

Abstract
In this report, we describe a series of 4-substituted piperidine and piperazine compounds based on tetrahydroquinoline 1, a compound that shows balanced, low nanomolar binding affinity for the mu opioid receptor (MOR) and the delta opioid receptor (DOR). We have shown that by changing the length and flexibility profile of the side chain in this position, binding affinity is improved at both receptors by a significant degree. Furthermore, several of the compounds described herein display good efficacy at MOR, while simultaneously displaying DOR antagonism. The MOR agonist/DOR antagonist has shown promise in the reduction of negative side effects displayed by selective MOR agonists, namely the development of dependence and tolerance.
Although opioid analgesics represent the gold standard for the treatment of acute and chronic pain, their usage is often accompanied by undesirable side effects such as the development of dependence and tolerance. A considerable amount of research has thus been done to find a potent analgesic that does not display these negative attributes. In general, clinically used opioid analgesics such as morphine evoke both the desired and undesired effects through activation of the mu opioid receptor (MOR). Numerous reports have indicated that the undesired MOR-related side effects may be ameliorated by concomitant ligand interaction with the delta opioid receptor (DOR). It has been shown that the co-administration of DOR-selective agonists1 or antagonists2 with a MOR agonist can attenuate the dependence and tolerance typically associated with the latter.
A ligand displaying good binding affinity for both MOR and DOR represents a significant advantage over the co-administration of multiple drugs, due to both increased pharmacokinetic simplicity as well as improved patient compliance. For these reasons, the development of small molecule MOR/DOR bifunctional opioid ligands has attracted much attention. MOR agonist/DOR antagonist compounds have been shown to be effective analgesics with a diminished tolerance and dependence profile3,4 and have found use in other areas, such as for the treatment of irritable bowel syndrome.5 Recently, we showed that a MOR agonist/DOR antagonist compound was an effective analgesic after interperitoneal administration, with a duration of action comparable to morphine.6
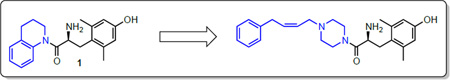
In an effort to further develop drug-like MOR/DOR bifunctional ligands, we turned our attention to compound 1 (Figure 1) a compound previously synthesized by our lab that displays equal binding affinity for both MOR and DOR, as well as for the kappa opioid receptor (KOR) (Ki = 25.8 nM (MOR); 33.0 nM (DOR); 36.5 nM (KOR), unpublished observations). Given the relative simplicity of the compound and its nonselective binding profile, we reasoned it would be a good starting point for derivatization. Computational modeling suggested that position 4 would be the optimal point for diversification, as an aromatic moiety at this position would be ideally situated to interact with Asn125, Thr218, and Lys303 in the MOR active site, and the resulting compound would thus function as a MOR agonist.7.
Figure 1.

Compound 1
1 was initially substituted with a benzyl group at the 4 position (2, Table 1). The synthesis of 2 began by subjecting ketone 13 to a Wittig reaction to yield alkene 14, which was subsequently hydrogenated and deprotected to give amine 15, to which was coupled Boc-protected L-2,6-dimethyltyrosine (Boc-L-Dmt) and deprotected (Scheme 1). Binding affinity (Ki) was obtained by competitive displacement of radiolabeled [3H]diprenorphine in C6 cells stably expressing MOR or DOR or CHO cells stably expressing KOR. Efficacy was assessed by agonist-stimulated [35S] GTPγS binding in the same cells.6,8,9
Table 1.
Binding affinity and efficacy data for analogues 2–12a
| Structure | Ki (nM) | EC50 (nM) | % stimulation | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MOR | DOR | KOR | MOR | DOR | KOR | MOR | DOR | KOR | ||
| 2 | 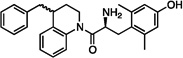 |
29±9 | 14±2 | 310±50 | dns | dns | dns | 11±6 | dns | dns |
| 3 | 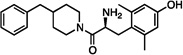 |
1.4±0.1 | 7.8±0.9 | 140±24 | dns | dns | dns | dns | dns | dns |
| 4 | 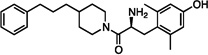 |
2.0±0.8 | 12±2 | 110±11 | 119±39 | dns | dns | 26±2 | dns | dns |
| 5 | 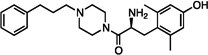 |
3.8±0.8 | 36±4 | 250±51 | 150±49 | dns | dns | 23±2 | dns | dns |
| 6 | 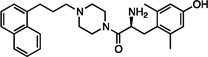 |
1.1±0.3 | 6.6±2 | 78±15 | 85±10 | dns | dns | 17±3 | dns | dns |
| 7 | 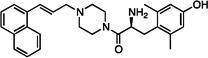 |
6.4±0.1 | 11±0.65 | 330±92 | dns | dns | dns | dns | dns | dns |
| 8 | 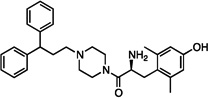 |
1.1±0.2 | 21±7 | 150±42 | dns | dns | dns | dns | dns | dns |
| 9 | 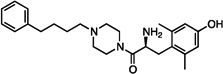 |
0.45±0.3 | 6.6±2 | 78±15 | 64±3 | dns | dns | 43±6 | dns | 12±1 |
| 10 | 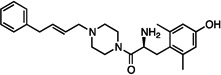 |
0.29±0.07 | 23±8.9 | 89±23 | 20±3.6 | dns | 1500±120 | 36±4 | dns | 16±3 |
| 11 | 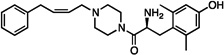 |
0.3±0.1 | 28±6.8 | 54±3.2 | 41±15 | dns | dns | 49±5 | dns | 11±7 |
| 12 | 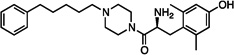 |
0.42±0.15 | 6.9±1.7 | 39±15 | 53±23 | dns | 660±80 | 42±1 | dns | 12±5 |
All values are expressed as the mean ± SEM of three separate assays performed in duplicate. dns: does not stimulate. Binding affinities (Ki) were obtained by competitive displacement of radiolabeled [3H]diprenorphine in membrane preparations. Efficacy data were obtained using agonist induced stimulation of [35S]GTPγS binding assay. Efficacy is represented as EC50 (nM) and percent maximal stimulation relative to standard agonist DAMGO (MOR), DPDPE (DOR), or U69,593 (KOR) at 10 µM.
Scheme 1. Synthesis of compound 2a.

a. Reagents and conditions: (a) triphenylphosphinebenzyl bromide, n-BuLi, THF, reflux; (b) H2, 10% Pd/C, MeOH, 50 psi; (c) TFA, DCM; (d) Boc-L-Dmt, HATU, HOBt-Cl, DIEA, DMF, 4Å molecular sieves, 40°C; (e) TFA, DCM.
Compared to 1, the resulting compound 2 displayed no significant change in binding affinity for MOR and DOR, but showed decreased affinity for KOR (Table 1). Unfortunately, 2 also displayed no notable efficacy at MOR as determined by the [35S] GTPγS assay. Because the synthesis of 2 proved somewhat laborious, and the resulting diastereomers could not be resolved by RP-HPLC, we reasoned that synthesis of further analogues could be simplified by the replacement of the tetrahydroquinoline (THQ) core of 2 with a piperidine, effectively eliminating a stereocenter. The resulting compound 3 displayed roughly a tenfold increase in binding affinity for MOR and DOR, but still lacked any efficacy at MOR. The remainder of our SAR campaign was focused on changing the length and flexibility profile of the side chain in an attempt to not only retain strong binding affinity for both MOR and DOR, but to increase efficacy at MOR. For purposes of synthetic utility as well as increased solubility, the piperidine core was also replaced with a piperazine for most of the analogues, the results of which are summarized in Table 1.
Compounds 3–5, and 9 were synthesized by coupling a commercially available piperidine or piperazine derivative with Boc-L-Dmt, followed by TFA-mediated deptrotection and HPLC purification to yield the final compounds. In the case of 8 and 12, a commercially available primary alcohol was first mesylated and refluxed with excess piperazine to give intermediates 16 and 17, which were then coupled with Boc-L-Dmt and deprotected under similar conditions (Scheme 2). The remainder of the compounds were synthesized as shown in Scheme 3. The appropriate commercially available aldehyde was subjected to a Horner-Wadsworth-Emmons type olefination to give alkenes 18, 19 and 27, which were then reduced to the corresponding alcohols using either DIBAL (for the formation of allylic alchols) or LAH (for the formation of saturated alcohols). Before reduction, alkene 19 was first hydrogenated to give saturated ester 24. All intermediates were then carried forward in a similar manner as in Scheme 2 to give finished products.
Scheme 2. Synthesis of analogues 3–5, 8, 9, 12a.

a. Reagents and conditions: (a) Boc-L-Dmt, PyBOP or HATU, HOBt-Cl, DIEA, DMF; (b) TFA, DCM; (c) MsCl, Et3N, DCM, 0°C; (d) piperazine, THF, reflux.
Scheme 3. Synthesis of analogues 6, 7, 10, 11a.

a. Reagents and conditions: (a) O=P(CH3CH2O)2CH2CO2CH3, NaH, THF, 0°C; (b) O=P(CF3CH2O)2CH2CO2CH3, NaH, THF, 0°C; (c) DIBAL, DCM, −78°C; (d) H2, 10% Pd/C, MeOH, 15 psi; (e) LAH, THF, 0°C; (f) MsCl, Et3N, DCM, 0°C; (g) piperazine, THF, reflux.
The synthesized analogues in Table 1 display a broad range of binding affinities for MOR (29 nM to 0.29 nM), and to a lesser extent, DOR (150 nM to 6.6 nM). Extension of the side chain of 3 from 1 to 3 methylene units did little to change binding at MOR or DOR, but encouragingly, the resulting compound (4) behaved as a weak partial agonist at MOR. Replacement of the piperidine core of 4 with a piperazine (5) proved inconsequential, and the continued balanced MOR/DOR binding profile of this analogue led us to pursue other aromatic moieties separated by three methylene units from the piperazine core. Analogue 6 in particular showed an improved balanced MOR/DOR binding profile, and also displayed a partial agonist profile at MOR. Interestingly, compound 7, in which the 1-naphthyl side chain of 6 is constrained with an additional double bond, showed no efficacy in the [35S]GTPγS assay at all three receptors, with an additional loss of binding affinity for KOR. The insertion of an extra aromatic moiety as in the case of the diphenylmethyl analogue 8 did little to increase binding affinity for either MOR or DOR. Further extension of the distance between the aromatic side chain and the piperazine core (9) resulted in a boost in MOR binding, without drastically affecting DOR. Although these 4 carbon analogues (9–11) suffered a slight loss of MOR/DOR affinity balance, all displayed good efficacy at MOR, particularly the unsaturated analogues 10 and 11 (20 and 41 nM, respectively). Side chain extension to 5 methylene units (12) did little to improve upon the profile of 10 or 11.
Structurally, these analogues exhibit some similarities to the class of trans-3,4-dimethyl- 4-(3-hydroxyphenyl)piperidine opioid antagonists originally described by Zimmerman10 and explored by others.11 In our series, the 3-hydroxyphenyl moiety is replaced by 2,6-L-dimethyltyrosine, and the piperidine (or piperazine) core is left unsubstituted. In both series, receptor selectivity is modulated by the nature of the lipophilic side chain attached para to the phenolic component of the molecule. The 11 piperidine and piperazine analogues of tetrahydroquinoline 1 described here display a favorable balance between binding affinity at MOR and DOR, and several (4–6, 9–12) display improved potency at MOR as compared to morphine (Ki (MOR) = 6.3 nM, (DOR) = 171 nM; EC50 (MOR) = 194 nM).12,13 These analogues are therefore promising leads for further derivatization and in vivo studies.
Supplementary Material
Acknowledgements
This work was supported by NIH grants DA003910 (H.I.M) and DA004087 (J.R.T). The authors thank Lisa Rosenthal and Alisha Griffin for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Lowery JJ, Raymond TJ, Giuvelis D, Bidlack JM, Polt R, Bilsky EJ. In Vivo Characterization of MMP-2200, a Mixed δ/µ Opioid Agonist, In Mice. J. Pharmacol. Exp. Ther. 2011;336:767–778. doi: 10.1124/jpet.110.172866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdelhamid EE, Sultana M, Portoghese PS, Takemori AE. Selective Blockage of the Delta Opioid Receptors Prevents the Development of Morphine Tolerance and Dependence in Mice. J. Pharmacol. Exp. Ther. 1991;258:299–303. [PubMed] [Google Scholar]
- 3.Schiller PW, Fundytus ME, Merovitz L, Weltrowska G, Nguyen TM, Lemieux C, Chung NN, Coderre TJ. The Opioid mu Agonist/delta Antagonist DIPP-NH(2)[Psi] Produces a Potent Analgesic Effect, No Physical Dependence, and Less Tolerance than Morphine in Rats. J. Med. Chem. 1999;42:3520–3526. doi: 10.1021/jm980724+. [DOI] [PubMed] [Google Scholar]
- 4.Ananthan S, Saini SK, Dersch CM, Xu H, McGlinchey N, Giuvelis D, Bilsky EJ, Rothman RB. 14-Alkoxy- and 14- Acyloxypyridomorphinans: µ Agonist/δ Antagonist Opioid Analgesics with Diminished Tolerance and Dependence Side Effects. J. Med. Chem. 2012;55:8350–8363. doi: 10.1021/jm300686p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breslin HJ, Diamond CJ, Kavash RW, Cai C, Dyatkin AB, Miskowski TA, Zhang S, Wade PR, Hornby PJ, He W. Identification of a Dual δ OR Antagonist/µ OR Agonist as a Potential Therapeutic for Diarrhea-Predominant Irritable Bowel Syndrome (IBS-d) Bioorg. Med. Chem. Lett. 2012;22:4869–4872. doi: 10.1016/j.bmcl.2012.05.042. [DOI] [PubMed] [Google Scholar]
- 6.Mosberg HI, Yeomans L, Harland AA, Bender AM, Sobczyk-Kojiro K, Anand JP, Clark MJ, Jutkiewicz EM, Traynor JR. Opioid Peptidomimetics: Leads for the Design of Bioavailable Mixed Efficacy µ Opioid Receptor (MOR) Agonist/δ Opioid Receptor (DOR) Antagonist Ligands. J. Med. Chem. 2013;56:2139–2149. doi: 10.1021/jm400050y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anand JP, Purington LC, Pogozheva ID, Traynor JR, Mosberg HI. Modulation of Opioid Receptor Ligand Affinity and Efficacy Using Active and Inactive State Receptor Models. Chem. Biol. Drug Des. 2012;80:763–770. doi: 10.1111/cbdd.12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radioactive compounds were purchased from Perkin-Elmer (Waltham, MA, U.S.). Opioid ligand- binding assays were performed using competitive displacement of 0.2 nM [3H]diprenorphine (50Ci/mmol) by the test compound from membrane preparations containing opioid receptors. The assay mixture, containing membrane suspension (20 µg protein/ tube) in 50 mM Tris-HCl buffer (pH 7.4), [3H]diprenorphine, and various concentrations of test peptide, was incubated at room temperature for 1 h to allow binding to reach equilibrium. The samples were rapidly filtered through Whatman GF/C filters using a Brandel harvester (Brandel, Gaithersburg, MD, U.S.) and washed three times with 50 mM Tris-HCl buffer. The radioactivity retained on dried filters was determined by liquid scintillation counting after saturation with EcoLume liquid scintillation cocktail in a Wallac 1450 MicroBeta (Perkin-Elmer, Waltham, MA, U.S.). Nonspecific binding was determined using 10 µM naloxone. Ki values were calculated using nonlinear regression analysis to fit a logistic equation to the competition data using GraphPad Prism, version 5.01, for Windows (GraphPad Software Inc., La Jolla, CA). The results presented are the mean ± standard error from at least three separate assays performed in duplicate.
- 9.Membranes (10–20 µg of protein/tube) were incubated 1 h at room temperature in GTPγS buffer (50 mM Tris-HCl, 100 mM NaCl, 5 mM MgCl, pH 7.4) containing 0.1 nM [35S]GTPγS, 30 µM guanosine diphosphate (GDP), and varying concentrations of test peptides. Peptide stimulation of [35S]GTPγS was compared with 10 µM standard compounds [D-Ala2,N-MePhe4,Gly-ol]enkephalin (DAMGO) at MOR, D-Pen2,5-enkephalin (DPDPE) at DOR, or U69,593 at KOR. The reaction was terminated by rapidly filtering through GF/C filters and washing 10 times with GTPγS buffer, and retained radioactivity was measured as described above. The results presented are the mean ± standard error from at least three separate assays performed in duplicate; maximal stimulation was determined using nonlinear regression analysis with GraphPad Prism (GraphPad Software Inc., La Jolla, CA).
- 10.Zimmerman DM, Nickander R, Horng JS, Wong DT. New Structural Concepts for Narcotic Antagonists Defined in a 4-Phenylpiperidine Series. Nature. 1978;275:332–334. doi: 10.1038/275332a0. [DOI] [PubMed] [Google Scholar]
- 11.Le Bourdonnec B, Goodman AJ, Michaut M, Ye H, Graczyk TM, Belanger S, Herbertz T, Yap GPA, DeHaven RN, Dolle RE. Elucidation of the Bioactive Conformation of the N-Substituted trans-3,4-Dimethyl-4-(3-hydroxyphenyl)piperidine Class of mu-Opioid Receptor Antagonists. J. Med. Chem. 2006;49:7278–7289. doi: 10.1021/jm060486f. [DOI] [PubMed] [Google Scholar]
- 12.Purington LC, Sobczyk-Kojiro K, Pogozheva ID, Traynor JR, Mosberg HI. Development and in Vitro Characterization of a Novel Bifunctional mu-Agonist/delta- Antagonist Opioid Tetrapeptide. ACS Chem. Biol. 2011;6:1375–1381. doi: 10.1021/cb200263q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neilan CL, Husbands SM, Breeden S, Ko MC, Aceto MD, Lewis JW, Woods JH, Traynor JR. Characterization of the Complex Morphinan Derivative BU72 as a High Efficacy, Long- Lasting mu-Opioid Receptor Agonist. Eur. J. Pharmacol. 2004;499:107–116. doi: 10.1016/j.ejphar.2004.07.097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.