

Abstract
Metachromatic leukodystrophy (MLD) is a lysosomal storage disorder of the brain caused by mutations in the gene encoding the lysosomal sulfatase, arylsulfatase A (ASA). It is not possible to treat the brain in MLD with recombinant ASA, because the enzyme does not cross the blood-brain barrier (BBB). In the present investigation, a BBB-penetrating IgG-ASA fusion protein is engineered and expressed, where the ASA monomer is fused to the carboxyl terminus of each heavy chain of an engineered monoclonal antibody (MAb) against the human insulin receptor (HIR). The HIRMAb crosses the BBB via receptor-mediated transport on the endogenous BBB insulin receptor, and acts as a molecular Trojan horse to ferry the ASA into brain from blood. The HIRMAb-ASA is expressed in stably transfected Chinese hamster ovary cells grown in serum free medium, and purified by protein A affinity chromatography. The fusion protein retains high affinity binding to the HIR, EC50 = 0.34 ± 0.11 nM, and retains high ASA enzyme activity, 20 ± 1 units/mg. The HIRMAb-ASA fusion protein is endocytosed and triaged to the lysosomal compartment in MLD fibroblasts. The fusion protein was radio-labeled with the Bolton-Hunter reagent, and the [125I]-HIRMAb-ASA rapidly penetrates the brain in the Rhesus monkey following intravenous administration. Film and emulsion autoradiography of primate brain shows global distribution of the fusion protein throughout the monkey brain. These studies describe a new biological entity that is designed to treat the brain of humans with MLD following non-invasive, intravenous infusion of an IgG-ASA fusion protein.
Keywords: arylsulfatase A, monoclonal antibody, drug delivery, insulin receptor, blood-brain barrier
Introduction
Metachromatic leukodystrophy (MLD) is a lysosomal storage disorder caused by mutations in the gene encoding the arylsulfatase A (ASA) enzyme (Fluharty, 2006). ASA deficiency in the brain leads to sulfatide glycolipid accumulation in the brain (Molander-Melin et al, 2004; Blomqvist et al, 2011). The most common form of MLD is the infantile form, and afflicted children suffer from severe abnormalities of the central nervous system (CNS), including gait disturbances, convulsions, and dementia, and die before 5 years of age (Fluharty, 2006). A potential treatment for MLD is enzyme replacement therapy (ERT) with recombinant ASA. However, ERT is not effective for the CNS, because the ASA enzyme does not cross the blood-brain barrier (BBB) (Matzner et al, 2005). To circumvent the BBB, the recombinant ASA enzyme has been administered by the intra-thecal route with direct injection into the cerebrospinal fluid (CSF) of mice (Stroobants et al, 2011). Whereas, the CSF route may deliver adequate amounts of enzyme to the brain in small animals, such as the mouse, there is little penetration into the brain from CSF in large animals. The intra-thecal injection of a massive amount, 960 mg, of a lysosomal enzyme, acid sphingomyelinase, in the Rhesus monkey resulted in distribution only to the ependymal surface of the brain (Ziegler et al, 2011). Drug penetration into the brain from the CSF is limited by diffusion, which is slow compared to the rapid rate of CSF exit from brain to blood via bulk flow (Pardridge, 2005).
The entire volume of the human brain can be treated by delivery of ASA via the trans-vascular route across the BBB. However, this approach requires the re-engineering of ASA as a molecule that is BBB-penetrating. This is possible with molecular Trojan horse (MTH) technology, which involves genetic fusion of ASA to another protein that does cross the BBB (Pardridge and Boado, 2012). A MTH is an endogenous peptide or peptidomimetic monoclonal antibody (MAb) that crosses the BBB via receptor-mediated transport. The most active MTH is a genetically engineered MAb against the human insulin receptor (HIR). The HIRMAb cross reacts with the insulin receptor of Old World primates such as the Rhesus monkey, but not with the insulin receptor of New World monkeys (Pardridge et al, 1995) or the mouse (Zhou et al, 2012). The purpose of the present investigation was first the genetic engineering, host cell expression, and biochemical validation of a fusion protein of the HIRMAb and human ASA. The amino terminus of the mature ASA is fused to the carboxyl terminus of each heavy chain of the HIRMAb, and this fusion protein is designated the HIRMAb-ASA fusion protein (Figure 1). The dimeric configuration of the ASA domain of the fusion protein replicates the normal dimeric configuration of ASA at neutral pH (Lukatela et al 1998). The second goal of the present study was to characterize the plasma pharmacokinetics, and brain uptake of the HIRMAb-ASA fusion protein in vivo in the Rhesus monkey. The study shows the HIRMAb-ASA fusion protein retains both high affinity binding to the HIR and high ASA enzyme activity, and is rapidly transported across the primate BBB in vivo.
Figure 1.

Structure of the HIRMAb-ASA fusion protein, which is formed by 2 light chains and 2 heavy chains, where the ASA monomer is fused to the carboxy terminus of each heavy chain. The domains of the light chain include the variable region of the light chain (VL) and the kappa constant region of the light chain (CL). The domains of the heavy chain include the variable region of the heavy chain (VH), the 3 sub-domains of the human IgG1 constant region (CH1, CH2, CH3), and the ASA enzyme.
Materials and Methods
Engineering of tandem vector and production of CHO line
A 1.5 kb cDNA encoding the human ASA cDNA was produced by reverse transcriptase polymerase chain reaction using the following forward and reverse oligodeoxynucleotide primers: 5′-phosphate-CCCGTCCGCCCAACATCGTGCT-3′ and 5′-phosphate-TCAGGCATGGGGATCTGGGCAATG-3′, and polyA+RNA from human liver. The ASA sequence, absent the enzyme signal peptide, was fused to the carboxyl terminus of the CH3 region of the heavy chain (HC) of the chimeric HIRMAb. A tandem vector was engineered in which the expression cassettes encoding this fusion HC, as well as the HIRMAb light chain (LC), and the murine dihydrofolate reductase, were placed on a single plasmid DNA (Boado et al, 2007). DNA sequencing showed the 3 expression cassettes spanned 10,003 nucleotides. The light chain was comprised of 234 amino acids (AA), which included a 20 AA signal peptide. The predicted molecular weight of the light chain is 23,398 Da with a predicted isoelectric point (pI) of 5.45. The fusion protein of the HIRMAb heavy chain and ASA was comprised of 953 AA, which included a 19 AA signal peptide, the 443 AA HIRMAb heavy chain, a 2 AA linker (Ser-Ser), and the 489 AA mature ASA enzyme. The predicted pI of the heavy chain is 6.43, and the predicted molecular weight of the heavy chain, without glycosylation, is 100,637 Da, which includes 51,900 Da within the ASA domain. The AA sequence of the ASA domain is 100% identical with the sequence of mature human ASA (Genbank NP_000478). The predicted molecular weight of the hetero-tetramer, without glycosylation, is 248,035 Da.
The tandem vector plasmid DNA was linearized and CHO cells, adapted to serum free medium (SFM), were electroporated with the tandem vector, selected with G418 and hypoxanthine-thymine deficient medium, and amplified with graded increases of methotrexate up to 80 nM. The CHO line underwent 2 successive rounds of 1 cell/well dilutional cloning, and positive clones were selected by measurement of medium human IgG concentrations by enzyme-linked immunosorbent assay (ELISA). The CHO line was stable through multiple generations, and produced medium IgG levels of 10 mg/L in shake flasks at a cell density of 1–2 million cells/mL in SFM.
Purification and analysis of fusion protein
The CHO cells were propagated in 1 L bottles, until 2.4L of conditioned SFM was collected. The medium was ultra-filtered with a 0.2 um Sartopore-2 sterile-filter unit (Sartorius Stedim Biotech, Goettingen, Germany), and applied to a 25 mL protein A Sepharose 4 Fast Flow (GE Life Sciences, Chicago, IL) column equilibrated in 25 mM Tris/25 mM NaCl/5 mM EDTA/pH=7.1. Following application of the sample, the column was washed with 25 mM Tris/1 M NaCl/5 mM EDTA/pH=7.1, and the fusion protein was eluted with 0.1 M sodium acetate/pH=3.7. The acid eluate was pooled, Tris base was added to raise the pH to 5.5, NaCl was added to 0.15 M, and the solution was stored sterile-filtered at 4C or −70C.
The identity of the HIRMAb-ASA fusion protein was verified by human IgG and human ASA Western blotting. For the human IgG Western blot, the primary antibody was a goat anti-human IgG (H+L) (Vector Labs, Burlingame, CA). For the human ASA Western blot, the primary antibody was a mouse anti-human ASA antibody (R&D Systems, Minneapolis, MN). The secondary antibody was a biotinylated horse anti-goat IgG or biotinylated horse anti-mouse IgG antibody (Vector Labs). The purity of the HIRMAb-ASA fusion protein was verified by reducing sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) using a 4–15% gradient gel. The prestained molecular weight (MW) standards were obtained from Fermentas (Glen Burnie, MD). The potency of the HIRMAb-ASA fusion protein was evaluated by measurement of the affinity of binding of the fusion protein to the HIR extracellular domain (ECD) using an ELISA. The HIR ECD was purified from the serum free conditioned medium produced by a stably transfected CHO line using lectin affinity chromatography, as described previously (Boado et al, 2007). ASA enzyme activity was measured using a spectrophotometric enzymatic assay using 4-nitrocatechol sulfate as the substrate at pH=5.0 at 37°C (Baum et al, 1955). The formation of 4-nitro catechol was followed by measurement of absorbance at 515 nm. The ASA enzyme activity was reported as units/mg protein where 1 unit=1 umol/min.
HIRMAb-ASA fusion protein uptake by MLD fibroblasts
MLD fibroblasts (GM00243) were obtained from the Coriell Institute for Medical Research (Camden, NJ), were grown overnight in DMEM with 10% FBS to >80% confluency in Lab-Tek chamber slide plates (Thermo Scientific, Rochester, NY). The medium was aspirated, the wells washed well with PBS, and the cells were treated with fresh DMEM with no serum and containing 10 ug/mL of the HIRMAb-ASA fusion protein. Following a 24 hr incubation at 37C, the medium was aspirated, the wells washed extensively with cold PBS, and the cells were fixed with 100% cold methanol for 40 min at −20C. Following a PBS wash, the plates were blocked with 10% donkey serum, and then co-labeled with 10 ug/mL of a goat anti-ASA antibody (R&D Systems), and 10 ug/ml of a mouse MAb to human lysosomal associated membrane protein (LAMP)-1 (Santa Cruz Biotechnology, Santa Cruz, CA). Negative control antibodies were the same concentrations of either goat or mouse IgG. The secondary antibodies (Molecular Probes/Invitrogen, Carlsbad, CA) were 5 ug/mL each of Alexa Fluor-488 conjugated donkey anti-mouse IgG (green channel) and Alexa Fluor-594 conjugated donkey anti-goat IgG (red channel). The washed slides were mounted with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Labs). Confocal microscopy was performed with a Leica TCS SP2 AOBS inverted fluorescence microscope with a Leitz P1 Apo 100X oil immersion objective and a Leica confocal laser scanning adapter utilizing argon (476 and 488 nm), new diode (561 nm) and helium-neon (633 nm) lasers, respectively. Optical sections (1 um, resolution 300 nm) were obtained sequentially through the z-plane of each sample.
Rhesus monkey brain uptake and pharmacokinetics
The HIRMAb-ASA fusion protein was radiolabeled with [125I]-Bolton-Hunter reagent (American Radiolabeled Chemicals, St. Louis, MO), which had a specific activity of 2200 uCi/nmol. Prior to labeling, the fusion protein was buffer exchanged with 0.01 M sodium acetate/140 mM NaCl/pH=5.5/0.001% Tween-80 and an Amicon Ultra-15 centrifugal filter unit (EMD Millipore, Billerica, MA). The labeled HIRMAb-ASA fusion protein was purified by gel filtration with a 1×28 cm column of Sephadex G-25 and an elution buffer of 0.01 M sodium acetate/140 mM NaCl/pH=5.5/0.001% Tween-80. An adult male Rhesus monkey, 8.2 kg, was investigated at MPI Research (Mattawan, MI). The animal was injected intravenously (IV) with 2042 uCi of [125I]-HIRMAb-ASA fusion protein by bolus injection over 30 seconds in the left femoral vein. The injection dose (ID) of the HIRMAb-ASA fusion protein was 55 ug/kg. The animal was anesthetized with intramuscular ketamine. All procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health. Following intravenous drug administration, femoral venous plasma was obtained at 2, 5, 15, 30, 60, 90, and 120 min for determination of total plasma [125I] radioactivity (DPM/mL) and plasma radioactivity that is precipitated by 10% cold trichloroacetic acid (TCA). The animal was euthanized, and samples of major organs (heart, liver, spleen, lung, skeletal muscle, and omental fat) were removed, weighed, and processed for determination of radioactivity. The cranium was opened and the brain was removed. Samples of frontal cortical gray matter, frontal cortical white matter, cerebellar gray matter, cerebellar white matter, and choroid plexus were removed for radioactivity determination.
Samples (~2 gram) of frontal cortex were removed for capillary depletion analysis to confirm transport of the HIRMAb-ASA fusion protein across the BBB. The capillary depletion method separates the vascular tissue in brain from the post-vascular compartment (Triguero et al, 1990). Based on measurements of the specific activity of brain capillary-specific enzymes, such as γ-glutamyl transpeptidase or alkaline phosphatase, the post-vascular supernatant is >95% depleted of brain vasculature (Triguero et al, 1990). To separate the vascular and post-vascular compartments, the brain was homogenized in 8 mL cold PBS in a tissue grinder. The homogenate was supplemented with 8 mL cold 40% dextran (70 kDa, Sigma Chemical Co.), and an aliquot of the homogenate was taken for radioactivity measurement. The homogenate was centrifuged at 3200 g at 4C for 10 min in a fixed angle rotor. The brain microvasculature quantitatively sediments as the pellet, and the post-vascular supernatant is a measure of capillary depleted brain parenchyma (Triguero et al, 1990). The vascular pellet and supernatant were counted for 125I radioactivity in parallel with the homogenate. The volume of distribution (VD) was determined for each of the 3 fractions from the ratio of total [125I] radioactivity in the brain fraction (DPM/gram brain) divided by the total [125I] radioactivity in the 120 min terminal plasma (DPM/uL plasma). The percent of radioactivity in the post-vascular supernatant that was precipitable with 10% cold TCA was determined. Plasma and tissue samples were analyzed for 125I radioactivity with a Wizard model 1470 gamma counter (Perkin Elmer, Shelton, CT).
The TCA precipitable [125I] radioactivity in plasma, DPM/mL, was converted to % injected dose (ID)/mL, and the %ID/mL was fit to a bi-exponential equation,
The intercepts (A1, A2) and the slopes (k1, k2) were used to compute the median residence time (MRT), the central volume of distribution (Vc), the steady state volume of distribution (Vss), the area under the plasma concentration curve (AUC), and the systemic clearance (CL), as described previously (Pardridge et al, 1995). Non-linear regression analysis used the AR subroutine of the BMDP Statistical Software (Statistical Solutions Ltd, Cork, Ireland). Data were weighted by 1/(%ID/mL)2.
The BBB permeability-surface area (PS) product was computed from the brain uptake at 2 hours, %ID/gram, divided by the 2 hour plasma AUC, %ID·min/mL, and reported as uL/min/gram.
Brain autoradiography
The fresh brain was cut into coronal slabs, and immediately frozen in liquid nitrogen. Frozen sections (20 um) were cut with a cryostat at −15°C; the sections were air dried and exposed to Kodak Biomax MR X-ray film (Carestream Health, Rochester, MN) for up to 7 days followed by x-ray film development. The films were scanned and the image was saved in Photoshop, and colorized with NIH Image software. Frozen sections of brain were also subbed to slides coated with HyperCoat LM-1 emulsion (GE Amersham, Chicago, IL); the slides were developed in the dark for 8 weeks, and processed with D-19 developer and fixer (Kodak, Rochester, NY). The unstained slides were examined with light microscopy under either dark field or bright field illumination with a Nikon Microphot FXA microscope and 100X, 200X, and 400X magnification. Digital images were taken with an Optixcam Summit OCS-13 digital microscope camera and transferred to Adobe Photoshop 6.0.
Results
The CHO-derived HIRMAb-ASA fusion protein was purified by protein A affinity chromatography to homogeneity on SDS-PAGE (Figure 2). The HIRMAb-ASA fusion protein and the chimeric HIRMAb use the same LC, which migrates with a molecular weight (MW) of 27 kDa, whereas the size of the HC of the HIRMAb-ASA fusion protein, 124 kDa, is larger than the size of the HC of the HIRMAb, 56 kDa (Figure 2), owing to fusion of the ASA to the antibody HC. The anti-human IgG antibody reacts with the LC and HC of both the HIRMAb and the HIRMAb-ASA fusion protein (Figure 3, left panel). The anti-human ASA antibody reacts only with the HC of the HIRMAb-ASA fusion protein, but not with the HC of the HIRMAb (Figure 3, right panel).
Figure 2.

Reducing SDS-PAGE and Coomassie blue staining of the chimeric HIRMAb and the HIRMAb-ASA fusion protein. Based on the migration of the molecular weight (MW) standards, the calculated MW of the light chain is 27 kDa, and the calculated MW of the heavy chain of the HIRMAb and the HIRMAb-ASA fusion protein is 56 kDa and 124 kDa, respectively.
Figure 3.

Western blot with a primary antibody against human IgG (left panel) or human ASA (right panel). The proteins tested are the chimeric HIRMAb, and the HIRMAb-ASA fusion protein.
The affinity for binding to the HIR of the HIRMAb-ASA fusion protein is comparable to the affinity of binding of the HIRMAb. The EC50 of the HIRMAb-ASA fusion protein binding to the HIR, 0.34 ± 0.11 nM, is equal to the EC50, 0.23 ± 0.06 nM, of the HIRMAb binding to the HIR (Figure 4). The ASA enzyme specific activity of the HIRMAb-ASA fusion protein was 20 ± 1 umol/min/mg protein, as measured with the spectrophotometric assay (Methods).
Figure 4.

Binding to the HIR is saturable for the chimeric HIRMAb and the HIRMAb-ASA fusion protein. The EC50 was determined by non-linear regression analysis and the value in ng/mL was converted to nM, based on a molecular weight of 150 kDa for the HIRMAb and a molecular weight of 300 kDa for the HIRMAb-ASA fusion protein.
MLD fibroblasts were incubated with 10 ug/mL HIRMAb-ASA fusion protein for 24 hours followed by fixation and immune labeling with a mouse monoclonal antibody to LAMP1, a lysosomal marker, and a goat polyclonal antibody to human ASA. The LAMP1 immunoreactivity within the cell is detected in the green channel (Figure 5A), and the ASA immunoreactivity is detected in the red channel (Figure 5B). The overlap of the ASA and LAMP1 immunoreactivity is shown in Figure 5C. There was no immunoreactivity in the cells labeled with mouse or goat isotype control antibodies.
Figure 5.
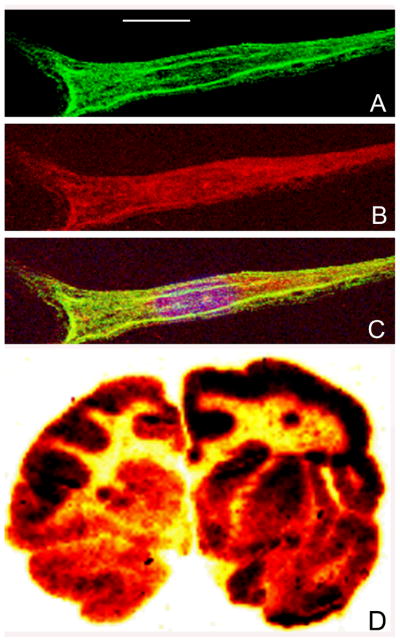
Panels A, B, and C are confocal micrographs of MLD fibroblasts incubated with the HIRMAb-ASA fusion protein (10 ug/mL) for 24 hours. The cells were fixed with 100% methanol and immune stained for confocal microscopy. The fixed cells were stained with a mouse monoclonal antibody to human lysosomal associated membrane protein (LAMP)-1 (panel A: green channel) and a goat polyclonal antibody to human ASA (panel B: red channel). The overlap image in panel C shows sequestration of the HIRMAb-ASA fusion protein within lysosomes. The magnification bar in panel A is 15 microns. Panel D is film autoradiography of a coronal section of Rhesus monkey brain removed 2 hours after the IV administration of the [125I]-HIRMAb-ASA fusion protein. The study shows global distribution of the fusion protein throughout the brain with higher uptake in gray matter as compared to white matter.
The HIRMAb-ASA fusion protein was radio-labeled with the [125I]-Bolton-Hunter reagent to a specific activity of 4.5 uCi/ug and a TCA precipitation of 99%. The [125I]-HIRMAb-ASA fusion protein (2042 uCi, 450 ug) was injected IV in an 8.2 kg male Rhesus monkey. The time course of plasma radioactivity is shown in Figure 6, which is expressed either as %ID/mL (Figure 6A) or ng/mL (Figure 6B). The percent of total plasma radioactivity that was precipitable by TCA was 98 ± 1%, 97 ± 1%, 88 ± 1%, 65 ± 1%, 45 ± 2%, 43 ± 2%, and 42 ± 2%, respectively at 2, 5, 15, 30, 60, 90, and 120 min after IV injection. The plasma profile of TCA-precipitable radioactivity was fit to a 2-exponential equation (Methods) to yield the pharmacokinetics (PK) parameters shown in Table I. The [125I]-HIRMAb-ASA fusion protein is rapidly cleared from blood with a mean residence time of 59 ± 12 minutes, a systemic volume of distribution (Vss) that is 5-fold greater the central compartment volume (Vc), and a high rate of systemic clearance, 3.9 ± 0.2 mL/min/kg (Table I).
Figure 6.

The plasma TCA-precipitable radioactivity is expressed as %ID/mL (A) and ng/mL (B) in the adult Rhesus monkey over a 120 min period after a single IV injection of 55 ug/kg [125I]-HIRMAb-ASA fusion protein.
Table I.
Pharmacokinetic parameters of the HIRMAb-ASA fusion protein
| parameter | units | value |
|---|---|---|
| A1 | %ID/mL | 0.243 ± 0.034 |
| A2 | %ID/mL | 0.018 ± 0.003 |
| k1 | min-1 | 0.185 ± 0.024 |
| k2 | min-1 | 0.010 ± 0.002 |
| MRT | min | 59 ± 12 |
| Vc | mL/kg | 46 ± 6 |
| Vss | mL/kg | 233 ± 39 |
| AUC|120 | %ID·min/mL | 2.57 ± 0.12 |
| AUCss | %ID·min/mL | 3.10 ± 0.19 |
| AUCss | ug·min/mL | 14.2 ± 0.8 |
| CL | mL/min/kg | 3.9 ± 0.2 |
Parameters computed from the plasma profile in Figure 6.
The volume of distribution (VD) of the HIRMAb-ASA fusion protein in brain homogenate at 2 hours after injection is high, 526 ± 23 uL/gram, compared to the brain VD of a non-specific human IgG1 isotype control antibody, 20 ± 6 ul/gram (Table II). The brain VD of the IgG1 isotype control antibody represents the brain uptake of a molecule that is sequestered within the blood volume of brain, and which does not cross the BBB. The high brain VD for the HIRMAb-ASA fusion protein indicates the fusion protein is either sequestered by the brain vasculature, or has penetrated the BBB and entered brain parenchyma. The VD of the HIRMAb-ASA fusion protein in the post-vascular supernatant, 341 ± 33 uL/gram, is greater than the VD of the HIRMAb-ASA fusion protein in the vascular pellet of brain, 277 ± 30 uL/gram (Table II), which indicates that the majority of the HIRMAb-ASA fusion protein has traversed the BBB and penetrated the brain parenchyma. The radioactivity in the post-vascular supernatant represents intact HIRMAb-ASA fusion protein, and not labeled metabolites, as the TCA precipitation of the post-vascular supernatant radioactivity is 95.2 ± 1.4% (Table II).
Table II.
Capillary depletion analysis for brain uptake of the HIRMAb-ASA fusion protein
| Molecule | Brain fraction | VD (μL/g) |
|---|---|---|
| HIRMAb-ASA fusion protein | Brain homogenate | 526 ± 23 |
| Post-vascular supernatant | 341 ± 33 | |
| Vascular pellet | 277 ± 30 | |
| Human IgG1 isotype control | Brain homogenate | 20 ± 6 |
Mean ± S.D. The fusion protein was administered by IV injection, and brain measurements made 120 min following injection. The radioactivity in the post-vascular supernatant was 95.2 ± 1.4 % precipitable by cold 10% trichloroacetic acid. The homogenate VD for the human IgG1 isotype control antibody is from Boado and Pardridge (2009).
The organ uptake of the HIRMAb-ASA fusion protein, expressed as % of injected dose (ID) per 100 gram wet organ weight, in the Rhesus monkey is listed in Table III for brain and peripheral organs. The major organs accounting for the removal of the HIRMAb-ASA fusion protein from plasma are liver and spleen (Table III). The brain uptake of the HIRMAb-ASA fusion protein is 1.1 ± 0.1 % ID/100 gram brain for the frontal cortex (Table III). The film autoradiography of the entire primate brain shows that the uptake in the frontal cortex is representative of the uptake of the whole brain (Figure 5D). The BBB PS product for the HIRMAb-ASA fusion protein is 4.2 ± 0.3 μL/min/gram.
Table III.
Organ uptake of the HIRMAb-ASA fusion protein in the Rhesus monkey
| organ | Organ uptake (%ID/100 grams) |
|---|---|
| Frontal gray | 1.08 ± 0.09 |
| Frontal white | 0.32 ± 0.10 |
| Cerebellar gray | 0.97 ± 0.03 |
| Cerebellar white | 0.59 ± 0.07 |
| Choroid plexus | 2.19 ± 0.68 |
| liver | 22.4 ± 1.1 |
| spleen | 14.7 ± 0.3 |
| lung | 3.4 ± 0.2 |
| heart | 1.1 ± 0.1 |
| fat | 0.33 ± 0.01 |
| Skeletal muscle | 0.25 ± 0.05 |
Data are mean ± SD of triplicates samples from one monkey.
Film autoradiography of the primate brain removed 2 hours after IV injection shows global distribution of the HIRMAb-ASA fusion protein throughout brain with higher uptake in gray matter as compared to white matter (Figure 5D). The frozen brain cortical tissue was also examined with light microscopic emulsion autoradiography (Figure 7). Panel 7A is an image taken with 100X magnification and dark field microscopy. The image shows a concentration of silver grains over brain microvessels, as well as silver grains throughout the parenchyma of brain. The area of the slide that contains no brain tissue shows very few silver grains, which indicates the background of the autoradiography is very low. Panel 7B is an image taken with 200X magnification and dark field microscopy, and the silver grains within the brain parenchyma are more visible at the higher magnification. Panel 7C was photographed with a 400X magnification under bright field microscopy, and corresponds to the rectangular area of Panel 7B; this image shows silver grains evenly distributed throughout the brain parenchyma.
Figure 7.
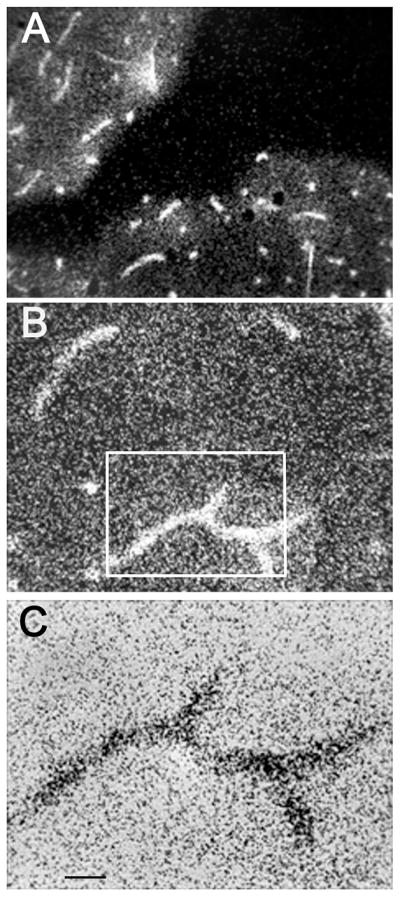
(A, B, C) Light microscopic emulsion autoradiography of primate brain removed 2 hours after the IV injection of the [125I]-HIRMAb-ASA fusion protein. Panels A and B dark field micrographs taken with a 100X and 200X magnifications, respectively. Panel C is bright field micrograph taken with 400X magnification, and corresponds to the rectangular area in panel B. The magnification bar in panel C is 11 microns.
Discussion
The results of this study are consistent with the following conclusions. First, an IgG-ASA fusion protein has been genetically engineered (Figure 1), and is comprised of two 27 kDa light chains, and two 124 kDa heavy chains (Figures 2 and 3). Second, the HIRMAb-ASA fusion protein is bi-functional, and binds the HIR with high affinity (Figure 4) and retains ASA enzyme activity (Results). Third, the HIRMAb-ASA fusion protein is endocytosed into MLD fibroblasts and triaged to the lysosomal compartment, as shown by confocal microscopy (Figure 5C). Fourth, the HIRMAb-ASA fusion protein is rapidly cleared from plasma with a high systemic volume of distribution (Table I), owing to uptake by peripheral tissues (Table III). Fifth, the HIRMAb-ASA fusion protein penetrates the brain (Table III), and film autoradiography shows global distribution of the fusion protein throughout brain (Figure 5D). Sixth, the HIRMAb-ASA fusion protein penetrates the BBB and distributes to the post-vascular parenchyma of brain, as shown by the capillary depletion method (Table II) and light microscopic emulsion autoradiography of monkey brain (Figure 7).
The nucleotide sequence of the cDNA encoding human ASA was reported by Stein et al (1989), which enabled the subsequent expression of recombinant ASA. The gene encoding ASA is mutated in MLD, and ASA enzyme replacement therapy (ERT) is a potential treatment of MLD. However, the ASA enzyme is a large molecule that does not penetrate the BBB, and ERT of MLD does not treat the serious CNS manifestations of MLD in either mouse models (Matzner et al, 2005) or in patients with MLD (Dali et al, 2009). In order to produce a BBB-penetrating form of ASA, the HIRMAb-ASA fusion protein was engineered. The ASA monomer is fused to the carboxyl terminus of each heavy chain of a genetically engineered HIRMAb (Figure 1). The HIRMAb traverses the BBB via receptor-mediated transport on the endogenous insulin receptor, and carries the fused ASA into the brain from blood. A biologically active HIRMAb-ASA fusion protein must be a bi-functional fusion protein and retain both high affinity binding to the insulin receptor and high ASA enzyme activity. With respect to the insulin receptor function, the affinity of the HIRMAb-ASA fusion protein for the HIR, as represented by the EC50 of 0.34 ± 0.11 nM, is equal to the affinity of the HIRMAb alone (Figure 4). The retention of high affinity binding of the fusion protein for the insulin receptor enables high BBB penetration. The BBB PS product of the HIRMAb-ASA fusion protein, 4.2 ± 0.3 uL/min/g, is comparable to the BBB PS product of the murine form of the HIRMAb, 5.4 ± 0.6 uL/min/g (Results), in the Rhesus monkey (Pardridge et al, 1995). The ASA enzyme activity of the HIRMAb-ASA fusion protein is 20 ± 1 units/mg (Results), as compared to the specific activity of recombinant ASA, which is 60 units/mg (Matzner et al, 2005). Based on molecular weights derived from the amino acid sequence, the molecular weight of the AGT-183 fusion protein half-tetramer is 124,035 Da, as opposed to the molecular weight of the ASA monomer, 51,900 Da (Methods). Therefore, on a molar basis, the ASA specific activity of the HIRMAb-ASA fusion protein is normalized to 48 units/mg protein, which is 80% of the specific activity of recombinant ASA.
The enzyme activity of ASA is dependent on the post-translational conversion of a cysteine residue in the amino terminal portion of the enzyme to an N-formylglycine residue, and this modification is mediated via sulfatase modifying factor 1 (SUMF1) (Schmidt et al, 1995). Transient expression of ASA in COS cells requires co-transfection of cells with both ASA and SUMF1 for maximal ASA enzyme activity and secretion (Cosma et al, 2003). However, the IgG domain drives secretion of the HIRMAb-ASA fusion protein in stably transfected CHO cells, and the HIRMAb-ASA fusion protein is secreted with ASA enzyme activity without co-transfection with the SUMF1 gene (Methods).
The HIRMAb domain of the HIRMAb-ASA fusion protein cross reacts with the insulin receptor of Old World primates such as the Rhesus monkey, but does not cross-react with the insulin receptor of New World monkeys (Pardridge et al, 1995) or the mouse (Zhou et al, 2012). Therefore, in vivo investigations are performed in the Rhesus monkey, and the present study describes the plasma PK of the HIRMAb-ASA fusion protein in this primate (Figure 6, Table I). The HIRMAb-ASA fusion protein was radio-iodinated with the [125I]-Bolton-Hunter reagent (Methods). This form of radio-labeling is preferred over oxidative methods, such as chloramine T or Iodogen, because the Bolton-Hunter reagent reaction is non-oxidative and does not damage the protein. A second advantage of the Bolton-Hunter reagent over the oxidative methods is that the Bolton-Hunter reagent places a large bulky (3,5-diiodo, 4-hydroxy-benzene propionyl) substituent on the ε-amino group of surface lysine residues, whereas the oxidative methods introduce the radioactive iodine at tyrosine residues. Peripheral degradation of the protein labeled with the oxidative method results in the release of TCA-soluble metabolite, iodotyrosine, which can cross the BBB on the amino acid transporter. The lack of BBB transport of blood-borne Bolton-Hunter-conjugated lysine residue is shown in the present studies. At 120 minutes after IV injection of the [125I]-HIRMAb-ASA fusion protein, the plasma radioactivity is 42 ± 2% TCA-precipitable (Results), whereas the radioactivity in the post-vascular supernatant of brain is 95 ± 1% TCA-precipitable (Table II). This finding means that radioactivity that distributes to brain from blood is HIRMAb-ASA fusion protein, and not radiolabeled metabolites.
The HIRMAb-ASA fusion protein is rapidly cleared from plasma (Figure 6), owing to uptake by peripheral tissues (Table III). The systemic volume of distribution, 233 ± 39 mL/kg, is 5-fold greater than the central volume of distribution, 46 ± 6 mL/kg (Table I). The plasma mean residence time in the Rhesus monkey, 59 ± 12 minutes (Table I), is comparable to the half-time in blood of recombinant ASA in the mouse, 40 minutes (Matzner et al, 2005). The rapid removal of the HIRMAb-ASA fusion protein is due to uptake of the protein by peripheral organs (Table III). The most active uptake is via the liver, 22.4 ± 1.1 % ID/100 grams (Table III). Since the liver weight in an adult Rhesus monkey is 170 grams (Penniston and Tanumihardjo, 2001), the amount of fusion protein cleared by the liver is 38% of the injected dose. Similarly, the amount of recombinant ASA cleared by the liver in the mouse is approximately 40% of injected dose (Matzner et al, 2005).
ASA does not penetrate the brain in the mouse following an injection dose of 20 mg/kg (Matzner et al, 2005), because the enzyme, like other large molecules does not cross the BBB. However, the HIRMAb-ASA fusion protein is rapidly transported into brain in the Rhesus monkey. The brain uptake is 1.1% ID/100 grams and 0.32 % ID/100 grams in gray matter and white matter in the cerebrum, respectively (Table III). Brain uptake is expressed per 100 grams brain, because the brain weight in the Rhesus monkey is 100 grams (Bourne, 1975). The peak brain uptake of fallypride, a lipid soluble small molecule, in the Rhesus monkey is 4% ID/brain (Christian et al, 2009). Therefore, the brain uptake of the HIRMAb-ASA fusion protein in the Rhesus monkey is comparable to the brain uptake of a lipid soluble small molecule.
The HIRMAb-ASA fusion protein distributes to all parts of brain, as shown by the film autoradiography (Figure 5C). There is higher uptake in gray matter as compared to white matter, owing to the higher vascular density in gray matter of brain (Lierse and Horstmann, 1959). The HIRMAb-ASA fusion protein penetrates the BBB and distributes to the post-vascular parenchyma of brain, as shown by the capillary depletion method (Table II). The VD of the fusion protein in the post-vascular supernatant exceeds the VD in the capillary pellet (Table II), which indicates the majority of fusion protein has penetrated the BBB, and entered brain parenchyma. The results of the capillary depletion method are confirmed with light microscopic emulsion autoradiography (Figure 7). The emulsion autoradiography shows clustering of silver grains within the microvascular compartment (Figure 7A), as well as uniform distribution of the fusion protein throughout the brain parenchyma (Figure 7B, C, D). The silver grains in brain parenchyma represent fusion protein, and not metabolites, since the TCA precipitation of radioactivity in the post-vascular supernatant is 95% (Table II). Following transport across the BBB, the fusion protein can be endocytosed by brain cells, since there is high expression of the insulin receptor on cells in the brain (Zhao et al, 1999). The confocal microscopy study shows that the HIRMAb-ASA fusion protein is endocytosed by target cells and triaged to the lysosomal compartment (Figure 5). Once inside the lysosomal compartment of the target cell, the lysosomal enzyme domain of the HIRMAb fusion protein is enzymatically active. Glycosoaminoglycans are reduced in either Hurler or Hunter fibroblasts following treatments with the respective HIRMAb-lysosomal enzyme fusion protein (Boado et al, 2008; Lu et al, 2010). The lysosomal inclusion bodies are reduced in both brain and peripheral organs following the twice-weekly treatment of Hurler mice with the IgG-lysosomal enzyme fusion protein (Boado et al, 2011).
The brain uptake of the HIRMAb-ASA fusion protein, 1.1% ID/brain, in the Rhesus monkey (Table III) allows for estimation of the amount of ASA that might distribute to the human brain with the IV administration of the HIRMAb-ASA fusion protein. Following the IV infusion of 2.5 mg/kg in a 50 kg human, the concentration of the fusion protein in brain is predicted to be 1375 ug fusion protein/brain. This is equivalent to 1.4 ug/gram brain in a 1,000 gram human brain, and 14 ng/mg protein, given 100 mg protein per gram brain. This level of fusion protein is 14% of the concentration of immunoreactive ASA in the human brain, which is 100 ng/mg protein (Sevin et al, 2006). However, it is likely that replacement of only 1–2% of normal ASA enzyme activity in brain will be therapeutic, as is observed in other lysosomal storage disorders (Muenzer and Fisher, 2004). About 5–20% of the population has ASA pseudo-deficiency (Fluharty, 2006), where with cellular ASA enzyme activity is 3–8% of normal without symptoms of MLD (Penzien et al, 1993).
In summary, the present investigation describes the genetic engineering, expression, and pharmacokinetics and brain uptake in the Rhesus monkey of the HIRMAb-ASA fusion protein. The protein is a bi-functional IgG-sulfatase fusion protein, which retains high affinity binding to the human insulin receptor and high ASA enzyme activity. The fusion protein is generated from stably transfected CHO cells which are cultivated in serum free medium. The manufacturing of the HIRMAb-ASA fusion protein will be facilitated by the use of a protein A capture column in the downstream processing. The HIRMAb-ASA is a new biological entity for the treatment of the brain in humans with MLD.
Acknowledgments
This research was supported by NIH grant R43-HD-074272-01. The authors are indebted to Winnie Tai and Phuong Tram for technical support, and to MPI Research, Inc. (Mattawan, MI) for collaboration with the primate study. Drs. Boado, Lu, and Hui are employees, and Dr. Pardridge is a consultant, of ArmaGen Technologies. Confocal laser scanning microscopy was performed at the CNSI Advanced Light Microscopy/Spectroscopy Shared Resource Facility at UCLA, supported with funding from a NIH-NCRR shared resources grant (CJX1-443835-WS-29646) and a NSF Major Research Instrumentation grant (CHE-0722519).
Abbreviations
- BBB
blood-brain barrier
- HIR
human insulin receptor
- MAb
monoclonal antibody
- TfR
transferrin receptor
- MLD
Metachromatic Leukodystrophy
- ASA
arylsulfatase A
- SUMF1
sulfatase modifying factor 1
- CHO
Chinese hamster ovary
- ERT
enzyme replacement therapy
- IV
intravenous
- PK
pharmacokinetics
- MTH
molecular Trojan horse
- HC
heavy chain
- LC
light chain
- AA
amino acid
- SFM
serum free medium
- MW
molecular weight
- ECD
extracellular domain
- FBS
fetal bovine serum
- LAMP
lysosomal associated membrane protein
- VD
volume of distribution
- TCA
trichloroacetic acid
- BCA
bicinchoninic acid
References
- Baum H, Dodgson KS, Spencer B. The assay of arylsulphatases A and B in human urine. Clin Chim Acta. 1959;4:453–5. doi: 10.1016/0009-8981(59)90119-6. [DOI] [PubMed] [Google Scholar]
- Blomqvist M, Gieselmann V, Mansson JE. Accumulation of lysosulfatide in the brain of arylsulfatase A-deficient mice. Lipids Health Dis. 2011;10:28. doi: 10.1186/1476-511X-10-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boado RJ, Pardridge WM. Comparison of blood-brain barrier transport of GDNF and an IgG-GDNF fusion protein in the Rhesus monkey. Drug Metab Disp. 2009;37:2299–2304. doi: 10.1124/dmd.109.028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boado RJ, Zhang Y, Pardridge WM. Genetic engineering, expression, and activity of a fusion protein of a human neurotrophin and a molecular Trojan horse for delivery across the human blood-brain barrier. Biotechnol Bioeng. 2007;97:1376–86. doi: 10.1002/bit.21369. [DOI] [PubMed] [Google Scholar]
- Boado RJ, Zhang Y, Xia CF, Wang Y, Pardridge WM. Genetic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood-brain barrier. Biotechnol Bioeng. 2008;99:475–484. doi: 10.1002/bit.21602. [DOI] [PubMed] [Google Scholar]
- Boado RJ, Hui EKW, Lu JZ, Zhou QH, Pardridge WM. Reversal of lysosomal storage in brain of adult MPS-I mice with intravenous Trojan horse-iduronidase fusion protein. Mol Pharm. 2011;8:1342–1350. doi: 10.1021/mp200136x. [DOI] [PubMed] [Google Scholar]
- Bourne GH. Anatomy and Physiology. In: Bourne GH, editor. The Rhesus Monkey. New York: Academic Press; 1975. pp. 6–10. [Google Scholar]
- Christian BT, Vandehey NT, Fox AS, Murali D, Oakes TR, Converse AK, Nickles RJ, Shelton SE, Davidson RJ, Kalin NH. The distribution of D2/D3 receptor binding in the adolescent rhesus monkey using small animal PET imaging. Neuroimage. 2009;44:1334–44. doi: 10.1016/j.neuroimage.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosma MP, Pepe S, Annunziata I, Newbold RF, Grompe M, Parenti G, Ballabio A. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell. 2003;113:445–56. doi: 10.1016/s0092-8674(03)00348-9. [DOI] [PubMed] [Google Scholar]
- Fluharty AL. Arylsulfatase A Deficiency. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 2006. [Google Scholar]
- Dali C, Lund AM. Intravaneous enzyme replacement therapy for metachromatic leukodystrophy (MLD). In Abstracts from the American College of Medical Genetics Annual Meeting; 2009. Abstract #195. [Google Scholar]
- Lierse W, Horstmann E. Quantitative anatomy of the cerebral vascular bed with especial emphasis on homogeneity and inhomogeneity in small parts of the gray and white matter. Acta Neurol. 1959;14:15–19. doi: 10.1111/j.1600-0404.1965.tb01946.x. [DOI] [PubMed] [Google Scholar]
- Lu JZ, Hui EKW, Boado RJ, Pardridge WM. Genetic engineering of a bi-functional IgG fusion protein with iduronate 2-sulfatase. Bioconj Chem. 2010;21:151–156. doi: 10.1021/bc900382q. [DOI] [PubMed] [Google Scholar]
- Lukatela G, Krauss N, Theis K, Selmer T, Gieselmann V, von Figura K, Saenger W. Crystal structure of human arylsulfatase A: the aldehyde function and the metal ion at the active site suggest a novel mechanism for sulfate ester hydrolysis. Biochemistry. 1998;37:3654–64. doi: 10.1021/bi9714924. [DOI] [PubMed] [Google Scholar]
- Matzner U, Herbst E, Hedayati KK, Lullmann-Rauch R, Wessig C, Schroder S, Eistrup C, Moller C, Fogh J, Gieselmann V. Enzyme replacement improves nervous system pathology and function in a mouse model for metachromatic leukodystrophy. Hum Mol Genet. 2005;14:1139–52. doi: 10.1093/hmg/ddi126. [DOI] [PubMed] [Google Scholar]
- Molander-Melin M, Pernber Z, Franken S, Gieselmann V, Mansson JE, Fredman P. Accumulation of sulfatide in neuronal and glial cells of arylsulfatase A deficient mice. J Neurocytol. 2004;33:417–27. doi: 10.1023/B:NEUR.0000046572.53905.2c. [DOI] [PubMed] [Google Scholar]
- Muenzer J, Fisher A. Advances in the treatment of mucopolysaccharidosis type I. N Engl J Med. 2004;350:1932–1934. doi: 10.1056/NEJMp048084. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM, Boado RJ. Reengineering biopharmaceuticals for targeted delivery across the blood-brain barrier. Methods Enzymol. 2012;503:269–92. doi: 10.1016/B978-0-12-396962-0.00011-2. [DOI] [PubMed] [Google Scholar]
- Pardridge WM, Kang YS, Buciak JL, Yang J. Human insulin receptor monoclonal antibody undergoes high affinity binding to human brain capillaries in vitro and rapid transcytosis through the blood-brain barrier in vivo in the primate. Pharm Res. 1995;12:807–816. doi: 10.1023/a:1016244500596. [DOI] [PubMed] [Google Scholar]
- Penniston KL, Tanumihardjo SA. Subtoxic hepatic vitamin A concentrations in captive rhesus monkeys (Macaca mulatta) J Nutr. 2001;131:2904–9. doi: 10.1093/jn/131.11.2904. [DOI] [PubMed] [Google Scholar]
- Penzien JM, Kappler J, Herschkowitz N, Schuknecht B, Leinekugel P, Propping P, Tonnesen T, Lou H, Moser H, Zierz S, et al. Compound heterozygosity for metachromatic leukodystrophy and arylsulfatase A pseudodeficiency alleles is not associated with progressive neurological disease. Am J Hum Genet. 1993;52:557–64. [PMC free article] [PubMed] [Google Scholar]
- Schmidt B, Selmer T, Ingendoh A, von Figura K. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell. 1995;82:271–8. doi: 10.1016/0092-8674(95)90314-3. [DOI] [PubMed] [Google Scholar]
- Sevin C, Benraiss A, Van Dam D, Bonnin D, Nagels G, Verot L, Laurendeau I, Vidaud M, Gieselmann V, Vanier M, et al. Intracerebral adeno-associated virus-mediated gene transfer in rapidly progressive forms of metachromatic leukodystrophy. Hum Mol Genet. 2006;15:53–64. doi: 10.1093/hmg/ddi425. [DOI] [PubMed] [Google Scholar]
- Stein C, Gieselmann V, Kreysing J, Schmidt B, Pohlmann R, Waheed A, Meyer HE, O’Brien JS, von Figura K. Cloning and expression of human arylsulfatase A. J Biol Chem. 1989;264:1252–9. [PubMed] [Google Scholar]
- Stroobants S, Gerlach D, Matthes F, Hartmann D, Fogh J, Gieselmann V, D’Hooge R, Matzner U. Intracerebroventricular enzyme infusion corrects central nervous system pathology and dysfunction in a mouse model of metachromatic leukodystrophy. Hum Mol Genet. 2011;20:2760–9. doi: 10.1093/hmg/ddr175. [DOI] [PubMed] [Google Scholar]
- Triguero D, Buciak J, Pardridge WM. Capillary depletion method for quantification of blood-brain barrier transport of circulating peptides and plasma proteins. J Neurochem. 1990;54:1882–1888. doi: 10.1111/j.1471-4159.1990.tb04886.x. [DOI] [PubMed] [Google Scholar]
- Zhao W, Chen H, Xu H, Moore E, Meiri N, Quon MJ, Alkon DL. Brain insulin receptors and spatial memory. J Biol Chem. 1999;274:34893–34902. doi: 10.1074/jbc.274.49.34893. [DOI] [PubMed] [Google Scholar]
- Zhou QH, Boado RJ, Pardridge WM. Selective plasma pharmacokinetics and brain uptake in the mouse of enzyme fusion proteins derived from species-specific receptor-targeted antibodies. J Drug Targeting. 2012;20:715–719. doi: 10.3109/1061186X.2012.712132. [DOI] [PubMed] [Google Scholar]
- Ziegler RJ, Salegio EA, Dodge JC, Bringas J, Treleaven CM, Bercury SD, Tamsett TJ, Shihabuddin L, Hadaczek P, Fiandaca M, et al. Distribution of acid sphingomyelinase in rodent and non-human primate brain after intracerebroventricular infusion. Exp Neurol. 2011;231:261–71. doi: 10.1016/j.expneurol.2011.06.019. [DOI] [PubMed] [Google Scholar]