow Metab. 2012;32:560–569. doi: 10.1038/jcbfm.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateen FJ, Josephs KA, Trenerry MR, Felmlee-Devine MD, Weaver AL, Carone M, White RD. Long-term cognitive outcomes following out-of-hospital cardiac arrest: a population-based study. Neurology. 2011;77:1438–1445. doi: 10.1212/WNL.0b013e318232ab33. [DOI] [PubMed] [Google Scholar]
- Mody M, Cao Y, Cui Z, Tay KY, Shyong A, Shimizu E, Pham K, Schultz P, Welsh D, Tsien JZ. Genome-wide gene expression profiles of the developing mouse hippocampus. Proc Natl Acad Sci U S A. 2001;98:8862–8867. doi: 10.1073/pnas.141244998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan DT, Holets VR, Toy DW, Cotman CW. Anatomical distributions of four pharmacologically distinct 3H-L-glutamate binding sites. Nature. 1983;306:176–179. doi: 10.1038/306176a0. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu K, Fukuda A, Togari H, Wada Y, Nishino H. Vulnerability to cerebral hypoxic-ischemic insult in neonatal but not in adult rats is in parallel with disruption of the blood-brain barrier. Stroke. 1997;28:2281–2288. doi: 10.1161/01.str.28.11.2281. discussion 8–9. [DOI] [PubMed] [Google Scholar]
- Nolan JP, Morley PT, Vanden Hoek TL, Hickey RW, Kloeck WG, Billi J, Bottiger BW, Okada K, Reyes C, Shuster M, Steen PA, Weil MH, Wenzel V, Carli P, Atkins D. Therapeutic hypothermia after cardiac arrest: an advisory statement by the advanced life support task force of the International Liaison Committee on Resuscitation. Circulation. 2003;108:118–121. doi: 10.1161/01.CIR.0000079019.02601.90. [DOI] [PubMed] [Google Scholar]
- O'Reilly SM, Grubb NR, O'Carroll RE. In-hospital cardiac arrest leads to chronic memory impairment. Resuscitation. 2003;58:73–79. doi: 10.1016/s0300-9572(03)00114-x. [DOI] [PubMed] [Google Scholar]
- Onodera H, Sato G, Kogure K. GABA and benzodiazepine receptors in the gerbil brain after transient ischemia: demonstration by quantitative receptor autoradiography. J Cereb Blood Flow Metab. 1987;7:82–88. doi: 10.1038/jcbfm.1987.12. [DOI] [PubMed] [Google Scholar]
- Peskine A, Rosso C, Picq C, Caron E, Pradat-Diehl P. Neurological sequelae after cerebral anoxia. Brain injury : [BI] 2010;24:755–761. doi: 10.3109/02699051003709581. [DOI] [PubMed] [Google Scholar]
- Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Critical care medicine. 2009;37:S186–S202. doi: 10.1097/CCM.0b013e3181aa5241. [DOI] [PubMed] [Google Scholar]
- Prass K, Dirnagl U. Glutamate antagonists in therapy of stroke. Restor Neurol Neurosci. 1998;13:3–10. [PubMed] [Google Scholar]
- Pulsinelli WA. Selective neuronal vulnerability: morphological and molecular characteristics. Prog. Brain Res. 1985;63:29–37. doi: 10.1016/S0079-6123(08)61973-1. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann. Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Buchan AM. The four-vessel occlusion rat model: method for complete occlusion of vertebral arteries and control of collateral circulation. Stroke. 1988;19:913–914. doi: 10.1161/01.str.19.7.913. [DOI] [PubMed] [Google Scholar]
- Rice D, Barone S., Jr Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environmental health perspectives. 2000;108(Suppl 3):511–533. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart Disease and Stroke Statistics--2012 Update: A Report From the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safar PJ, Kochanek PM. Therapeutic hypothermia after cardiac arrest. N. Engl. J. Med. 2002;346:612–613. doi: 10.1056/NEJM200202213460811. [DOI] [PubMed] [Google Scholar]
- Shoykhet M, Simons DJ, Alexander H, Hosler C, Kochanek PM, Clark RS. Thalamocortical dysfunction and thalamic injury after asphyxial cardiac arrest in developing rats. J Neurosci. 2012;32:4972–4981. doi: 10.1523/JNEUROSCI.5597-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–129. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Tang M, Alexander H, Clark RS, Kochanek PM, Kagan VE, Bayir H. Minocycline reduces neuronal death and attenuates microglial response after pediatric asphyxial cardiac arrest. J Cereb Blood Flow Metab. 2010;30:119–129. doi: 10.1038/jcbfm.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towfighi J, Mauger D, Vannucci RC, Vannucci SJ. Influence of age on the cerebral lesions in an immature rat model of cerebral hypoxia-ischemia: a light microscopic study. Brain research. Developmental brain research. 1997;100:149–160. doi: 10.1016/s0165-3806(97)00036-9. [DOI] [PubMed] [Google Scholar]
- Walson KH, Tang M, Glumac A, Alexander H, Manole MD, Ma L, Hsia CJ, Clark RS, Kochanek PM, Kagan VE, Bayr H. Normoxic versus hyperoxic resuscitation in pediatric asphyxial cardiac arrest: effects on oxidative stress. Critical care medicine. 2011;39:335–343. doi: 10.1097/CCM.0b013e3181ffda0e. [DOI] [PubMed] [Google Scholar]
- Yager JY, Shuaib A, Thornhill J. The effect of age on susceptibility to brain damage in a model of global hemispheric hypoxia-ischemia. Brain research. Developmental brain research. 1996;93:143–154. doi: 10.1016/0165-3806(96)00026-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.