

Abstract
Soluble amyloid-beta (Aβ) oligomers are hypothesized to be the pathogenic species in Alzheimer's disease (AD), and increased levels of oligomers in the brain subsequent to traumatic brain injury (TBI) may exacerbate secondary injury pathways and underlie increased risk of developing AD in later life. To determine whether TBI causes Aβ aggregation and oligomerization in the brain, we exposed triple transgenic AD model mice to controlled cortical impact injury and measured levels of soluble, insoluble, and oligomeric Aβ by enzyme-linked immunosorbent assay (ELISA) at 1, 3, and 7 days postinjury. TBI rapidly increased levels of both soluble and insoluble Aβ40 and Aβ42 in the injured cortex at 1 day postinjury. We confirmed previous findings that identified damaged axons as a major site of Aβ accumulation using both immunohistochemistry and biochemistry. We also report that soluble Aβ oligomers were significantly increased in the injured cortex, as demonstrated by both ELISA and Western blot. Interestingly, the mouse brain is able to rapidly clear trauma-induced Aβ, with both soluble and insoluble Aβ species returning to sham levels by 7 days postinjury. In conclusion, we demonstrate that TBI causes acute accumulation and aggregation of Aβ in the brain, including the formation of low- and high-molecular-weight Aβ oligomers. The formation and aggregation of Aβ into toxic species acutely after injury may play a role in secondary injury cascades after trauma and, chronically, may contribute to increased risk of developing AD in later life.
Key words: : adult brain injury, axonal injury, beta amyloid
Introduction
Traumatic brain injury (TBI) increases the likelihood of developing Alzheimer's disease (AD) in later life.1–3 Though the underlying mechanism is not known, TBI does cause accumulation of amyloid-beta (Aβ) in axons of the injured brain4–6 as well as acute deposition of insoluble Aβ plaques in severe TBI cases.7–9 Because accumulation and aggregation of Aβ in the brain is hypothesized to play a major role in AD pathogenesis,10,11 induction of these processes after injury may explain increased risk of AD after TBI. Further, formation of toxic Aβ species after injury may contribute to secondary injury cascades and contribute to neuronal cell death post-trauma.
Aβ peptides are generated by sequential cleavage of amyloid precursor protein (APP) by β- and γ-secretases.12 Monomeric forms of Aβ can aggregate to form oligomers, protofibrils, and, finally, the fibrils that deposit as amyloid plaque.10 Although Aβ plaques are the hallmark pathology of AD, it is now widely accepted that plaques are not the pathogenic, disease-causing species.13 Instead, overwhelming evidence suggests that it is soluble Aβ oligomers, the intermediate species formed during aggregation of monomeric soluble Aβ peptides into plaque, that are the pathogenic species causing neurotoxicity and synaptic dysfunction in AD.13–15 Indeed, the soluble pool of Aβ, containing soluble monomers and oligomers, correlates better with AD symptoms and severity than Aβ plaque levels.16,17 Further, soluble Aβ oligomers impair synaptic transmission in mouse models of AD18,19 and are toxic to neurons in vitro.20–22 In addition to the potential long-term implication that aggregation can have on increasing the risk of AD pathogenesis, the formation of toxic soluble oligomers may contribute to secondary injury processes after TBI. Indeed, a recent study detected the presence of a high-molecular-weight (HMW) Aβ oligomer species in human cerebrospinal fluid (CSF) acutely after TBI,23 and the presence of these oligomers appears to predict patient outcome after injury.23
Several groups, including ours, have reported increased levels of soluble Aβ in the brain after experimental TBI using either wild-type (WT) or AD transgenic (Tg) mouse models.4,24–31 To study insoluble Aβ species and Aβ plaque formation after injury, the use of Tg mouse models overexpressing human APP is required because human Aβ is more aggressive at forming fibrils than mouse Aβ.32 Studies on insoluble Aβ species in Tg mice have focused primarily on amyloid plaque formation, and the results have been equivocal—with independent reports of experimental TBI increasing,4,26,30,33,34 decreasing,35,36 or having no effect37–39 on insoluble Aβ levels or amyloid plaque formation. To date, we are not aware of any studies on the formation of Aβ oligomers after injury in animal models of TBI.
Because many of the animal studies referenced above focused either on a single time point after injury, or on multiple time points after injury separated by long periods, greater temporal resolution may be necessary to determine Aβ dynamics after TBI. Therefore, to determine whether TBI causes acute Aβ aggregation and formation of soluble oligomers in the brain, we exposed triple Tg (3×Tg) AD model mice to controlled cortical impact (CCI) injury and measured levels of soluble, insoluble, and oligomeric Aβ in the injured cortex at 1, 3, and 7 days after injury.
Methods
Controlled cortical impact model of experimental traumatic brain injury
All procedures were carried out in accord with protocols approved by the Georgetown University Animal Care and Use Committee (Washington, DC). CCI injury was induced using a Leica Impact One Stereotaxic Impactor device (Leica Microsystems, Richmond, IL), as previously described.40 Briefly, mice were anesthetized with isoflurane (induction at 4% and maintenance at 2%) evaporated in oxygen and administered through a nose mask. Anesthesia depth was monitored by assessing respiration rate and pedal withdrawal reflexes, and mice were placed on a custom-made stereotaxic frame with a built-in heating bed to maintain body temperature at 37°C. The head was mounted in the stereotaxic frame, and the surgical site was clipped and cleaned with Nolvasan and ethanol scrubs. A 10-mm mid-line incision was made over the skull, and the skin and fascia were reflected to make a 4-mm craniotomy on the central aspect of the left parietal bone. The 3.5-mm-diameter impactor tip of the injury device was lowered to the surface of the exposed dura until contact was made. The impactor tip was then retracted and lowered 2.0 mm before inducing impact (impact velocity, 5.25 m/s) to cause a moderate injury. After injury, the incision was closed with staples, anesthesia was terminated, and the animal was placed in a heated cage to maintain normal core temperature for 45 min postinjury. Sham injury consisted of exposure to anesthesia, stereotaxic mounting, skin and fascia reflection, and closing of incision with staples.
Animals
AD disease model mice (3×Tg) were originally generated by comicroinjection of human APP(Swe) and human tau(P301L) transgenes into the PS1(M146V) knock-in mouse.41 The colony at Georgetown University was established in 2004 and has been previously characterized.42 This colony has seen substantial drift from the pathology originally described by Oddo and colleagues41 and does not deposit any amyloid plaque until at least 18 months of age.42 We used two cohorts of mice for our studies, both at preplaque ages: Time-course experiments were performed in 10-month-old male mice (sacrificed 1, 3, or 7 days after sham or CCI injury; n=4/time point), and a second cohort of 8-week-old male mice was sacrificed 1 day after sham or CCI injury (n=3/time point). We chose two different time points to determine whether older mice would have a greater affinity toward Aβ aggregation after TBI.
Tissue homogenization and sequential extraction of proteins
Mice were anesthetized and transcardially perfused with ice-cold 1×phosphate-buffered saline (PBS). The extracted brain was rapidly dissected into ipsi- and contralateral hemispheres, and a 5-mm-diameter punch of cortical tissue was taken from the ipsilateral side in TBI samples, centered around the lesion, and from the corresponding area in shams and contralateral cortex. The fimbria and surrounding white matter (WM) tracts were also dissected from the ipsilateral side before collecting cortical samples in the time-course experiment. Samples were snap-frozen and kept at −80°C until homogenization. Proteins were sequentially extracted from samples to obtain separate fractions containing proteins of differing solubility. Samples were homogenized in 10 volumes of ice-cold tissue homogenization buffer, containing 250 mM of sucrose, 20 mM of Tris base, 1 mM of ethylenediaminetetraacetic acid (EDTA), and 1 mM of ethylene glycol tetraacetic acid (pH 7.4), using a ground glass pestle in a Dounce homogenizer. After six strokes, the homogenate was mixed 1:1 with 0.4% diethylamine (DEA) in a 100-mM NaCl solution before an additional six strokes and then centrifuged at 135,000g at 4°C for 45 min. The supernatant (DEA-soluble fraction containing extracellular and cytosolic proteins) was collected and neutralized with 10% of 0.5 M of Tris-HCl (pH 6.8). The pellet was resuspended in 1×radioimmunoprecipitation assay (RIPA) buffer, containing 50 mM of Tris-HCl, 150 mM of NaCl, 0.25% deoxycholic acid, 1% nonyl phenoxypolyethoxylethanol, and 1 mM of EDTA (pH 7.4), by sonication for 10 sec and spun at 135,000g at 4°C for 45 min. After collecting the supernatant (RIPA-soluble fraction containing membrane-bound proteins), a third sequential extraction in formic acid was performed to obtain insoluble proteins. The RIPA-insoluble pellet was resuspended in one volume of 70% formic acid, sonicated, and spun at 14,000 rpm for 15 min at 4°C. The supernatant was collected (formic acid fraction containing insoluble proteins) and neutralized in 20 volumes of neutralization buffer containing 1 M of Tris base and 0.5 M of Na2PO4. DEA, RIPA, and formic acid fractions were frozen and kept at −80°C until needed for biochemistry. All buffers were prepared fresh using chemicals from Sigma-Aldrich (St. Louis, MO), and mammalian tissue protease inhibitor cocktail was added before use. All procedures were carried out on ice.
Measurement of amyloid-beta by enzyme-linked immunosorbent assay
Human Aβ40 and Aβ42 were individually measured in DEA (soluble Aβ) and formic acid (insoluble Aβ) fractions using commercially available kits from Invitrogen (Carlsbad, CA) per the manufacturer's instructions. These kits utilize neoepitope-specific antibodies (Abs) to specifically detect either human Aβ40 or Aβ42 and have no cross-reactivity with full-length APP.43,44 All readings were converted to pg per mg protein, except for Aβ measured in the formic acid fraction, which is expressed as pg per mL.
Oligomeric Aβ was measured using a commercially available kit (IBL International GmbH, Hamburg, Germany). This ELISA uses the same N-terminus-specific anti-Aβ Ab for both capture and detection of Aβ (82E1), requiring at least a dimer to be present to generate a signal.45 This ELISA cannot differentiate between oligomers of different sizes, and because the result is calculated in molarity standardized on Aβ dimers, it is not in direct proportion to the weight or number of detected oligomers. The results are expressed as pmol/L.
Western blot analyses of proteins
Protein levels of APP, beta-site amyloid precursor protein cleaving enzyme 1 (BACE1), and APP carboxyl terminal fragment (CTF; α and β) were measured in the RIPA fraction, as previously described.28 Briefly, protein concentration was first determined by bicinchoninic acid assay (Pierce, Rockford, IL) to prepare samples of equal protein concentration. Samples were prepared in Laemelli buffer containing beta-mercaptoethanol (BME) or lithium dodecylsulfate and reducing agent and then resolved on either 7% or 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels or 4–12% precast Bis-Tris gels (Invitrogen), transferred onto nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA), and blocked for a minimum of 1 h in blocking buffer (5% nonfat dry milk in 1×PBS containing 0.05% Tween 20 [PBS-T]) before incubation overnight at 4°C in primary Ab prepared in PBS-T containing 1% nonfat dry milk. The following primary Abs were used: C1/6.1 to detect full-length APP and APP CTFs (monoclonal Ab against the C-terminus of APP, a kind gift from Dr. Paul Mathews, Nathan S. Kline Institute, Orangeburg, NY), BACE1 (AB5940, polyclonal; Millipore, Billerica, MA), and myelin basic protein (SMI-99P, monoclonal; Covance, Princeton, NJ). Membranes were washed five times for 5 min in 1×PBS-T and incubated in the appropriate HRP-conjugated secondary Ab (anti-mouse immunoglobulin G [IgG] or anti-rabbit IgG, 1:10,000; Jackson ImmunoResearch, West Grove, PA) for 1 h at room temperature. Membranes were washed again (5×5 min in 1×PBS-T) before visualizing protein complexes using SuperSignal West Pico Chemiluminescent Substrate (Pierce). Protein bands were quantified by densitometric analysis of scanned film using QuantityOne Basic software (Bio-Rad). Data are presented as percent of sham.
Immunohistochemical detection of amyloid-beta
Mice were anesthetized and transcardially perfused with 4% paraformaldehyde (PFA) and brains were drop-fixed in 4% PFA overnight before cryoprotection in 30% sucrose. Frozen brains were sliced into 50-μm floating coronal sections using a microtome. Before immunostaining, sections were incubated in 0.3% H202 to quench endogenous peroxidases, blocked with 3% goat serum in 0.25% Triton X and 1×PBS, and incubated overnight in rabbit anti-Aβ42 (1:500, catalog no. 700254; Invitrogen) or rabbit anti-APP (1:1000, catalog no. 51-2700; Invitrogen). After three 5-min washes in 1×PBS, sections were incubated in goat anti-rabbit secondary Ab at a 1:1000 dilution made in 0.25% Triton X and 1×PBS, followed by adenosine-triphosphate–binding cassette ABC (1:400, catalog no. PK-6100; Vector Laboratories, Burlingame, CA) and 3,3'-diaminobenzidine (Sigma-Aldrich) substrate to visualize primary Abs.
Western blot detection of amyloid-beta oligomers
Non-reduced DEA samples (100 ug of protein/sample) were prepared in Laemelli buffer without the addition of BME and separated by SDS-PAGE on five-well 12% Bis-Tris gels. As a positive control, Aβ oligomers (100 mM) were prepared in vitro from human Aβ42 peptide, as described in Stine and colleagues.46 Briefly, lyophilized Aβ42 peptide (American Peptide Company, Sunnyvale, CA) was dissolved in 1,1,1,3,3,3,-hexafluoro-2-propanol (HFIP; Sigma-Aldrich) until clear and then air-dried overnight to allow the HFIP to evaporate and a peptide film to form, which was stored at −20°C until use. Peptide films were brought to room temperature before preparing 5 mM of Aβ stock by resuspension in anhydrous dimethyl sulfoxide (Sigma-Aldrich) at room temperature. This was diluted in ice-cold Opti-MEM® (Invitrogen) for a final concentration of 100 μM of Aβ, vortexed for 15 sec, and then kept at 4°C for 24 h. A final concentration of 50 mM of Aβ oligomer preparation was loaded as the positive control. Western blotting was carried out as described above, with the exception that blots were incubated overnight at 4°C with the monoclonal anti-human Aβ antibody, 6E10, recognizing amino acid residue 1–16 of Aβ (1:1000, catalog no. SIG-39320; Covance) and probed with HRP-conjugated anti-mouse secondary Ab (1:10,000; Jackson ImmunoResearch).
Slot blot detection of amyloid-beta oligomers
For detection of oligomers in their native form by the oligomer-specific Ab, A11 (AHB0052; Invitrogen, and AB9234; Millipore), 5 μL of DEA brain homogenate was loaded into the Whatman Minifold II System slot blot apparatus (GE Healthcare Life Sciences, Piscataway, NJ) to apply sample directly to nitrocellulose membrane. The membrane was treated as described above, except it was incubated in A11 primary Ab (1:1000) and rabbit secondary Ab (1:10,000).
Statistical analysis
Data obtained from independent measurements are presented as the mean±standard error of the mean and were analyzed using either one-way analysis of variance (ANOVA) followed by post-hoc analysis by Dunnett's multiple comparison test, comparing all 1-, 3-, and 7-day time points to sham (for time-course experiments), or by unpaired t-test for comparison of sham versus injured (for characterization studies with only one time point). All statistical tests were performed using GraphPad Prism statistical software (version 5.0c; GraphPad Software, Inc., San Diego, CA). Differences were considered significant when p<0.05.
Results
Traumatic brain injury causes increased amyloid precursor protein processing, production of amyloid-beta, and aggregation of amyloid-beta into insoluble species
We have previously reported that TBI causes an increase in APP processing and Aβ production in C57/Bl6 mice.27,28 Here, we repeat those findings in 10-month-old 3×Tg AD model mice. APP processing can occur through either an α-secretase- or a β-secretase-mediated pathway, with the latter pathway leading to production of Aβ. We found that TBI caused a significant increase in both α- and β-mediated processing of APP (Fig. 1A). Levels of the β-secretase enzyme, BACE1, and the APP cleavage product, β-CTF, were increased in the injured cortex at 1 and 3 days after TBI (342% and 196% increase above sham levels, respectively; p<0.05), indicating activated β-secretase-mediated cleavage of APP at both of these time points (Fig. 1A). Levels of α-CTF, generated by α-secretase cleavage of APP in the nonamyloidogenic pathway, were also increased by 86–93% at 1, 3, and 7 days after injury (p<0.05; Fig. 1A).
FIG. 1.

TBI increases APP processing and both soluble and insoluble Aβ in the ipsilateral cortex of 10-month-old 3×Tg mice. (A) Quantification of amyloid precursor protein (APP), beta-site APP cleaving enzyme 1 (BACE1), and APP C-terminal fragments (APP-CTF) from Western blots. Representative blots are shown. (B) Levels of soluble and insoluble Aβ40 and Aβ42, quantified by ELISA. Percentage of insoluble Aβ indicates the percent of insoluble Aβ in the entire brain homogenate and was calculated from raw Aβ levels (pg/mL). ANOVA with Dunnett's post-hoc analysis comparing 1, 3, and 7 days to sham. *p<0.05; **p<0.01; ***p<0.001 (n=4 mice/time point). TBI, traumatic brain injury; Aβ, amyloid-beta; ELISA, enzyme-linked immunosorbent assay; ANOVA, analysis of variance.
The increased processing of APP through the β-secretase pathway after TBI causes increased production of Aβ. We found that injury caused a significant increase in levels of soluble Aβ40 (166% increase, compared to sham; p<0.001) and soluble Aβ42 (58% increase, compared to sham; p<0.05) in the ipsilateral cortex at 1 day postinjury (Fig. 1B), but both species had returned to sham levels at 3 days postinjury. We also extracted insoluble Aβ species using formic acid as part of a multi-step extraction procedure. We found that TBI caused a 120% increase in insoluble Aβ40 (p<0.001) and a 112% increase in insoluble Aβ42 (p<0.01) in the injured cortex, peaking at 1 day postinjury (Fig. 1B). Insoluble Aβ40 levels returned to sham levels by 3 days after injury, similar to the time course of soluble Aβ. However, insoluble Aβ42 remained elevated at 3 days postinjury (74% increase over sham levels; p<0.05), which may indicate a slower clearance of this insoluble species.
We further analyzed our data by performing a pools analysis to determine whether the increased levels of Aβ after TBI is caused by aggregation of soluble Aβ into insoluble Aβ or whether the changes merely reflect increased Aβ levels in all pools (Fig. 1B). Before injury, the majority of Aβ is available in the soluble fraction (79% of total Aβ40 and 84% of total Aβ42), with only the remainder in the insoluble fraction (21% of Aβ40 and 16% of Aβ42). After injury, however, the percentage of Aβ40 and Aβ42 that is in the insoluble pool increases over time as excess soluble Aβ is resolved, so that by 7 days postinjury 33% of the total Aβ40 pool is insoluble (p<0.01). The shift in Aβ42 occurs earlier, with the percentage of Aβ42 in the insoluble pool increasing from 16% in sham mice to 32% at 3 days (p<0.01) and remaining increased at 7 days (33%; p<0.01). These data indicate that although total Aβ levels may return to sham levels by 7 days postinjury, an imbalance remains in the ratio of soluble to insoluble Aβ species, with insoluble Aβ species making up a greater proportion of total Aβ 1 week postinjury.
Amyloid-beta peptides aggregate in the brain after traumatic brain injury in young triple transgenic mice
To examine whether increased aggregation after TBI in our 10-month-old 3×Tg mice is related to their advanced age, we also measured Aβ after injury in a cohort of 8-week-old 3×Tg mice. Levels of soluble Aβ40 (155% increase, p<0.001; Fig. 2) and Aβ42 (122% increase, p<0.05; Fig. 2) and insoluble Aβ40 and Aβ42 (248 and 153% increase, respectively, p<0.05; Fig. 2) were all significantly increased in the injured cortex at 1 day postinjury (Fig. 2), indicating that acute injury-induced aggregation was not dependent on age. Comparison of ipsi- and contralateral cortices found that the TBI-induced increase in Aβ occurred in the ipsilateral side only.
FIG. 2.

TBI increases levels of soluble and insoluble Aβ40 and Aβ42 in the injured cortex of young triple transgenic (3×Tg) mice. Quantification of soluble and insoluble Aβ40 and Aβ42 from brains of 8-week-old 3×Tg mice 1 day postinjury. Ipsi- and contralateral cortices are shown. Unpaired t-test for comparison of sham versus injured. *p<0.05; ***p<0.001 (n=3 mice/group). TBI, traumatic brain injury; Aβ, amyloid-beta.
Amyloid-beta 42 accumulates in damaged axons after injury
Immunohistochemistry (IHC) was performed using a cleavage-site–specific Ab against Aβ42 on coronal slices from injured and sham animals. To ensure that the Aβ42-specific Ab was not cross-reacting with APP, we performed IHC on adjacent sections of 3-day post-TBI tissue. We found that the APP-stained tissue presented an abundance of immunoreactive swellings in the ipsilateral corpus callosum and injured cortex (Fig. 3A). In contrast, there were only a small number of Aβ42-positive accumulations in the corpus callosum (Fig. 3A). These data demonstrate that the site-specific Aβ42 Ab is not detecting full-length APP.
FIG. 3.

Aβ accumulates in damaged axons after injury. (A) Comparison of anti-APP and neoepitope-specific anti-Aβ42 antibodies on contiguous sections from a 3-day post-TBI mouse show large amounts of APP staining, but very little Aβ42 staining. Scale bar, 100 μM. (B) In the injured cortex 1 day postinjury, there was Aβ42-positive plaque-like staining, along with intracellular Aβ accumulation in cells directly adjacent to the lesion. Scale bar, 20 μM. (C) Intense Aβ42 immunoreactivity in damaged axons of the external capsule and fimbria. Scale bar, 20 μM. (D) Representative Western blots for myelin basic protein (MBP) confirm successful isolation of white matter tracts from cortical tissue from sham and TBI brain. These white matter homogenates were probed for soluble Aβ40 by ELISA. One-way ANOVA with Dunnett's post-hoc analysis comparing 1, 3, and 7 days to sham. **p<0.01. (n=4 mice/time point). Aβ, amyloid-beta; APP, amyloid precursor protein; TBI, traumatic brain injury; ELISA, enzyme-linked immunosorbent assay; ANOVA, analysis of variance.
At 1 day postinjury, we found increased intracellular accumulation of Aβ42 and diffuse accumulations in the ipsilateral cortex near the area of lesion in injured mice (Fig. 3B). However, although diffuse plaque was detected in the ipsilateral cortex of 1 injured mouse, this was a rare occurrence and was observed in only 1 of 7 injured mice at this time point.
We found that the majority of Aβ42 staining occurred in WM tracts in areas of traumatic axonal injury, with the most intense staining in the ipsilateral external capsule and in the fimbria of the hippocampus (Fig. 3C). Aβ-positive swellings were visible along the lengths of damaged axons, with multiple varicosities visible within a single axon. We verified this increase in WM Aβ by measuring soluble Aβ40 levels in dissected WM by ELISA, showing that levels of soluble Aβ40 are increased by 144% at 1 day after injury (p<0.01), but return to sham levels by 3 days (Fig. 3D). Myelin basic protein levels were measured in the RIPA fraction of WM and ipsilateral cortical samples from the same animals to verify enrichment of axons in the WM dissection.
Traumatic brain injury increases levels of soluble amyloid-beta oligomers in the brain
Oligomerization of Aβ monomers is an obligatory, intermediate step in Aβ aggregation. To determine whether injury increases Aβ oligomerization in the brain, we first looked for the presence of soluble Aβ oligomers by Western blot and found multiple bands immunoreactive for the anti-Aβ Ab, 6E10, in our injured TBI samples that were not present in our sham samples or in the negative control lacking primary Ab (Fig. 4A). To confirm this, we measured levels of Aβ oligomers in the DEA fraction by ELISA and found that levels of soluble Aβ oligomers were increased in the injured cortex by 53% at 1 day after injury, compared to sham (p<0.05; Fig. 4B). Soluble Aβ oligomer levels also returned to sham levels by 3 days after injury.
FIG. 4.
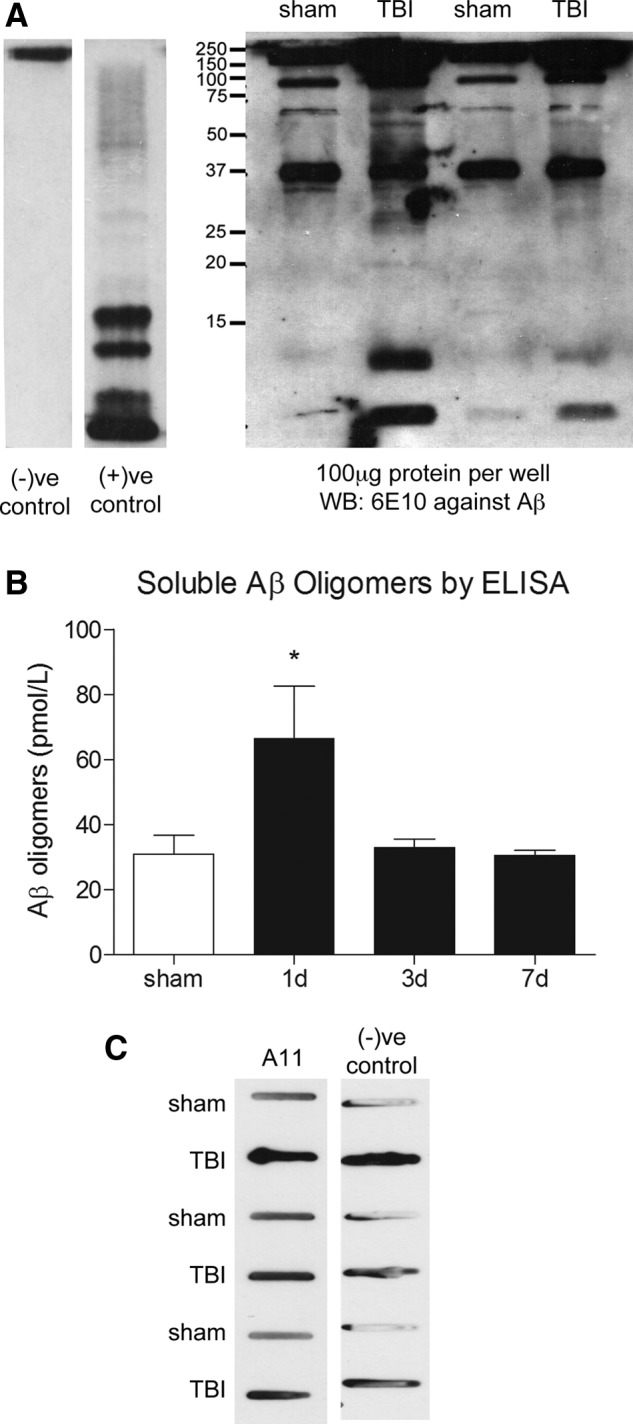
TBI increases levels of Aβ oligomers in the injured cortex of 3×Tg mice. (A) Representative Western blot of 100 μg of nonreduced DEA-soluble protein resolved on 12% gels and probed with the anti-Aβ antibody, 6E10. Multiple 6E10-positive bands are present in 1-day TBI samples, compared to sham samples. These bands correspond to multiple bands from the positive control, which contains Aβ oligomers prepared in vitro. The negative control consists of 100 μg of 1-day TBI DEA homogenate probed with secondary antibody alone. (B) Quantification of soluble Aβ oligomers by ELISA confirms that soluble Aβ oligomers are increased in the ipsilateral cortex at 1 day postinjury. (C) Representative slot blot showing attempt to detect Aβ oligomers in unresolved samples by slot blot assay and the oligomer-specific antibody, A11, and the corresponding negative control blot that did not contain A11 antibody. ANOVA with Dunnett's post-hoc analysis comparing 1, 3, and 7 days to sham. *p<0.05 (n=4 mice/time point). Aβ, amyloid-beta; DEA, diethylamine; ELISA, enzyme-linked immunosorbent assay; ANOVA, analysis of variance.
Another technique commonly used for detecting Aβ oligomers is probing unresolved proteins on a dot or slot blot with the oligomeric-specific Ab, A11. We used A11 purchased from both Invitrogen and Millipore, but found that the slot blot technique could not be used for detection of Aβ oligomers in our samples because of the presence of endogenous autoAbs produced after TBI. These endogenous autoAbs appear to bind to both anti-mouse and -rabbit secondary Abs, producing immunoreactivity in the TBI samples that can be mistaken for a specific signal. So, although we saw an increase in A11 immunoreactivity in our TBI samples, these blots exactly resembled the negative control, indicating that the presumed A11 immunoreactivity was actually the result of binding of secondary Ab to endogenous TBI-induced Abs.
Discussion
We and others have previously demonstrated that experimental TBI increases levels of soluble Aβ in the brain using either WT or human APP Tg mice.4,24–31 Here, we repeat and expand upon those findings, using 3×Tg AD model mice to show that TBI increases levels of both soluble and insoluble Aβ in the brain and alters the ratio of insoluble to soluble Aβ, so that even after total levels return to baseline, a greater proportion of Aβ in the injured brain is insoluble. For the first time, we also report the presence of soluble oligomeric Aβ species after TBI. Our data demonstrate that the accumulation of soluble Aβ acutely after TBI can lead to the oligomerization and fibrilization of Aβ in the mouse brain—however, the mouse brain does appear to be particularly proficient at clearing these abnormal Aβ accumulations within 7 days of injury.
There is a large amount of evidence indicating soluble oligomers as the pathogenic species of Aβ in AD.13 Recently, application of nanomolar concentrations of soluble Aβ oligomers to hippocampal slice cultures for 30 min was shown to induce synaptotoxicity and impair hippocampal synaptic transmission.47 Given the rapid ability of oligomers to induce these changes, it is possible that increased levels of soluble Aβ oligomers acutely after TBI could contribute to synaptic breakdown and induction of secondary injury pathways. Interestingly, a recent study of human CSF of TBI patients found that an HMW soluble Aβ oligomer is detectable in a subset of patients within 72 h of injury.23 Further, this group found that patients with higher levels of Aβ oligomers had poorer outcome 6 months after injury, compared to patients with high levels of just Aβ42 monomers in CSF, suggesting a relationship between Aβ oligomer formation and impairment after injury.
Here, we report the accumulation of Aβ oligomers after experimental TBI. We identified oligomers using two distinct methods: SDS-PAGE coupled with Western blot analysis and an oligomer-specific ELISA. SDS-PAGE is one of the most common methods used for Aβ oligomer size determination, and combining this with Western blot detection of Aβ with 6E10 Ab enhances sensitivity and resolution. In our blots, we can visualize the presence of numerous low-molecular-weight (LMW) and HMW 6E10-positive bands. The strongest of these appear to be a band below 10 kDa, which corresponds to a mono- and dimeric Aβ band, and a second band just above 10 kDa that corresponds to Aβ trimers on our positive control. We can also detect higher molecular weight species at approximately 30 kDa and between 45 and 100 kDa. Because the addition of SDS to samples may accelerate Aβ aggregation during the SDS-PAGE procedure,48 we confirmed the presence of oligomers in our samples using a second method. The Aβ oligomer-specific ELISA uses the 82E1 N-terminus-specific Ab for both capture and detection and so requires two or more epitopes to be present in the same peptide sequence to generate a positive signal.45 Similar to our Western blot data, we found that Aβ oligomers were significantly increased at 1 day postinjury. However, the brain was able to rapidly resolve the excess oligomers, which returned to sham levels by 3 days post-trauma. Finally, we wanted to use an oligomer-specific Ab, A11, to detect oligomers in brain homogenate after TBI. A11 is an oligomeric confirmation-specific Ab that will detect oligomeric species of several amyloidogenic peptides (including prion peptide, human insulin, α-synuclein, polyglutamine, lysozyme, and islet amyloid polypeptide in addition to Aβ) once they undergo a conformational change49 and was demonstrated to not cross-react with soluble LMW Aβ or Aβ fibrils.49 We used a slot blot assay that has been commonly employed to detect oligomers in nondenatured samples in conjunction with A11 Ab48 and probed the blots with A11 sourced from two different companies. We found that both Abs showed a strong increase in signal in our TBI samples. However, despite the secondary Ab being anti-rabbit, these data were proven to be a false positive when we probed blots with just secondary Ab alone. We include these data here because this technique is commonly used in the AD field; however, as our data demonstrate, care should be taken when using this type of assay to probe TBI samples.
Previous studies of Aβ aggregation after experimental injury have focused primarily on the formation of amyloid plaque, because the principal objective has been to replicate the acute formation of plaque after TBI reported in humans. Initial studies in non-Tg rodents did not detect plaque formation after injury50–56; however, it should be noted that levels of Aβ in mouse brain, even after injury, are well below the normal thresholds for detection by IHC. This lack of plaque formation in WT mice may also be the result of the fact that rodent Aβ has a lower ability to aggregate at low peptide concentrations, compared to human Aβ.57 This problem is circumvented by the use of mice overexpressing human APP. Whereas TBI does not induce acute plaque formation in these mice,34 it may accelerate chronic plaque deposition after a single33 or repeat TBI.30 Unfortunately, it is difficult to draw definitive conclusions because experimental TBI has also been reported to decrease,35,36 or have no effect on,37–39 insoluble Aβ or amyloid plaque formation.
A potential confounding factor in previous animal studies may be the length of time between injury induction and Aβ analysis, because different single time points ranging from acute to chronic have been explored, depending on the study. Data from human TBI studies indicate that plaques can appear as rapidly as 24 h after injury,7 and an increased number of TBI brains have plaques present many years after a single TBI, compared to noninjured controls.58 Understanding Aβ dynamics acutely, chronically, and in the intermediate phase after injury are critical to our understanding of how the occurrence of a single TBI might be leading to the development of AD in later life. In this study, we have limited our investigations to the acute phase after TBI to obtain a more complete picture of Aβ production, accumulation, and aggregation in the mouse brain. We chose this time period because plaque development occurs very rapidly in the human brain after TBI,7–9 and it is also the time frame in which we have observed peak increases in soluble Aβ in WT TBI mouse brain.27,28 This peak in Aβ production occurs because of increased β-secretase cleavage of APP, as indicated by an accumulation of BACE1 and β-CTF. Interestingly, this occurs simultaneously with an increase in α-secretase cleavage of APP. This simultaneous trafficking of APP through both the α-secretase and β-secretase cleavage pathways is not normal, because, usually, levels of APP remain constant and the proportion of APP sent through either secretase pathway is inversely related. This can be explained by the accumulation of APP in the injured brain, which would result in both α- and β-mediated processing. In our study, we found no increase in total protein levels of APP at 7 days postinjury; however, we do see a large accumulation of APP in injured axons throughout the entire ipsilateral cortex, hippocampus, and thalamus. We believe this abnormally accumulating APP is acting as the excess substrate required to produce excess α- and β-CTF simultaneously.
Previous studies have identified axons as primary sites of Aβ accumulation after TBI in both humans5,6 and animals.4,34,56,59 Colocalization of Aβ with accumulated APP, BACE1, and presenilin 14,6 within axons indicate axons as a hot spot for Aβ generation after injury. Here, we provide additional evidence that axons appear to be a major site of Aβ accumulation after injury using both IHC and ELISA data from isolated WM. Rapid generation of Aβ after injury causes accumulation of soluble Aβ40 and Aβ42 peptides, generation of soluble Aβ oligomers, and formation of insoluble Aβ species. We did not detect any fibrillar Aβ deposits in the majority of our mice, but did observe a cloudy Aβ42-positive accumulation in just one mouse cortex. There was insufficient tissue to fully determine whether this deposition was thioflavin S positive. One other study investigated levels of insoluble Aβ in the injured cortex of 3×Tg mice 24 h after TBI and found an increase in insoluble Aβ40, but not soluble Aβ40, at 24 h after injury.34 However, the study did not report any amyloid plaque deposition.
Our data reveal that the mouse brain rapidly resolves excess levels of TBI-induced soluble, oligomeric, and insoluble Aβ within 3–7 days after injury, suggesting that Aβ clearance pathways are very active in the mouse brain after TBI. This occurs despite the continued elevation of both BACE1 and β-CTF at 3 days postinjury, indicating increased Aβ production at 3 days. This robust Aβ clearance in mouse brain may explain why there has been such difficulty reproducing acute TBI-induced amyloid deposition in rodents.
One factor that could influence Aβ clearance is apolipoprotein E (APOE) genotype. A retrospective study of human TBI postmortem brains revealed a strong influence of APOE4 genotype on plaque formation after injury.60 Whereas only 10% of non-APOE4 brains had amyloid plaques, 35% of heterozygous APOE4 brains and 100% of homozygous APOE4 brains had amyloid deposits after TBI. In animal studies, the presence of the human APOE4 gene in PDAPP mice caused an increase in amyloid burden 6 months after experimental TBI.33 However, although another study using similar mice did not report a significant effect of APOE4 genotype on Aβ levels after TBI by ELISA, there was an increase in intracellular Aβ staining in APOE4 TBI mice, compared to APOE3 TBI mice.26
A potential limitation of our study is that we are investigating acute aggregation of Aβ after injury, whereas it is chronic accumulation and aggregation of Aβ that is thought to cause AD. Investigation of chronic time points after injury in 3×Tg mice will be needed to determine whether TBI causes pathological and cognitive changes associated with AD in these mice, such as Aβ deposition, neurofibrillary tangle formation, synaptic dysfunction or loss, and memory deficits. However, our data report the detection of soluble Aβ oligomers in an animal model of TBI for the first time, supporting the finding of these oligomers after human TBI. Increased levels of soluble Aβ oligomers in the brain during the acute phase after injury is relevant outside of their potential role in AD development, because the particular toxicity of this Aβ species may contribute to secondary injury after TBI.
Acknowledgments
The project described above was supported by R01NS081068 and R03NS067417 (to M.P.B.) from the National Institute of Neurological Disorders and Stroke (NINDS) and T32NS041218 (to P.M.W.; principal investigators, Drs. Jean Wrathall and Kathleen Maguire-Zeiss), the Cosmos Club Foundation's Young Scholars Grant Program (to P.M.W.), and the Georgetown University Medical Center Graduate Student Organization's Student Research Grants Program (to P.M.W.). The authors thank Drs. R. Scott Turner and Hyang-Sook Hoe for providing 3×Tg AD model mice and Dr. Paul Mathews (Nathan S. Kline Institute, Orangeburg, NY) for C1/6.1 Ab.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Mortimer J.A., van Duijn C.M., Chandra V., Fratiglioni L., Graves A.B., Heyman A., Jorm A.F., Kokmen E., Kondo K., Rocca W.A., Shalat S.L., Soininen H., and Hofman A. (1991). Head trauma as a risk factor for Alzheimer's disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group Int. J. Epidemiol. 20, Suppl.2, S28–S35 [DOI] [PubMed] [Google Scholar]
- 2.Plassman B.L., Havlik R.J., Steffens D.C., Helms M.J., Newman T.N., Drosdick D., Phillips C., Gau B.A., Welsh-Bohmer K.A., Burke J.R., Guralnik J.M., and Breitner J.C. (2000). Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology 55, 1158–1166 [DOI] [PubMed] [Google Scholar]
- 3.Schofield P.W., Tang M., Marder K., Bell K., Dooneief G., Chun M., Sano M., Stern Y., and Mayeux R. (1997). Alzheimer's disease after remote head injury: an incidence study. J. Neurol. Neurosurg. Psychiatry 62, 119–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen X.H., Siman R., Iwata A., Meaney D.F., Trojanowski J.Q., and Smith D.H. (2004). Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am. J. Pathol. 165, 357–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith D.H., Chen X.H., Iwata A., and Graham D.I. (2003). Amyloid beta accumulation in axons after traumatic brain injury in humans. J. Neurosurg. 98, 1072–1077 [DOI] [PubMed] [Google Scholar]
- 6.Uryu K., Chen X.H., Martinez D., Browne K.D., Johnson V.E., Graham D.I., Lee V.M., Trojanowski J.Q., and Smith D.H. (2007). Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 208, 185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ikonomovic M.D., Uryu K., Abrahamson E.E., Ciallella J.R., Trojanowski J.Q., Lee V.M., Clark R.S., Marion D.W., Wisniewski S.R., and Dekosky S.T. (2004). Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp. Neurol. 190, 192–203 [DOI] [PubMed] [Google Scholar]
- 8.Roberts G.W., Gentleman S.M., Lynch A., and Graham D.I. (1991). beta A4 amyloid protein deposition in brain after head trauma. Lancet 338, 1422–1423 [DOI] [PubMed] [Google Scholar]
- 9.Roberts G.W., Gentleman S.M., Lynch A., Murray L., Landon M., and Graham D.I. (1994). Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 57, 419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardy J., and Selkoe D.J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 11.Hardy J.A., and Higgins G.A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185 [DOI] [PubMed] [Google Scholar]
- 12.Selkoe D.J. (1998). The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell. Biol. 8, 447–453 [DOI] [PubMed] [Google Scholar]
- 13.Walsh D.M., and Selkoe D.J. (2007). A beta oligomers—a decade of discovery. J. Neurochem. 101, 1172–1184 [DOI] [PubMed] [Google Scholar]
- 14.Benilova I., Karran E., and De Strooper B. (2012). The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 [DOI] [PubMed] [Google Scholar]
- 15.Selkoe D.J. (2002). Alzheimer's disease is a synaptic failure. Science 298, 789–791 [DOI] [PubMed] [Google Scholar]
- 16.McLean C.A., Cherny R.A., Fraser F.W., Fuller S.J., Smith M.J., Beyreuther K., Bush A.I., and Masters C.L. (1999). Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46, 860–866 [DOI] [PubMed] [Google Scholar]
- 17.Lue L.F., Kuo Y.M., Roher A.E., Brachova L., Shen Y., Sue L., Beach T., Kurth J.H., Rydel R.E., and Rogers J. (1999). Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am. J. Pathol. 155, 853–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh D.M., Klyubin I., Fadeeva J.V., Cullen W.K., Anwyl R., Wolfe M.S., Rowan M.J., and Selkoe D.J. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539 [DOI] [PubMed] [Google Scholar]
- 19.Shankar G.M., Li S., Mehta T.H., Garcia-Munoz A., Shepardson N.E., Smith I., Brett F.M., Farrell M.A., Rowan M.J., Lemere C.A., Regan C.M., Walsh D.M., Sabatini B.L., and Selkoe D.J. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert M.P., Barlow A.K., Chromy B.A., Edwards C., Freed R., Liosatos M., Morgan T.E., Rozovsky I., Trommer B., Viola K.L., Wals P., Zhang C., Finch C.E., Krafft G.A., and Klein W.L. (1998). Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U. S. A. 95, 6448–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dahlgren K.N., Manelli A.M., Stine W.B., Jr., Baker L.K., Krafft G.A., and LaDu M.J. (2002). Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053 [DOI] [PubMed] [Google Scholar]
- 22.Deshpande A., Mina E., Glabe C., and Busciglio J. (2006). Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J. Neurosci. 26, 6011–6018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gatson J.W., Warren V., Abdelfattah K., Wolf S., Hynan L.S., Moore C., Diaz-Arrastia R., Minei J.P., Madden C., and Wigginton J.G. (2013). Detection of beta-amyloid oligomers as a predictor of neurological outcome after brain injury. J. Neurosurg. 118, 1336–1342 [DOI] [PubMed] [Google Scholar]
- 24.Abrahamson E.E., Ikonomovic M.D., Ciallella J.R., Hope C.E., Paljug W.R., Isanski B.A., Flood D.G., Clark R.S., and DeKosky S.T. (2006). Caspase inhibition therapy abolishes brain trauma-induced increases in Abeta peptide: implications for clinical outcome. Exp. Neurol. 197, 437–450 [DOI] [PubMed] [Google Scholar]
- 25.Abrahamson E.E., Ikonomovic M.D., Dixon C.E., and Dekosky S.T. (2009). Simvastatin therapy prevents brain trauma-induced increases in beta-amyloid peptide levels. Ann. Neurol. 66, 407–414 [DOI] [PubMed] [Google Scholar]
- 26.Laskowitz D.T., Song P., Wang H., Mace B., Sullivan P.M., Vitek M.P., and Dawson H.N. (2010). Traumatic brain injury exacerbates neurodegenerative pathology: improvement with an apolipoprotein E-based therapeutic. J. Neurotrauma 27, 1983–1995 [DOI] [PubMed] [Google Scholar]
- 27.Loane D.J., Pocivavsek A., Moussa C.E., Thompson R., Matsuoka Y., Faden A.I., Rebeck G.W., and Burns M.P. (2009). Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat. Med. 15, 377–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loane D.J., Washington P.M., Vardanian L., Pocivavsek A., Hoe H.S., Duff K.E., Cernak I., Rebeck G.W., Faden A.I., and Burns M.P. (2011). Modulation of ABCA1 by an LXR agonist reduces beta-amyloid levels and improves outcome after traumatic brain injury. J. Neurotrauma 28, 225–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mannix R.C., Zhang J., Park J., Zhang X., Bilal K., Walker K., Tanzi R.E., Tesco G., and Whalen M.J. (2011). Age-dependent effect of apolipoprotein E4 on functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 31, 351–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uryu K., Laurer H., McIntosh T., Pratico D., Martinez D., Leight S., Lee V.M., and Trojanowski J.Q. (2002). Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J. Neurosci. 22, 446–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H., Durham L., Dawson H., Song P., Warner D.S., Sullivan P.M., Vitek M.P., and Laskowitz D.T. (2007). An apolipoprotein E-based therapeutic improves outcome and reduces Alzheimer's disease pathology following closed head injury: evidence of pharmacogenomic interaction. Neuroscience 144, 1324–1333 [DOI] [PubMed] [Google Scholar]
- 32.Fraser P.E., Nguyen J.T., Inouye H., Surewicz W.K., Selkoe D.J., Podlisny M.B., and Kirschner D.A. (1992). Fibril formation by primate, rodent, and Dutch-hemorrhagic analogues of Alzheimer amyloid beta-protein. Biochemistry 31, 10716–10723 [DOI] [PubMed] [Google Scholar]
- 33.Hartman R.E., Laurer H., Longhi L., Bales K.R., Paul S.M., McIntosh T.K., and Holtzman D.M. (2002). Apolipoprotein E4 influences amyloid deposition but not cell loss after traumatic brain injury in a mouse model of Alzheimer's disease. J. Neurosci. 22, 10083–10087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tran H.T., Sanchez L., Esparza T.J. and Brody D.L. (2011). Distinct temporal and anatomical distributions of amyloid-beta and tau abnormalities following controlled cortical impact in transgenic mice. PLoS One 6, e25475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakagawa Y., Nakamura M., McIntosh T.K., Rodriguez A., Berlin J.A., Smith D.H., Saatman K.E., Raghupathi R., Clemens J., Saido T.C., Schmidt M.L., Lee V.M., and Trojanowski J.Q. (1999). Traumatic brain injury in young, amyloid-beta peptide overexpressing transgenic mice induces marked ipsilateral hippocampal atrophy and diminished Abeta deposition during aging. J. Comp. Neurol. 411, 390–398 [PubMed] [Google Scholar]
- 36.Nakagawa Y., Reed L., Nakamura M., McIntosh T.K., Smith D.H., Saatman K.E., Raghupathi R., Clemens J., Saido T.C., Lee V.M., and Trojanowski J.Q. (2000). Brain trauma in aged transgenic mice induces regression of established abeta deposits. Exp. Neurol. 163, 244–252 [DOI] [PubMed] [Google Scholar]
- 37.Conte V., Uryu K., Fujimoto S., Yao Y., Rokach J., Longhi L., Trojanowski J.Q., Lee V.M., McIntosh T.K., and Pratico D. (2004). Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J. Neurochem. 90, 758–764 [DOI] [PubMed] [Google Scholar]
- 38.Murai H., Pierce J.E., Raghupathi R., Smith D.H., Saatman K.E., Trojanowski J.Q., Lee V.M., Loring J.F., Eckman C., Younkin S., and McIntosh T.K. (1998). Twofold overexpression of human beta-amyloid precursor proteins in transgenic mice does not affect the neuromotor, cognitive, or neurodegenerative sequelae following experimental brain injury. J. Comp. Neurol. 392, 428–438 [DOI] [PubMed] [Google Scholar]
- 39.Smith D.H., Nakamura M., McIntosh T.K., Wang J., Rodriguez A., Chen X.H., Raghupathi R., Saatman K.E., Clemens J., Schmidt M.L., Lee V.M., and Trojanowski J.Q. (1998). Brain trauma induces massive hippocampal neuron death linked to a surge in beta-amyloid levels in mice overexpressing mutant amyloid precursor protein. Am. J. Pathol. 153, 1005–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Washington P.M., Forcelli P.A., Wilkins T., Zapple D., Parsadanian M., and Burns M.P. (2012). The effect of injury severity on behavior: a phenotypic study of cognitive and emotional deficits after mild, moderate and severe controlled cortical impact injury in mice. J. Neurotrauma 29, 2283–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oddo S., Caccamo A., Shepherd J.D., Murphy M.P., Golde T.E., Kayed R., Metherate R., Mattson M.P., Akbari Y., and LaFerla F.M. (2003). Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421 [DOI] [PubMed] [Google Scholar]
- 42.Hirata-Fukae C., Li H.F., Hoe H.S., Gray A.J., Minami S.S., Hamada K., Niikura T., Hua F., Tsukagoshi-Nagai H., Horikoshi-Sakuraba Y., Mughal M., Rebeck G.W., LaFerla F.M., Mattson M.P., Iwata N., Saido T.C., Klein W.L., Duff K.E., Aisen P.S., and Matsuoka Y. (2008). Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res. 1216, 92–103 [DOI] [PubMed] [Google Scholar]
- 43.Luo Y., Bolon B., Kahn S., Bennett B.D., Babu-Khan S., Denis P., Fan W., Kha H., Zhang J., Gong Y., Martin L., Louis J.C., Yan Q., Richards W.G., Citron M., and Vassar R. (2001). Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 4, 231–232 [DOI] [PubMed] [Google Scholar]
- 44.Savage M.J., Trusko S.P., Howland D.S., Pinsker L.R., Mistretta S., Reaume A.G., Greenberg B.D., Siman R., and Scott R.W. (1998). Turnover of amyloid beta-protein in mouse brain and acute reduction of its level by phorbol ester. J. Neurosci. 18, 1743–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xia W., Yang T., Shankar G., Smith I.M., Shen Y., Walsh D.M., and Selkoe D.J. (2009). A specific enzyme-linked immunosorbent assay for measuring beta-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch. Neurol. 66, 190–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stine W.B., Jr., Dahlgren K.N., Krafft G.A., and LaDu M.J. (2003). In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278, 11612–11622 [DOI] [PubMed] [Google Scholar]
- 47.Li S., Jin M., Zhang D., Yang T., Koeglsperger T., Fu H., and Selkoe D.J. (2013). Environmental novelty activates beta2-adrenergic signaling to prevent the impairment of hippocampal LTP by Abeta oligomers. Neuron 77, 929–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pryor N.E., Moss M.A., and Hestekin C.N. (2012). Unraveling the early events of amyloid-beta protein (Abeta) aggregation: techniques for the determination of Abeta aggregate size. Int. J. Mol. Sci. 13, 3038–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kayed R., Head E., Thompson J.L., McIntire T.M., Milton S.C., Cotman C.W., and Glabe C.G. (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 50.Ciallella J.R., Ikonomovic M.D., Paljug W.R., Wilbur Y.I., Dixon C.E., Kochanek P.M., Marion D.W., and DeKosky S.T. (2002). Changes in expression of amyloid precursor protein and interleukin-1beta after experimental traumatic brain injury in rats. J. Neurotrauma 19, 1555–1567 [DOI] [PubMed] [Google Scholar]
- 51.Hoshino S., Tamaoka A., Takahashi M., Kobayashi S., Furukawa T., Oaki Y., Mori O., Matsuno S., Shoji S., Inomata M., and Teramoto A. (1998). Emergence of immunoreactivities for phosphorylated tau and amyloid-beta protein in chronic stage of fluid percussion injury in rat brain. Neuroreport 9, 1879–1883 [DOI] [PubMed] [Google Scholar]
- 52.Laurer H.L., Bareyre F.M., Lee V.M., Trojanowski J.Q., Longhi L., Hoover R., Saatman K.E., Raghupathi R., Hoshino S., Grady M.S., and McIntosh T.K. (2001). Mild head injury increasing the brain's vulnerability to a second concussive impact. J. Neurosurg. 95, 859–870 [DOI] [PubMed] [Google Scholar]
- 53.Lewen A., Li G.L., Olsson Y., and Hillered L. (1996). Changes in microtubule-associated protein 2 and amyloid precursor protein immunoreactivity following traumatic brain injury in rat: influence of MK-801 treatment. Brain Res. 719, 161–171 [DOI] [PubMed] [Google Scholar]
- 54.Masumura M., Hata R., Uramoto H., Murayama N., Ohno T., and Sawada T. (2000). Altered expression of amyloid precursors proteins after traumatic brain injury in rats: in situ hybridization and immunohistochemical study. J. Neurotrauma 17, 123–134 [DOI] [PubMed] [Google Scholar]
- 55.Pierce J.E., Trojanowski J.Q., Graham D.I., Smith D.H., and McIntosh T.K. (1996). Immunohistochemical characterization of alterations in the distribution of amyloid precursor proteins and beta-amyloid peptide after experimental brain injury in the rat. J. Neurosci. 16, 1083–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stone J.R., Okonkwo D.O., Singleton R.H., Mutlu L.K., Helm G.A., and Povlishock J.T. (2002). Caspase-3-mediated cleavage of amyloid precursor protein and formation of amyloid Beta peptide in traumatic axonal injury. J. Neurotrauma 19, 601–614 [DOI] [PubMed] [Google Scholar]
- 57.Otvos L., Jr., Szendrei G.I., Lee V.M., and Mantsch H.H. (1993). Human and rodent Alzheimer beta-amyloid peptides acquire distinct conformations in membrane-mimicking solvents. Eur. J. Biochem. 211, 249–257 [DOI] [PubMed] [Google Scholar]
- 58.Johnson V.E., Stewart W., and Smith D.H. (2012). Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 22, 142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith D.H., Chen X.H., Nonaka M., Trojanowski J.Q., Lee V.M., Saatman K.E., Leoni M.J., Xu B.N., Wolf J.A., and Meaney D.F. (1999). Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J. Neuropathol. Exp. Neurol. 58, 982–992 [DOI] [PubMed] [Google Scholar]
- 60.Nicoll J.A., Roberts G.W., and Graham D.I. (1995). Apolipoprotein E epsilon 4 allele is associated with deposition of amyloid beta-protein following head injury. Nat. Med. 1, 135–137 [DOI] [PubMed] [Google Scholar]