

Abstract
N-linked glycosylation is a central regulatory factor that influences the immune system in varied and profound ways, including leukocyte homing, T cell receptor signaling and others. Moreover, N-glycan branching has been demonstrated to change as a function of infection and inflammation. Our previous findings suggest that complex-type N-glycans on the class II major histocompatibility complex play an important role in antigen selection within antigen presenting cells (APCs) such that highly branched N-glycans promote polysaccharide (glycoantigen, GlyAg) presentation following Toll-like receptor 2 (TLR2)-dependent antigen processing. In order to explore the impact of N-glycan branching on the myeloid-derived APC population without the confounding problems of altering the branching of lymphocytes and non-hematopoietic cells, we created a novel myeloid-specific knockout of the β-1,2-N-acetylglucosaminyltransferase II (Mgat2) enzyme. Using this novel mouse, we found that the reduction in multi-antennary N-glycans characteristic of Mgat2 ablation had no impact on GlyAg-mediated TLR2 signaling. Likewise, no deficits in antigen uptake or cellular homing to lymph nodes were found. However, we discovered that Mgat2 ablation prevented GlyAg presentation and T cell activation in vitro and in vivo without apparent alterations in protein antigen response or myeloid-mediated protection from infection. These findings demonstrate that GlyAg presentation can be regulated by the N-glycan branching pattern of APCs, thereby establishing an in vivo model where the T cell-dependent activity of GlyAgs can be experimentally distinguished from GlyAg-mediated stimulation of the innate response through TLR2.
Keywords: antigen presentation, CDG-IIa, glycoantigen, MHC class II, T lymphocyte
Introduction
It is increasingly clear that the nature of the glycans on cell surface and secreted glycoproteins significantly impacts the function of the underlying protein. In some cases, change is achieved via alterations in glycan-binding protein affinity toward its glycan ligand. For example, loss of Mgat5 and therefore tetra-antennary N-glycans in T cells causes a loss of galectin binding within the T cell receptor (TCR) complex, ultimately leading to defects in normal TCR signaling (Demetriou et al. 2001). In other cases, the change in glycoform more directly shifts the activity of the glycoprotein itself. One emerging example is the impact of α2,6-linked sialic acids on the bi-antennary glycans found with the Fc domain of IgG molecules (Anthony et al. 2012). When present, the IgG molecules show anti-inflammatory activity (Kaneko et al. 2006; Anthony et al. 2008, 2011) and decreased capacity to suppress viral replication (Ackerman et al. 2013) compared with asialyl-IgG molecules.
We have demonstrated that the class II major histocompatibility complex (MHCII) relies upon complex-type N-glycans (cN-glycans) to bind and present polysaccharide antigens leading to T cell activation (Ryan et al. 2011). One such polysaccharide “glycoantigen” (GlyAg) is polysaccharide A1 (PSA) from the capsule of Bacteroides fragilis, a commensal gram-negative bacterium present in most mammalian gastrointestinal tracts (Kasper et al. 1977). PSA is a potent stimulator of Toll-like receptor 2 (TLR2), which is necessary to activate responding antigen presenting cells (APCs) to produce nitric oxide (NO) (Wang et al. 2006). NO is then responsible for cleaving the GlyAg into fragments of appropriate size (Kreisman et al. 2007) to bind MHCII at high affinity (Cobb and Kasper 2008; Velez et al. 2009) for presentation and T cell recognition. Importantly, the T cell response generated by GlyAg stimulation appears to be protective against fibrotic (Tzianabos et al. 1994, 2000; Chung et al. 2002, 2003) and neurodegenerative autoimmunity (Ochoa-Reparaz, Mielcarz, Ditrio, et al. 2010; Ochoa-Reparaz, Mielcarz, Wang, et al. 2010). As a result, the T cell and potentially TLR2-mediated innate response to GlyAgs in the gut are critical factors behind the hygiene hypothesis whereby exposure to the appropriate microbial factors is thought to play a protective immunologic role that limits allergy and autoimmunity (Strachan, 1989, 2000). Although we have shown that cN-glycans play a role in GlyAg-mediated T cell activation, in vivo exploration of this pathway has been difficult due to lack of appropriate murine models.
In the present study, we used the Cre-Lox system to create a novel mouse strain that lacks branched cN-glycans in the myeloid lineage, including macrophages, dendritic cells and the granulocytes (e.g. neutrophils) in order to study the impact of cN-glycan branching in vivo without the complications surrounding the changes in N-glycan structure on B or T lymphocytes, or non-hematopoietic cells. We chose ablation of the murine Mgat2 locus because loss of Mgat2 function is the defining defect of the congenital disorders of glycosylation type IIa (CDG-IIa) and does not cause ER stress and in contrast to other early N-glycan processing enzyme knockout mice, the germline Mgat2-null mice remain viable (Wang et al. 2001). Humans with CDG-IIa are characterized by mutations in β-1,2-N-acetylglucosaminyltransferase II, a Golgi bound glycosyltransferase that catalyzes an essential transition step in the synthesis pathway in which hybrid N-glycans are modified to become multi-antennary cN-glycan structures (Figure 1A; Jaeken 2010). Mgat2-null mice, similar to CDG-IIa patients, exhibit serious malfunctions of multiple organ systems including the nervous, skeletomuscular and gastrointestinal systems (Wang et al. 2001; Jaeken 2010).
Fig. 1.
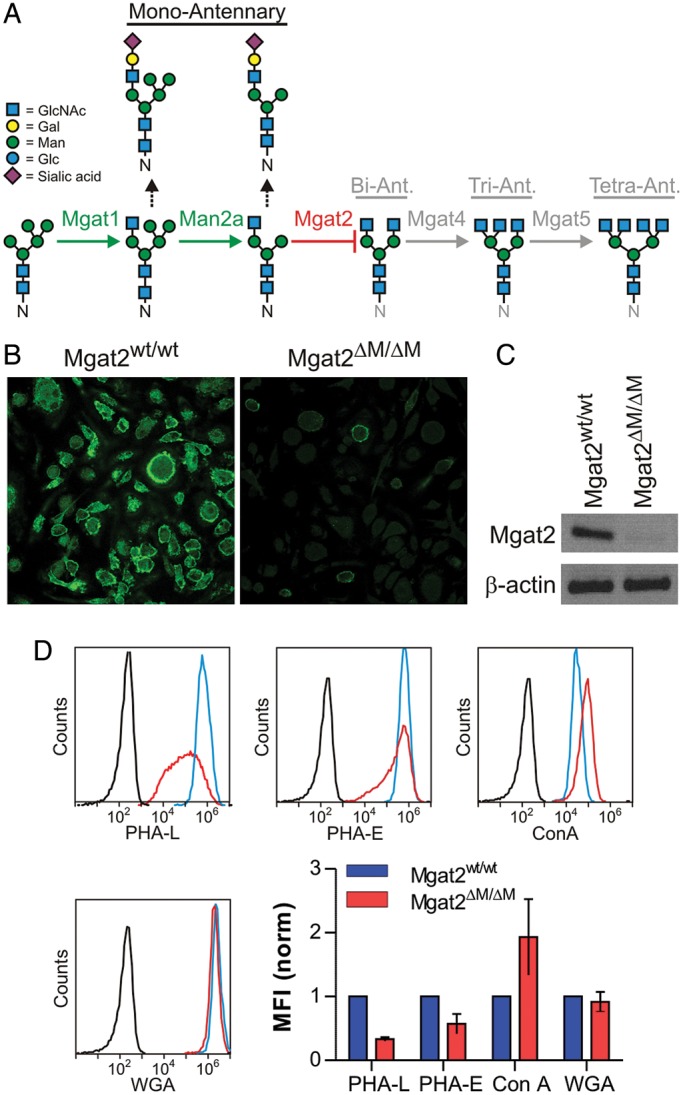
Mgat2ΔM/ΔM BMDCs lack branched cN-glycans. (A) Schematic representation of the N-glycosylation pathway and the blockade generated by Mgat2 ablation. (B) BMDCs derived from Mgat2wt/wt and Mgat2ΔM/ΔM mice stained with fluorescein-conjugated PHA-L and viewed by confocal microscopy, showing the loss of cN-glycans associated with Mgat2 loss. (C) RT-PCR from Mgat2wt/wt and Mgat2ΔM/ΔM BMDCs, demonstrating the loss of Mgat2 transcript in knockout cells. (D) Lectin flow cytometry of Mgat2wt/wt (blue) and Mgat2ΔM/ΔM (red) BMDCs compared with unstained controls (black), revealing loss of cN-glycans (PHA-L and PHA-E), an increase in available mannose (ConA), and consistent GlcNAc dimers and hybrid N-glycans (WGA). Mean fluorescence intensity (MFI) of multiple samples are shown and are consistent with a blockade in multi-antennary N-glycan synthesis. Bars represent the mean ± SD, with n ≥ 3 on all measurements.
Our study revealed that GlyAg presentation and T cell activation was severely depressed in vitro and in vivo when APCs lacked bi-, tri- or tetra-antennary cN-glycans; however, all other functions appeared normal, including antigen uptake and processing, cellular homing to the nearest draining lymph node and protection from fungal infection mediated by myeloid cells (i.e. neutrophils and macrophages). Finally, TLR2-mediated APC activation was not significantly impacted by the loss of Mgat2. These data demonstrate that the MHCII glycoform is a key component of the GlyAg presentation machinery and can serve as a regulatory feature under disease conditions known to significantly alter cN-glycan branching, such as CDG-IIa (Wang et al. 2001; Jaeken 2010), alcoholic liver disease (Mann et al. 1994; Gravel et al. 1996), breast and ovarian cancer (Goodarzi and Turner 1995; Turner et al. 1995; Saldova et al. 2007), rheumatoid arthritis (Thompson et al. 1989, 1993) and Crohn's disease (Goodarzi and Turner 1998).
Results
Ablation of the Mgat2 locus
Mice lacking a functional Mgat2 locus (Mgat2ΔM/ΔM) were created by crossing an animal carrying a Mgat2 locus with flanking LoxP sites described previously (Wang et al. 2001) with an animal expressing the CRE recombinase under the promoter for the myeloid marker lysozyme M (LyzM) described previously (Clausen et al. 1999). Mice homozygous for both loci were bred on the C57Bl/6 background and used herein to study the impact of blocking the synthesis of branched cN-glycans within myeloid-derived APCs (Figure 1A).
Bone marrow-derived dendritic cells (BMDCs) from both Mgat2ΔM/ΔM and Mgat2wt/wt control mice were derived and analyzed for surface glycoform by confocal microscopy. Using the branching-sensitive lectin Phaseolus vulgaris leucoagglutinin (PHA-L), we found that the relatively homogenous population of BMDCs derived from Mgat2ΔM/ΔM mice showed low binding compared with wild-type control BMDCs (Figure 1B), indicating a paucity of mature multi-antennary cN-glycans at the cell surface. As was expected, RT-PCR from BMDC populations confirmed the lack of Mgat2 transcript in PHA-L-low cells (Figure 1C). Finally, we used flow cytometry to quantify the glycosylation changes using a panel of lectins. The data demonstrate a significant loss in cN-glycan-dependent PHA-L and Phaseolus vulgaris erythroagglutinin (PHA-E) staining. Conversely, mannose-specific Concanavalin A (ConA) binding was increased, likely due to the increase in high-mannose and hybrid structures. Wheat germ agglutinin (WGA), which recognizes the core GlcNAc dimer of both N-glycans and sialic acids, showed no change in binding (Figure 1D). Given the known reduction in terminal sialic acids associated with Mgat2 ablation (Wada et al. 1992; Yamashita et al. 1993; Wang et al. 2001), the lack of change seen with WGA suggests that it is either associating with the core GlcNAc dimer which should be unchanged in this system or associating with the mono-antennary hybrid glycans which can still carry sialic acid.
Loss of Mgat2 limits GlyAg T cell activation in vitro
Our previous findings revealed that BMDCs treated with either castanospermine or kifunensine to limit the maturation of cN-glycans had a profound impact on the ability present GlyAg and stimulate T cells. Here, we used PHA-L+ and PHA-L− BMDCs and thioglycollate-recruited peritoneal macrophages (Mφ) from Mgat2ΔM/ΔM and Mgat2wt/wt controls to measure their ability to stimulate T cell cytokine production in response to either GlyAg or the conventional protein antigen ovalbumin (OVA). Responding T cells were isolated from wild-type or OVA-specific OT-II mice, respectively. We found that OVA responses remain intact for both Mgat2ΔM/ΔM APC populations (Figure 2A and C), but neither the BMDCs (Figure 2B) nor the Mφ (Figure 2D) from Mgat2ΔM/ΔM mice were able to support GlyAg-specific T cell activation even with the hybrid N-glycans characteristic of Mgat2 ablation.
Fig. 2.

Mgat2ΔM/ΔM APCs fail to present the GlyAg PSA. (A) Mgat2wt/wt and Mgat2ΔM/ΔM BMDCs were cultured with ovalbumin protein (OVA)-specific OT-II CD4+ T cells and 50 µg/mL OVA for 3 days to allow T cell activation and IFNγ production. No difference in stimulation was observed between BMDC genotypes (P > 0.05). (B) The same BMDCs were also cultured with wild-type CD4+ T cells and 50 µg/mL PSA GlyAg. The Mgat2ΔM/ΔM BMDCs failed to stimulate T cells using PSA as an antigen (P < 0.05). (C and D) These assays were repeated as before, only using peritoneal macrophages (Mφ) rather than BMDCs. As before, the response to OVA-loaded Mgat2ΔM/ΔM macrophages was intact (P > 0.05) but PSA failed to activate the T cells (P < 0.05). Bars represent the mean ± SD, with n ≥ 3 on all measurements.
A key step in the mechanism of GlyAg presentation is APC activation via TLR2 signaling, which generates both NO and TNFα (Wang et al. 2006). In order to determine whether loss of cN-glycans altered the ability of the APCs to respond to TLR2 agonists, PHA-L+ and PHA-L− BMDCs were stimulated with varied concentrations of GlyAg (Figure 3A) or the canonical TLR2 ligands Pam3CYSK4 (P3C; Figure 3B) and lipoteichoic acid (LTA; Figure 3C). In all cases, no significant difference in TLR2-mediated activation was seen, as measured by TNFα production. Likewise, BMDCs were able to endocytose fluorescent GlyAg (PSA-488) to an extent indistinguishable from wild-type cells (Figure 3D). Using DQ-OVA, a BODIPY-conjugated ovalbumin that is internally quenched as an intact protein but highly fluorescent upon proteolytic cleavage (Lewis and Cobb 2010), we found that conventional antigen endocytosis and processing was also unaffected by loss of cN-glycan branching (Figure 3D). Finally, using flow cytometry, we found no change in either the lineage markers CD11c and CD11b or surface MHCII and TLR2 concentrations in the mutant BMDCs (Figure 3E).
Fig. 3.

Mgat2ΔM/ΔM BMDC surface glycoprotein expression and TLR2 activity are normal. BMDCs were stimulated with a range of PSA (A), P3C (B) or LTA (C) for 24 h. Activation was measured by TNFα ELISA, which showed no change in TLR2-mediated APC response resulting from the lack of Mgat2. (D) BMDCs were incubated with fluorescent PSA (PSA-488) or DQ-OVA, an OVA conjugate that is fluorescent only after proteolytic cleavage. Cells were analyzed by flow cytometry after 24 h. Mgat2wt/wt (dotted line) and Mgat2ΔM/ΔM (grey line) BMDCs endocytosed PSA and OVA at equal efficiency, and both were able to process OVA to equal extents. (E) Common surface glycoprotein markers on DCs from Mgat2wt/wt (dotted line) and Mgat2ΔM/ΔM (grey line) BMDCs were analyzed by flow cytometry and compared with isotype controls (black). The absence of branched cN-glycans did not alter surface expression of CD11c, MHCII, CD11b or TLR2. Bars represent the mean ± SD, with n ≥ 3 and P > 0.05 for all measurements.
Mgat2ΔM/ΔM mice develop normally
Unlike global Mgat2-null mice (Wang et al. 2001), deletion of Mgat2 in myeloid cells did not adversely affect overall health or growth kinetics when compared with wild-type controls. We found no signs of histological pathology in the lung, brain, intestine, kidney or pancreas (Figure 4A), which is an important observation since LyzM has been shown to be expressed by epithelial cells in the lung (Lei et al. 2007) and intestine (Brandtzaeg et al. 1992) under some conditions. Immune system development, as measured by the number of circulating leukocytes (Figure 4B), weight gain (Figure 4C) and blood chemistry (Figure 4D), were statistically indistinguishable between wild-type and mutant mice.
Fig. 4.

Mgat2ΔM/ΔM mice show no overt tissue or blood pathology. (A) Hematoxylin and eosin (H&E) stained tissues from Mgat2wt/wt and Mgat2ΔM/ΔM mice, revealing a paucity of visible pathology associated with the loss of cN-glycans in the myeloid compartment. (B) Cellular analysis of circulating leukocytes, red blood cells, platelets and protein levels show no statistical difference between the mice. (C) Weight gain and overall growth of these mice are indistinguishable. (D) Blood chemistry analyses show very modest elevations in alanine aminotransferase and aspartate aminotransferase within the Mgat2ΔM/ΔM mice compared with controls. Bars represent the mean ± SD, with n = 4 and P > 0.05 for all measurements.
In addition, the total number of CD11b+ myeloid cells in the spleen, blood, mesenteric lymph nodes (mLNs), mediastinal (med) lymph nodes (lung LN) and inguinal lymph nodes (iLNs) were not different between strains (Figure 5A). The reduction in cN-glycans in Mgat2ΔM/ΔM mice was readily apparent in bone marrow and to a lesser extent the spleen but not the lung, intestine and liver tissue sections by confocal microscopy upon staining with PHA-L lectin (Figure 5B). Direct analysis of the heterogeneous population of CD11b+ myeloid cells by flow cytometry revealed that ∼30–50% of target cells lacked branched cN-glycans in the lung, bone marrow, spleen and blood (Figure 5C). In contrast to the PHA-L pattern seen with homogenous BMDC cultures in vitro (Figure 1D), the biphasic distribution of staining likely arises by virtue of the heterogeneous population of tissue myeloid cells at various stages of development, the lack of synchronized CD11b and LyzM expression (and thus Mgat2 ablation), and the fact that days are required to see glycoform change at the cell surface following loss of N-glycan processing enzyme activity (Ryan et al. 2011).
Fig. 5.

Mgat2ΔM/ΔM mice show defects in cN-glycan synthesis within the myeloid hematopoietic compartment. (A) The total number of CD11b+ myeloid cells was quantified by flow cytometry. No statistically significant differences were seen in the spleen, blood, mLN, medLN or iLN (P > 0.05 for all cells). (B) Tissue sections were stained with 7-AAD (blue) and PHA-L (green) to visualize nuclei and cN-glycans, respectively. Only the marrow and, to a lesser degree, the spleen showed clear differences in PHA-L staining, demonstrating the specificity of the knockout. (C) Approximately 30–50% of CD11b+ myeloid cells in the lung, spleen, marrow and blood showed the PHA-L− phenotype in Mgat2ΔM/ΔM mice (red) compared with PHA-L-unstained CD11b+ cells (blue) and control Mgat2wt/wt mice (black), as quantified by flow cytometry. Bars represent the mean ± SD.
Mgat2ΔM/ΔM mice clear fungal eye infection normally
Since loss of branched cN-glycans in myeloid cells affected neither the homeostatic cell numbers (Figure 5A) nor TLR2-mediated innate signaling in vitro (Figure 3A–C), we further examined the possible impact on leukocyte recruitment and antimicrobial activity in vivo using an established model of Aspergillus fumigatus corneal eye infection (Leal et al. 2010, 2012, 2013). Both of these innate immune processes are mediated by either resident macrophages and dectin-1 (i.e. cell recruitment; Leal et al. 2010) or neutrophils (i.e. fungal killing; Leal et al. 2012) in a T cell-independent fashion. A monomeric dsRED RFP-expressing A. fumigatus strain was used to visualize fungal growth during infection following corneal abrasion. At 24 and 48 h post-infection, no difference was detected between fungal growth in Mgat2wt/wt and Mgat2ΔM/ΔM mice based on brightfield opacity (Figure 6A) and relative dsRED RFP fluorescence (Figure 6A and B). Fungal burden, as measure by CFU, was quantified at 48 h and was similar in both Mgat2wt/wt and Mgat2ΔM/ΔM mice (Figure 6C). Although we cannot rule out the possibility that the PHA-L+ myeloid cells from various tissues (Figure 5C) account for this anti-microbial activity, the lack of any change whatsoever in protection suggests that leukocyte activity remains unchanged in Mgat2ΔM/ΔM mice.
Fig. 6.
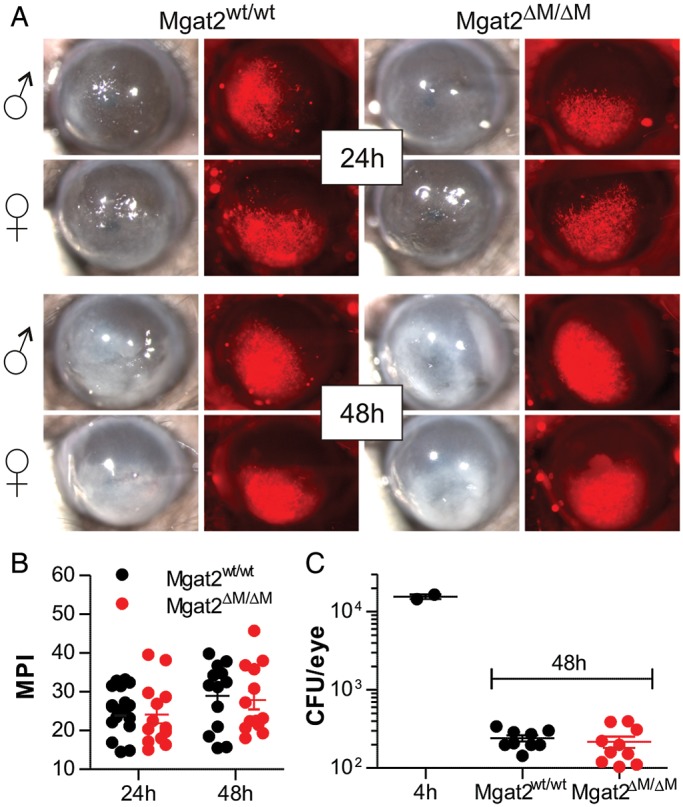
Mgat2ΔM/ΔM mice have intact innate responses to fungal infection. (A) The cornea of Mgat2wt/wt and Mgat2ΔM/ΔM mice were infected with a dsRED RFP-expressing A. fumigatus strain to induce keratitis, which is normally controlled by neutrophils and macrophages. Monitoring both males and females at 24 and 48 h post-infection, no difference was seen in overall opacity (brightfield) or fungal growth (red fluorescence). (B) Quantitation of the red signal (MPI) from infected corneas showing no difference between groups (n = 13–18). (C) CFU analysis at 48 h revealed that both wild-type and mutant mice equally controlled the infection by reducing fungal CFU more than two logs compared with the 4 h time point, collectively demonstrating intact innate cellular responses despite the loss of cN-glycans in Mgat2ΔM/ΔM mice. Bars represent the mean ± SD.
Mgat2ΔM/ΔM mice fail to respond to GlyAg stimulation
Our findings show that limiting N-glycans to high-mannose and mono-antennary hybrid structures on APCs such as dendritic cells and macrophages through Mgat2 ablation prevents GlyAg-mediated T cell activation without altering the underlying pathways of TLR2 stimulation and endocytosis in vitro. In order to test the response in the more complex in vivo environment, we examined the ability to elicit a T cell response against GlyAg in the lung. Mgat2wt/wt and Mgat2ΔM/ΔM mice were immunized intranasally with the GlyAg PSA or saline control. Two days following the last PSA dose, CD4+ T cells were isolated from the medLNs and tested for antigenic recall using APCs derived from a naïve wild-type mouse. We found that immunized Mgat2wt/wt mice exhibited robust recall responses to PSA ex vivo (Figure 7A), suggesting clonal expansion of PSA-responsive T cells during immunization; however, immunization of Mgat2ΔM/ΔM mice with PSA failed to expand PSA-specific T cells and thus did not generate responses above non-immunized background (Figure 7A).
Fig. 7.

Mgat2ΔM/ΔM DCs home to lymph nodes but fail to stimulate T cell responses to PSA in vivo. (A) Mgat2wt/wt and Mgat2ΔM/ΔM mice were intranasally immunized with PSA four times over 1 week. On day 9, CD4+ T cells were isolated from medLNs and re-stimulated ex vivo with fresh wild-type BMDCs and PSA at 50 µg/mL for 4 days. Wild-type mice showed a robust antigen recall over saline-treated controls, indicating clonal expansion during immunization, but Mgat2ΔM/ΔM mice failed to exhibit T cell expansion as seen by equal responses from PSA-treated and saline-treated mice. (B) Fluorescently-conjugated PSA (PSA-594) was administered to Mgat2wt/wt mice via the nasal route. After 24 h, the medLN were harvested and analyzed by flow cytometry. Newly immigrated DCs (CD103+CD11c+) were detected and the level of PSA uptake quantified by fluorescence. These cells were also stained with PHA-L lectin to characterize the cN-glycans. In wild-type mice, 25.8% of newly immigrated DCs carried PSA to the lymph node (compare with saline control), and essentially none of these PSA+ cells lacked cN-glycans. (C) Using the same experimental design, PSA-594 was also administered to Mgat2ΔM/ΔM mice. Like their wild-type counterparts, newly immigrated DCs carried PSA, and this was apparent in both PHA-L+ and PHA-L− cells, demonstrating a lack of any homing defect. (D) Data summary of repeated the uptake and migration experiments shown in (B) and (C), illustrating no difference (P > 0.05) in CD103+ DC migration or PSA uptake despite >40% of the PSA+ migrating cells lacking cN-glycans as measured by PHA-L staining in mutant mice (P < 0.05). Bars represent the mean ± SD, with n ≥ 3 for all measurements.
Although these findings suggest that the lack of T cell activation and expansion following PSA immunization is due to deficient MHCII-driven presentation by APCs that emigrate from the lung to the medLN for T cell recognition, it remains possible that APC homing via selectin interactions has been disrupted by the loss of Mgat2. We therefore measured the ability of lung dendritic cells to home to the draining lymph node following antigen capture to facilitate cognate interactions with responding T cells. Mice were administered fluorescently conjugated GlyAg (PSA-594) intranasally as before and dendritic cells from the medLNs were harvested for analysis. Surface expression of CD103 was used as a marker to differentiate CD11c+ DCs immigrating from the lung as opposed to lymph node resident DCs as reported previously (Jakubzick et al. 2008). In Mgat2wt/wt mice, ∼1% of the medLN DCs were CD103+CD11c+. Of these newly immigrated DCs, 25.8% were PSA-594 positive and 1.6% lacked branched N-glycans (Figure 7B and D). Similar to wild type, nearly 1% of the lymph node DCs were newly immigrated in Mgat2ΔM/ΔM mice, whereas over half (55.1%) carried fluorescent GlyAg and 43% did not express branched cN-glycans at the cell surface due to the loss of Mgat2 (Figure 7C and D). These data confirm the lack of a defect in both endocytosis and lymph node homing in the Mgat2ΔM/ΔM mice.
Discussion
MHCI and MHCII are cell surface glycoproteins that are central players in the establishment of T cell-mediated immunity through their roles in presenting endogenous and exogenous antigen, respectively. To date, every known sequence of MHCI and MHCII contains a universally conserved site of N-glycosylation near the end of the peptide binding groove (reviewed in Ryan and Cobb 2012). For MHCI, the presence of N-glycans is required for proper folding in the ER and trafficking to the cell surface (Barbosa et al. 1987), although the impact of altering the N-glycan structures (as opposed to removing the glycan completely) on MHCI function and antigen binding has yet to be explored in depth. Conversely, N-glycosylation of MHCII is not required for folding or trafficking (Hart 1982; Germain and Rinker 1993; Elliott et al. 1994), yet we have shown that high-mannose N-glycans characteristic of mannosidase inhibition by kifunensine on MHCII fail to support the presentation of at least one class of antigens: GlyAgs (Ryan et al. 2011). In the present study, we have extended this general discovery by creating a novel murine model in which the Mgat2 locus is conditionally ablated in the myeloid lineage of leukocytes, thereby limiting N-glycans to hybrid structures in these cells. Using this model, we found that these mono-antennary hybrid N-glycans also fail to support MHCII presentation of GlyAgs, revealing that multi-antennary N-glycans are required for the presentation and T cell activation mediated by GlyAgs in vitro and in vivo. This occurs without the loss of endocytosis, TLR2 signaling or leukocyte homing to the appropriate draining lymph node for T cell priming.
The ability of GlyAgs, particularly PSA from the commensal bacterium B. fragilis, to induce a potent and protective immune response is found in their ability to stimulate multiple pathways. We have shown that PSA is recognized by TLR2, leading to the production of NO and TNFα by APCs (Wang et al. 2006). This seemingly pro-inflammatory activity is required for the induction of a regulatory T cell response because upon endocytosis, PSA and other GlyAgs are processed to small fragment through oxidation by NO (Cobb et al. 2004; Velez et al. 2009). Once processed, GlyAg binds to MHCII at high affinity and is ultimately recognized by the αβ-TCR on responding T cells, thereby initiating a regulatory IL-10-dependent response which has been shown to inhibit pathologic inflammation (Tzianabos et al. 1995, 1998; Ochoa-Reparaz, Mielcarz, Ditrio, et al. 2010; Ochoa-Reparaz, Mielcarz, Wang, et al. 2010) through the inhibition of effector T cell responses (Kreisman and Cobb 2011). Our data demonstrate that branched cN-glycans on MHCII, whose synthesis requires the Mgat2 gene, are a necessary feature of MHCII on the APC that enables robust GlyAg binding, presentation and T cell recognition.
The precise biophysical requirement for branched cN-glycans on MHCII for GlyAg presentation remains unknown. MHCII molecules carrying fully mature cN-glycans are known to form poor quality crystals for structure determination, yet these cN-glycans are required for GlyAg binding, thus limiting progress on crystallization of the GlyAg–MHCII complex. However, molecular modeling may provide some insight. Given that GlyAg and peptide binding to MHCII is mutually exclusive (Cobb and Kasper 2008), peptide binding is unaffected by the N-glycans (Ryan et al. 2011), cN-glycans alone fail to bind to GlyAgs (Ryan et al. 2011) and the bound form of GlyAgs is at least three times larger than the average bound peptide (Kreisman et al. 2007), it is not unreasonable to suggest that the universally conserved cN-glycans add the surface area to the binding interface for the bulky GlyAgs. This would account for the need for large multi-antennary cN-glycans on MHCII for GlyAg binding. Indeed, our findings lead to a model in which the MHCII binding site for GlyAg is a combination of carbohydrate–carbohydrate interactions with N-glycans and carbohydrate–protein interactions with the peptide binding groove of MHCII (reviewed in Ryan and Cobb 2012).
One recent study utilized TLR2-null mice to suggest that the protective efficacy of GlyAgs is due to the stimulation of TLR2 on the T cell, which reprograms the T cell to be regulatory in nature (Round et al. 2011). However, this study neglected to account for a number of complicating factors in using TLR2-null mice, especially the loss of GlyAg processing concomitant with the loss of TLR2 in the APC (Wang et al. 2006). In the absence of TLR2, GlyAgs are not processed or presented by MHCII (Wang et al. 2006). The present study clearly demonstrates that in the absence of αβ-TCR recognition of MHCII-presented GlyAg, no T cell response is apparent despite the intact ability of GlyAgs to stimulate an innate response through TLR2.
From a more general view, ablation of Mgat2 in the germline as a model for human CDG-IIa has been described previously (Wang et al. 2001). Like many of the CDGs, loss of a functional Mgat2 gene leads to severe defects. In mice, most of the homozygous Mgat2-null animals die early during postnatal development, whereas the few that survive exhibit a similar phenotype to their human counterparts (Wang et al. 2001). Mgat2-null mice exhibited dysmorphic facial features, spinal scoliosis, seizures and locomotor deficits. In addition, intestinal defects were noted, including distended stomach and intestinal obstruction, possibly linked to altered mucin levels (Wang et al. 2001). In contrast, the myeloid-specific Mgat2 knockout mouse displayed none of these phenotypes, suggesting that the source of severe morbidity and high mortality rates observed in the Mgat2-null mouse are unlikely of hematopoietic origin. Indeed, Mgat2ΔM/ΔM tissue histology remains normal and the colony continues to breed without trouble. A small percentage of the Mgat2ΔM/ΔM mice (∼25%) show detectable anemia (not shown), but the etiology of this change remains unclear. All of the analyses herein excluded the anemic littermates, which are the subject of ongoing evaluation.
In summary, our findings demonstrate that N-glycan branching is a critical feature of MHCII cN-glycans that directly impacts the ability to present GlyAgs. Given the known propensity to alter N-glycan branching patterns in cells as a function of disease and inflammation (Thompson et al. 1989, 1993; Mann et al. 1994; Goodarzi and Turner 1995, 1998; Turner et al. 1995; Gravel et al. 1996; Saldova et al. 2007), these results suggest that regulation of N-glycosylation within the myeloid compartment may serve as a switch to utilize or not utilize commensal-derived GlyAgs to suppress ongoing inflammation via the induction of regulatory T cells. Moreover, our data establish a viable model in which to differentiate the function and impact of TLR2 stimulatory activity of GlyAgs which remains intact in Mgat2ΔM/ΔM mice, from GlyAg-mediated T cell activation which is lost due to a blockade of MHCII-dependent presentation.
Materials and methods
Antigens
PSA was expressed by a B. fragilis variant that expresses only PSA in the capsule (Krinos et al. 2001) and purified to homogeneity essentially as described previously (Tzianabos et al. 1992). Ovalbumin protein was purchased from Sigma. P3C and LTA were purchased from Invivogen.
Mice
Animal colonies were maintained in a specific pathogen-free environment at Case Western Reserve University and were treated under IACUC-approved guidelines in accordance with approved protocols. Mgat2ΔM/ΔM mice were generated by crossing the Mgat2 (B6.129-Mgat2tm1Jxm/J; stock 006892) and LyzM-cre (B6.129P2-Lyz2tm1(cre)Ifo/J; stock 004781) parental strains, which were purchased from The Jackson Laboratory. Wild-type C57BL/6J mice (stock 000664) and OT-II mice (B6.Cg-Tg(TcraTcrb)425Cbn/J; stock 004194) were purchased from the Jackson Laboratory. Mouse genotypes were confirmed using Jackson Laboratory PCR protocols.
Flow cytometry
Cells were stained for flow cytometry as described previously (Ryan et al. 2011). Briefly, cells were stained with fluorescein-conjugated (Vector Laboratories) or AlexaFluor647-conjugated (Life Technologies) lectins and/or the indicated antibodies for 30 min at 4°C. Lectins included were PHA-L, PHA-E, WGA and ConA. For antigen uptake, BMDCs were incubated for 24 h with 10 ng/mL GM-CSF (Invitrogen) supplemented media alone or plus 50 µg/mL AlexaFluor488-conjugated PSA or DQ-OVA (Invitrogen). FACS analysis was performed using a C6 flow cytometer (Accuri Cytometers). Analyses of FACS data were performed using FCS Express (De Novo Software).
In vitro T cell activation
T cell activation assays were performed as described previously (Ryan et al. 2011). Bone marrow cells were differentiated into BMDCs in culture using 10 ng/mL GM-CSF (Invitrogen) for 10 days. Peritoneal macrophages were obtained exactly as described previously using plastic adherence of harvested cells (Lewis and Cobb 2010). Cells lacking Mgat2 were selected and purified based on PHA-L (Vector Labs) staining. Briefly, Mgat2ΔM/ΔM BMDCs were labeled with 20 μg/mL biotinylated PHA-L and separated using anti-biotin magnetic microbeads (Miltenyi Biotec). The non-binding cells (PHA-L negative) found in the flow through were analyzed by RT-PCR as described previously (Ryan et al. 2011), and flow cytometry to confirm purity using a streptavidin-APC conjugate (BioLegend). CD4+ T cells were isolated from the spleen for PSA and OVA activation assays by CD4+ magnetic bead positive selection (Miltenyi Biotec) and verified by flow cytometry. CD4+ T cells (1.5 × 105) were cocultured with BMDCs (1.5 × 104) or macrophages (3 × 104) and incubated for 3 (OT-II) or 4 days (C57BL/6) with 50 µg/mL OVA or PSA, respectively. Culture supernatants were analyzed for IFNγ by sandwich ELISA according to the manufacturer's protocol (BioLegend).
Toll-like receptor activation
BMDCs (1 × 105) were cultured with a range of concentrations of PSA, P3C or LTA for 24 h. Culture supernatants were analyzed for TNFα production by sandwich ELISA according to the manufacturer's protocol (BioLegend).
Tissue histology and blood chemistry
Mice were sent to the Comparative Pathology Laboratory at the University of California at Davis for necropsy, tissue paraffin embedding and blood work. Hematoxylin and eosin tissue section staining was performed at UC Davis and analyzed by a trained pathologist at the Comparative Pathology Laboratory. For confocal microscopy, tissue sections were deparaffinized using sequential 10 min washes with xylene (×2); 3 min washes with 100% ethanol (×2), 75% ethanol (×1) and 50% ethanol (×1); followed by two washes in TBS (pH 7.6, 25 mM Tris base, 150 mM NaCl, 2 mM KCl) for 5 min each. Epitope retrieval was performed by microwave heating in 10 mM sodium citrate buffer (pH 6) and then allowed to cool to room temperature. Slides were blocked using 3% bovine serum albumin (Sigma) in TBS for 30 min. Tissue sections were stained with 2 μg/mL fluorescein-conjugated PHA-L for 1 h with 7-AAD nuclear stain (BD Biosciences) added for the final 10 min. Coverslips were mounted using Fluorogel with DABCO (Electron Microscopy Sciences). BMDCs were collected on day 9 and recultured on sterile coverslips. After 24 h, coverslips were fixed in 4% paraformaldehyde and then stained with 10 μg/mL fluorescein-conjugated PHA-L. Confocal microscopy was performed using an SP5 laser scanning confocal microscope (Leica).
Aspergillus corneal infection
As described previously (Leal et al. 2012), Aspergillus strain Af293.1 dsRed was cultured for 2–3 days in Vogel's minimal media and conidia were disrupted with a bacterial L-loop, harvested in PBS and filtered through sterile cotton gauze to obtain pure conidial suspensions. Conidia were quantified using a hemocytometer and adjusted to 20,000 conidia/µL in PBS. Mice were anaesthetized with 1.25% 2,2,2-tri-bromoethanol in PBS. The corneal epithelium was abraded using a 30-gage needle, through which a 2-µL injection containing conidia was released into the corneal stroma using a 33-gage Hamilton syringe. Mice were imaged under a stereomicroscope for corneal opacification (brightfield) and fungal growth (RFP fluorescence) as described (Leal et al. 2010). Mean pixel intensity (MPI) was collected from each image pixel histogram within CorelX4 Photo Paint. At each time point, animals were euthanized, and eyes placed in PBS and homogenized for CFU analysis (Leal et al. 2013).
In vivo T cell activation
To analyze transfer of PSA GlyAg by DCs from the lung to draining lymph nodes, mice were intranasally administered 200 µg AlexaFluor594-conjugated PSA in 20 µL saline or saline alone. After 24 h, lung draining lymph nodes (mediastinal) were collected. Isolated cells were labeled with PHA-L lectin and anti-CD103 and CD11c mAb for analysis on a LSRII flow cytometer (BD Biosciences). Data analysis was performed using FlowJo (Tree Star). For detection of T cell responses, mice were primed with four doses of 25 µg of PSA in 20 µL of saline or saline alone over the course of 1 week. Two days after the final dose, medLNs were collected. CD4+ T cells were isolated as described above for T cell activation assays. Mgat2wt/wt BMDCs (5 × 103) were cultured alone or with CD4+ T cells (5 × 104) and incubated for 4 days with 50 µg/mL PSA or media alone. Culture supernatants were analyzed for IFNγ by sandwich ELISA as before.
Statistics
All data are shown as the mean ± SD. Comparisons were generally performed using a Student's t-test for significance with a 95% confidence interval using GraphPad Instat software.
Funding
This work was supported by NIH (R01GM82916 and DP2OD004225 to BAC, F31EY019841 to SML and R01EY018612 and P30EY011373 to EP).
Conflict of interest
None declared.
Abbreviations
7-AAD, 7-amino-actinomycin D; APC, antigen presenting cell; BMDC, Bone marrow-derived dendritic cell; CDG, congenital disorders of glycosylation; CFU, colony forming units; cN-glycans, complex-type N-glycans; ConA, Concanavalin A; DC, dendritic cell; ER, endoplasmic reticulum; GlyAg, glycoantigen; GM-CSF, granulocyte macrophage colony stimulating factor; IFN, interferon; iLN, inguinal lymph node; LTA, lipoteichoic acid; LyzM, lysozyme M; medLN, mediastinal lymph node; MFI, mean fluorescence intensity; MHCII, class II major histocompatibility complex; mLN, mesenteric lymph node; MPI, mean pixel intensity; NO, nitric oxide; OVA, antigen ovalbumin; P3C, Pam3CYSK4; PBS, phosphate-buffered saline; PHA-E, Phaseolus vulgaris erythroagglutinin; PHA-L, Phaseolus vulgaris leucoagglutinin; PSA, polysaccharide A1; RFP, red fluorescent protein; TBS, Tris-buffered saline; TCR, T cell receptor; TLR2, Toll-like receptor 2; WGA, wheat germ agglutinin.
Acknowledgements
The authors thank Jenny Johnson for assistance with the lung model and Lori SC Kreisman for critical evaluation of this manuscript.
References
- Ackerman ME, Crispin M, Yu X, Baruah K, Boesch AW, Harvey DJ, Dugast AS, Heizen EL, Ercan A, Choi I, et al. Natural variation in Fc glycosylation of HIV-specific antibodies impacts antiviral activity. J Clin Invest. 2013;123:2183–2192. doi: 10.1172/JCI65708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature. 2011;475:110–113. doi: 10.1038/nature10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony RM, Nimmerjahn F, Ashline DJ, Reinhold VN, Paulson JC, Ravetch JV. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science. 2008;320:373–376. doi: 10.1126/science.1154315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony RM, Wermeling F, Ravetch JV. Novel roles for the IgG Fc glycan. Ann N Y Acad Sci. 2012;1253:170–180. doi: 10.1111/j.1749-6632.2011.06305.x. [DOI] [PubMed] [Google Scholar]
- Barbosa JA, Santos-Aguado J, Mentzer SJ, Strominger JL, Burakoff SJ, Biro PA. Site-directed mutagenesis of class I HLA genes. Role of glycosylation in surface expression and functional recognition. J Exp Med. 1987;166:1329–1350. doi: 10.1084/jem.166.5.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandtzaeg P, Halstensen TS, Huitfeldt HS, Krajci P, Kvale D, Scott H, Thrane PS. Epithelial expression of HLA, secretory component (poly-Ig receptor), and adhesion molecules in the human alimentary tract. Ann N Y Acad Sci. 1992;664:157–179. doi: 10.1111/j.1749-6632.1992.tb39758.x. [DOI] [PubMed] [Google Scholar]
- Chung DR, Chitnis T, Panzo RJ, Kasper DL, Sayegh MH, Tzianabos AO. CD4+ T cells regulate surgical and postinfectious adhesion formation. J Exp Med. 2002;195:1471–1478. doi: 10.1084/jem.20020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung DR, Kasper DL, Panzo RJ, Chtinis T, Grusby MJ, Sayegh MH, Tzianabos AO. CD4+ T cells mediate abscess formation in intra-abdominal sepsis by an IL-17-dependent mechanism. J Immunol. 2003;170:1958–1963. doi: 10.4049/jimmunol.170.4.1958. [DOI] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- Cobb BA, Kasper DL. Characteristics of carbohydrate antigen binding to the presentation protein HLA-DR. Glycobiology. 2008;18:707–718. doi: 10.1093/glycob/cwn050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb BA, Wang Q, Tzianabos AO, Kasper DL. Polysaccharide processing and presentation by the MHCII pathway. Cell. 2004;117:677–687. doi: 10.016/j.cell.2004.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature. 2001;409:733–739. doi: 10.1038/35055582. [DOI] [PubMed] [Google Scholar]
- Elliott EA, Drake JR, Amigorena S, Elsemore J, Webster P, Mellman I, Flavell RA. The invariant chain is required for intracellular transport and function of major histocompatibility complex class II molecules. J Exp Med. 1994;179:681–694. doi: 10.1084/jem.179.2.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain RN, Rinker AG. Peptide binding inhibits protein aggregation of invariant-chain free class II dimers and promotes surface expression of occupied molecules. Nature. 1993;363:725–728. doi: 10.1038/363725a0. [DOI] [PubMed] [Google Scholar]
- Goodarzi MT, Turner GA. Decreased branching, increased fucosylation and changed sialylation of alpha-1-proteinase inhibitor in breast and ovarian cancer. Clin Chim Acta. 1995;236:161–171. doi: 10.1016/0009-8981(95)06049-j. [DOI] [PubMed] [Google Scholar]
- Goodarzi MT, Turner GA. Reproducible and sensitive determination of charged oligosaccharides from haptoglobin by PNGase F digestion and HPAEC/PAD analysis: Glycan composition varies with disease. Glycoconj J. 1998;15:469–475. doi: 10.1023/a:1006930902625. [DOI] [PubMed] [Google Scholar]
- Gravel P, Walzer C, Aubry C, Balant LP, Yersin B, Hochstrasser DF, Guimon J. New alterations of serum glycoproteins in alcoholic and cirrhotic patients revealed by high resolution two-dimensional gel electrophoresis. Biochem Biophys Res Commun. 1996;220:78–85. doi: 10.1006/bbrc.1996.0360. [DOI] [PubMed] [Google Scholar]
- Hart GW. The role of asparagine-linked oligosaccharides in cellular recognition by thymic lymphocytes. Effects of tunicamycin on the mixed lymphocyte reaction. J Biol Chem. 1982;257:151–158. [PubMed] [Google Scholar]
- Jaeken J. Congenital disorders of glycosylation. Ann N Y Acad Sci. 2010;1214:190–198. doi: 10.1111/j.1749-6632.2010.05840.x. [DOI] [PubMed] [Google Scholar]
- Jakubzick C, Bogunovic M, Bonito AJ, Kuan EL, Merad M, Randolph GJ. Lymph-migrating, tissue-derived dendritic cells are minor constituents within steady-state lymph nodes. J Exp Med. 2008;205:2839–2850. doi: 10.1084/jem.20081430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- Kasper DL, Hayes ME, Reinap BG, Craft FO, Onderdonk AB, Polk BF. Isolation and identification of encapsulated strains of Bacteroides fragilis. J Infect Dis. 1977;136:75–81. doi: 10.1093/infdis/136.1.75. [DOI] [PubMed] [Google Scholar]
- Kreisman LS, Cobb BA. Glycoantigens induce human peripheral Tr1 cell differentiation with gut-homing specialization. J Biol Chem. 2011;286:8810–8818. doi: 10.1074/jbc.M110.206011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisman LS, Friedman JH, Neaga A, Cobb BA. Structure and function relations with a T-cell-activating polysaccharide antigen using circular dichroism. Glycobiology. 2007;17:46–55. doi: 10.1093/glycob/cwl056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krinos CM, Coyne MJ, Weinacht KG, Tzianabos AO, Kasper DL, Comstock LE. Extensive surface diversity of a commensal microorganism by multiple DNA inversions. Nature. 2001;414:555–558. doi: 10.1038/35107092. [DOI] [PubMed] [Google Scholar]
- Leal SM, Jr, Cowden S, Hsia YC, Ghannoum MA, Momany M, Pearlman E. Distinct roles for Dectin-1 and TLR4 in the pathogenesis of Aspergillus fumigatus keratitis. PLoS Pathog. 2010;6:e1000976. doi: 10.1371/journal.ppat.1000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal SM, Jr, Roy S, Vareechon C, Carrion SD, Clark H, Lopez-Berges MS, Dipietro A, Schrettl M, Beckmann N, Redl B, et al. Targeting iron acquisition blocks infection with the fungal pathogens Aspergillus fumigatus and Fusarium oxysporum. PLoS Pathog. 2013;9:e1003436. doi: 10.1371/journal.ppat.1003436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal SM, Jr, Vareechon C, Cowden S, Cobb BA, Latge JP, Momany M, Pearlman E. Fungal antioxidant pathways promote survival against neutrophils during infection. J Clin Invest. 2012;122:2482–2498. doi: 10.1172/JCI63239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei W, Jaramillo RJ, Harrod KS. Transactivation of lung lysozyme expression by Ets family member ESE-1. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1359–L1368. doi: 10.1152/ajplung.00130.2007. [DOI] [PubMed] [Google Scholar]
- Lewis CJ, Cobb BA. Carbohydrate oxidation acidifies endosomes, regulating antigen processing and TLR9 signaling. J Immunol. 2010;184:3789–3800. doi: 10.4049/jimmunol.0903168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann AC, Record CO, Self CH, Turner GA. Monosaccharide composition of haptoglobin in liver diseases and alcohol abuse: Large changes in glycosylation associated with alcoholic liver disease. Clin Chim Acta. 1994;227:69–78. doi: 10.1016/0009-8981(94)90136-8. [DOI] [PubMed] [Google Scholar]
- Ochoa-Reparaz J, Mielcarz DW, Ditrio LE, Burroughs AR, Begum-Haque S, Dasgupta S, Kasper DL, Kasper LH. Central nervous system demyelinating disease protection by the human commensal Bacteroides fragilis depends on polysaccharide A expression. J Immunol. 2010;185:4101–4108. doi: 10.4049/jimmunol.1001443. [DOI] [PubMed] [Google Scholar]
- Ochoa-Reparaz J, Mielcarz DW, Wang Y, Begum-Haque S, Dasgupta S, Kasper DL, Kasper LH. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol. 2010;3:487–495. doi: 10.1038/mi.2010.29. [DOI] [PubMed] [Google Scholar]
- Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan SO, Bonomo JA, Zhao F, Cobb BA. MHCII glycosylation modulates Bacteroides fragilis carbohydrate antigen presentation. J Exp Med. 2011;208:1041–1053. doi: 10.1084/jem.20100508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan SO, Cobb BA. Roles for major histocompatibility complex glycosylation in immune function. Semin Immunopathol. 2012;34:425–441. doi: 10.1007/s00281-012-0309-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldova R, Royle L, Radcliffe CM, Abd Hamid UM, Evans R, Arnold JN, Banks RE, Hutson R, Harvey DJ, Antrobus R, et al. Ovarian cancer is associated with changes in glycosylation in both acute-phase proteins and IgG. Glycobiology. 2007;17:1344–1356. doi: 10.1093/glycob/cwm100. [DOI] [PubMed] [Google Scholar]
- Strachan DP. Hay fever, hygiene, and household size. Br Med J. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan DP. Family size, infection and atopy: The first decade of the "hygiene hypothesis". Thorax. 2000;55(Suppl. 1):S2–S10. doi: 10.1136/thorax.55.suppl_1.s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson S, Dargan E, Griffiths ID, Kelly CA, Turner GA. The glycosylation of haptoglobin in rheumatoid arthritis. Clin Chim Acta. 1993;220:107–114. doi: 10.1016/0009-8981(93)90011-r. [DOI] [PubMed] [Google Scholar]
- Thompson S, Kelly CA, Griffiths ID, Turner GA. Abnormally-fucosylated serum haptoglobins in patients with inflammatory joint disease. Clin Chim Acta. 1989;184:251–258. doi: 10.1016/0009-8981(89)90058-2. [DOI] [PubMed] [Google Scholar]
- Turner GA, Goodarzi MT, Thompson S. Glycosylation of alpha-1-proteinase inhibitor and haptoglobin in ovarian cancer: Evidence for two different mechanisms. Glycoconj J. 1995;12:211–218. doi: 10.1007/BF00731322. [DOI] [PubMed] [Google Scholar]
- Tzianabos AO, Finberg RW, Wang Y, Chan M, Onderdonk AB, Jennings HJ, Kasper DL. T cells activated by zwitterionic molecules prevent abscesses induced by pathogenic bacteria. J Biol Chem. 2000;275:6733–6740. doi: 10.1074/jbc.275.10.6733. [DOI] [PubMed] [Google Scholar]
- Tzianabos AO, Gibson FC, III, Cisneros RL, Kasper DL. Protection against experimental intraabdominal sepsis by two polysaccharide immunomodulators. J Infect Dis. 1998;178:200–206. doi: 10.1086/515594. [DOI] [PubMed] [Google Scholar]
- Tzianabos AO, Kasper DL, Cisneros RL, Smith RS, Onderdonk AB. Polysaccharide-mediated protection against abscess formation in experimental intra-abdominal sepsis. J Clin Invest. 1995;96:2727–2731. doi: 10.1172/JCI118340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzianabos AO, Onderdonk AB, Zaleznik DF, Smith RS, Kasper DL. Structural characteristics of polysaccharides that induce protection against intra-abdominal abscess formation. Infect Immun. 1994;62:4881–4886. doi: 10.1128/iai.62.11.4881-4886.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzianabos AO, Pantosti A, Baumann H, Brisson JR, Jennings HJ, Kasper DL. The capsular polysaccharide of Bacteroides fragilis comprises two ionically linked polysaccharides. J Biol Chem. 1992;267:18230–18235. [PubMed] [Google Scholar]
- Velez CD, Lewis CJ, Kasper DL, Cobb BA. Type I Streptococcus pneumoniae carbohydrate utilizes a nitric oxide and MHC II-dependent pathway for antigen presentation. Immunology. 2009;127:73–82. doi: 10.1111/j.1365-2567.2008.02924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y, Nishikawa A, Okamoto N, Inui K, Tsukamoto H, Okada S, Taniguchi N. Structure of serum transferrin in carbohydrate-deficient glycoprotein syndrome. Biochem Biophys Res Commun. 1992;189:832–836. doi: 10.1016/0006-291x(92)92278-6. [DOI] [PubMed] [Google Scholar]
- Wang Q, McLoughlin RM, Cobb BA, Charrel-Dennis M, Zaleski KJ, Golenbock D, Tzianabos AO, Kasper DL. A bacterial carbohydrate links innate and adaptive responses through Toll-like receptor 2. J Exp Med. 2006;203:2853–2863. doi: 10.1084/jem.20062008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tan J, Sutton-Smith M, Ditto D, Panico M, Campbell RM, Varki NM, Long JM, Jaeken J, Levinson SR, et al. Modeling human congenital disorder of glycosylation type IIa in the mouse: Conservation of asparagine-linked glycan-dependent functions in mammalian physiology and insights into disease pathogenesis. Glycobiology. 2001;11:1051–1070. doi: 10.1093/glycob/11.12.1051. [DOI] [PubMed] [Google Scholar]
- Yamashita K, Ideo H, Ohkura T, Fukushima K, Yuasa I, Ohno K, Takeshita K. Sugar chains of serum transferrin from patients with carbohydrate deficient glycoprotein syndrome. Evidence of asparagine-N-linked oligosaccharide transfer deficiency. J. Biol. Chem. 1993;268:5783–5789. [PubMed] [Google Scholar]