

Abstract
Brain inflammation may play an important role in the pathophysiology of early brain injury after subarachnoid hemorrhage (SAH). Our aim was to demonstrate brain inflammation development and to determine whether isoflurane, a clinically available volatile anesthetic agent, prevents brain inflammation after SAH. This study used 162 8-week-old male CD-1 mice. We induced SAH with endovascular perforation in mice and randomly assigned animals to sham-operated (n=21), SAH+vehicle-air (n=35) and SAH+2% isoflurane (n=31). In addition to evaluation of brain injury (neurological scores, brain edema and Evans blue dye extravasation), brain inflammation was evaluated by means of expression changes in markers of inflammatory cells (ionized calcium binding adaptor molecule-1, myeloperoxidase), cytokines (tumor necrosis factor [TNF]-α, interleukin-1β), adhesion molecules (intercellular adhesion molecule [ICAM]-1, P-selectin), inducers of inflammation (cyclooxygenase-2, phosphorylated c-Jun N-terminal kinase [p-JNK]) and endothelial cell activation (von Willebrand factor) at 24 hours post-SAH. Sphingosine kinase inhibitor (N, N-dimethylsphingosine [DMS]) and sphingosine-1-phosphate receptor-1/3 antagonist (VPC23019) were used to block isoflurane’s effects (n=22, each). SAH caused early brain injury, which was associated with inflammation so that all evaluated markers of inflammation were increased. Isoflurane significantly inhibited both brain injury (P<0.001, respectively) and inflammation (myeloperoxidase, P=0.022; interleukin-1β, P=0.002; TNF-α, P=0.015; P-selectin, P=0.010; ICAM-1, P=0.016; p-JNK, P<0.001; cyclooxygenase-2, P=0.003, respectively). This beneficial effect of isoflurane was abolished with DMS and VPC23019. Isoflurane may suppress post-SAH brain inflammation possibly via the sphingosine-related pathway.
Keywords: Subarachnoid hemorrhage, early brain injury, isoflurane, inflammation
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) is a life-threatening disease carrying the risk of sudden death of 12.4% before receiving medical intervention (Huang and van Gelder, 2002) while a majority of deaths occur within the first 48 hours post ictus owing to the impact of the initial bleeding (Broderick et al., 1994). Subarachnoid blood, elevation of intracranial pressure, and reduced cerebral perfusion initiate an acute injury cascade such as microvascular disturbance and inflammatory reaction, leading to early brain injury (EBI), one of the important causes of unfavorable outcomes after SAH (Sehba and Bederson, 2006; Fujii et al., 2013).
Isoflurane is a volatile anesthetic and a lipophilic molecule (Antkowiak, 2001). We reported that 2% isoflurane prevented post-SAH neuronal apoptosis and blood-brain barrier (BBB) disruption through sphingosine-related pathway activation (Altay et al, 2012a, 2012b). Preconditioning with isoflurane was also reported to reduce lipopolysaccharide-induced inflammation in vivo (Plachinta et al., 2003). However, it remains undetermined whether isoflurane post-treatment prevents brain inflammation in an acute stage of SAH. In this study, thus, we examined effects of post-treatment isoflurane on brain inflammatory markers after SAH in mice and whether the treatmant mechanism involved the sphingosine-related pathway, which was verified by using a sphingosine kinase (SphK) inhibitor and a sphingosine-1-phosphate (S1P) receptor-1/3 (S1P1/3) antagonist (VPC23019).
Material and Methods
Experimental Design and Animal Groups
The animal and ethics review committee at Loma Linda University approved all protocols. One hundred sixty two 8-week-old male CD-1 mice (30–38g; Charles River, Wilmington, MA) were used.
To examine whether isoflurane attenuated EBI and brain inflammation after SAH, animals were randomly divided into 3 groups, and evaluated at 24 hours: sham-operated+30% O2+70% medical air (O2-medical air; sham group, n=21), SAH+O2-medical air (vehicle group, n=35) and SAH+2% isoflurane (isoflurane group, n=31).
To confirm that isoflurane had anti-inflammatory effects on brain after SAH, we used a potent and specific SphK inhibitor, N, N-dimethylsphingosine (DMS) and a S1P1- and S1P3- receptor antagonist, VPC23019, both of which were reported to block isoflurane’s action on post-SAH brain. Animals were randomly divided into 4 groups, and evaluated at 24 hours post-SAH: dimethyl sulfoxide (DMSO; a vehicle)+sham-operated+O2-medical air (n=13), DMSO+SAH+2% isoflurane (n=18), DMS+SAH+2% isoflurane (n=22), and VPC23019+SAH+2% isoflurane (n=22).
Mouse SAH Model
SAH endovascular monofilament model was produced as described previously (Altay et al., 2012a). Briefly, animals were anesthetized with an intraperitoneal injection of ketamine/xylazine (100/10mg/kg). A sharpened 4-0 monofilament nylon suture was advanced through the internal carotid artery (ICA) to perforate the anterior cerebral artery. In the sham surgery, the filament was advanced 5mm through the ICA without perforating the artery. Body temperature was kept constant (37.5±0.5°C) during the operation.
Drug Administration
One hour after SAH induction, 2% isoflurane (Baxter, Deerfield, IL) was continuously administered for 1 hour with O2-medical air.
DMS (Enzo, Plymouth Meeting, PA; final concentration, 0.17μg/0.5μL) and VPC23019 (Avanti Polar Lipids Inc., Alabaster, Alabama; final concentration, 0.26μg/0.5μL) were automatically infused at a rate of 0.1μL/minute intracerebroventricularly, 60 minutes before the SAH induction, because there was little information if the both drugs can pass the BBB. The dose for intracerebroventricular injections was determined based on our previous studies (Altay et al., 2012a, 2012b). The vehicle groups were given the same volume (0.5μL) of DMSO (1.1g/mL/kg) diluted in phosphate-buffered saline (PBS). Mice were placed in a head holder (Stoelting Stereotactic Instrument, Wood Dale, IL) and a 26s-gauge needle of a 10μL Hamilton syringe (Microliter #701; Hamilton, Reno, NV) was inserted through a burr hole perforated on the skull into the right lateral ventricle using the following coordinates relative to bregma: 0.1mm posterior; 0.9mm lateral; and 3.1mm below the horizontal plane of bregma (Hirt et al., 2004). The needle was removed 10 minutes after completion of the infusion, and the burr hole was quickly plugged with bone wax.
Severity of SAH
The severity of SAH was blindly evaluated using the SAH grading scale at sacrifice (Altay et al., 2012a, 2012b). The SAH grading system was as follows: the basal cistern was divided into six segments, and each segment was allotted a grade from 0 to 3 depending on the amount of subarachnoid blood clot in the segment; grade 0, no subarachnoid blood; grade 1, minimal subarachnoid blood; grade 2, moderate blood clot with recognizable arteries; and grade 3, blood clot obliterating all arteries within the segment. The animals received a total score ranging from 0 to 18 after adding the scores from all six segments. Thirteen mice with SAH grading scores ≤7, which had no significant brain injury (Altay et al., 2012a, 2012b) were excluded.
Mortality and Neurological Scores
We calculated mortality at 24 hours after SAH. Neurological score was blindly evaluated at 24 hours after SAH as previously described (Altay et al., 2012a, 2012b: Supplementary Material and Methods).
Brain Water Content and BBB Disruption
Brain water content (n=7 per group) (Altay et al., 2012a, 2012b) and Evans blue dye extravasation (n=6 per group) (Manaenko et al., 2011) were measured as previously described (Supplementary Material and Methods).
Western Blotting
We isolated and collected the left cerebral hemisphere (perforation side) at 24 hours after SAH (n=5 per group in the first study; n=6 per group in the second study). Western blotting was performed as previously described (Altay et al., 2012a, 2012b) using the following primary antibodies: anti-phospho-c-Jun N-terminal kinase (JNK), anti-myeloperoxidase (MPO), anti-P-selectin, anti-interleukin (IL)-1β, anti-tumor necrosis factor (TNF)-α (1:200, Santa Cruz Biotechnology, Santa Cruz, CA), anti-intercellular adhesion molecule (ICAM)-1 (1:1000, Millipore, Temecula, CA) and anti-cyclooxygenase (COX)-2 (1:1000, Abcam, Cambridge, MA) antibodies.
Immunofluorescence
Animals were euthanized 24 hours after surgery and brains were processed (n=3 per group) as previously described (Altay et al., 2012a). Ten-micron-thick coronal sections at the level of bregma 1mm (caudally) were cut on a cryostat (LM3050S; Leica Microsystems, Bannockburn, Ill). Double-fluorescence labeling was performed using the following primary antibodies: anti-von Willebrand factor (vWF; 1:400, Abcam, Cambridge, MA) and anti-P-selectin (1:200, Santa Cruz Biotechnology, Santa Cruz, CA) antibodies. Besides, anti-ionized calcium binding adaptor molecule (iba)-1 antibody (1:50, Abcam, Cambridge, MA) was applied and stained alone.
Statistics
Neurological scores were expressed as median±25th to 75th percentiles and other data were expressed as mean±SD. After confirming that each population being compared followed a normal distribution using Barlett’s tests, neurological scores were analyzed using Kruskal-Wallis test, followed by Tukey’s multiple comparisons. Other statistical differences were analyzed using unpaired t tests and one-way analysis of variance (ANOVA) with Tukey-Kramer post hoc tests. Differences in mortality were tested using Fisher’s exact tests or chi-square tests as appropriate. P<0.05 was considered statistically significant.
Results
Isoflurane prevents post-SAH brain injury and inflammation
Mortality rate was not significantly different between the SAH groups (vehicle, 32.3% [10 of 31 mice]; and isoflurane, 22.2% [6 of 27]). No sham-operated mice died. SAH grade was equivalent between the groups (P=0.657). SAH significantly aggravated neurological scores, brain edema and Evans blue dye extravasation in the left cerebral hemisphere compared with the sham group (P<0.05, respectively), which were all significantly ameliorated by the isoflurane treatment (P<0.001, respectively) as shown in our previous studies (Fig. S1) (Altay et al., 2012a, 2012b).
Western blot analyses showed that SAH increased expression levels of neutrophils marker MPO in the left cerebral hemisphere compared with the sham (P<0.001), which was significantly suppressed by isoflurane (P=0.022; Figure 1A). Immunofluorescence showed that SAH also significantly increased activated microglia/macrophages (immunopositive for iba-1) in the left cerebral hemisophere, which was also inhibited by isoflurane (Figure 1B). Consistent with these results, expression levels of cytokines (IL-1β, TNF-α; Figure 2), adhesion molecules (P-selectin, ICAM-1; Figure 3) and intracellular mediators of inflammation (p-JNK, COX-2; Figure 4) were significantly higher in the vehicle group than in the sham group (P<0.001, P<0.001, P=0.003, P=0.047, P<0.001, P<0.001, respectively), and were significantly suppressed in the isoflurane group (P=0.002, P=0.015, P=0.010, P=0.016, P<0.001, P=0.003, respectively). The expression of P-selectin was colocalized with endothelial cells, displaying vWF in the left cerebral hemisphere (Figure 3C).
Figure 1.

Representative Western blots, quantitative analysis of MPO expression (n=5 per group) (A), representative brain section and immunofluorescence for iba-1 (n=3 per group) (B) in the left cerebral hemisphere at 24 hours after SAH. The protein band density values are calculated as a ratio of that of β-actin. Vehicle, SAH+vehicle-air group; 2% ISO, SAH+2% isoflurane group; values, mean±SD; *P<0.05, ANOVA.
Figure 2.
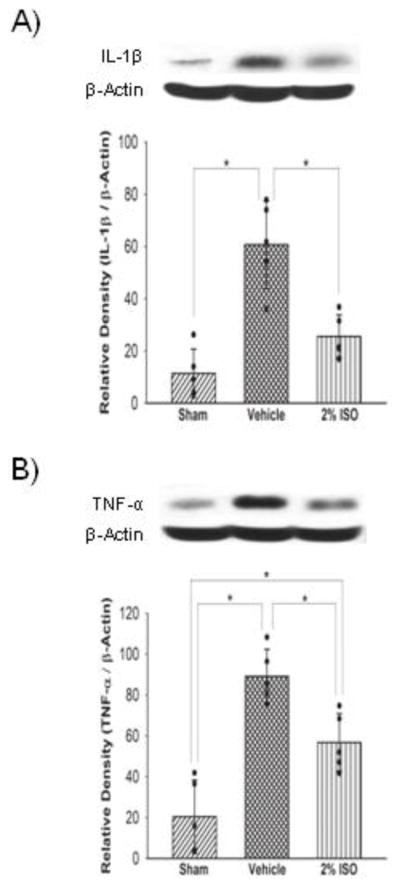
Representative Western blots and quantitative analysis of IL-1β (A) and TNF-α (B) expressions in the left cerebral hemisphere at 24 hours after SAH. The protein band density values are calculated as a ratio of that of β-actin. Vehicle, SAH+vehicle-air group; 2% ISO, SAH+2% isoflurane group; n=5 per group; values, mean±SD; *P<0.05, ANOVA.
Figure 3.

Representative Western blots, quantitative analysis of P-selectin (n=5 per group) (A) and ICAM-1 (n=5 per group) (B) expressions, representative brain section and double immunofluorescence images showing the colocalization of P-selectin (red) with vWF (green) (n=3 per group) (C) in the left cerebral hemisphere at 24 hours after SAH. The protein band density values are calculated as a ratio of that of β-actin. Vehicle, SAH+vehicle-air group; 2% ISO, SAH+2% isoflurane group; values, mean±SD; *P<0.05, ANOVA.
Figure 4.
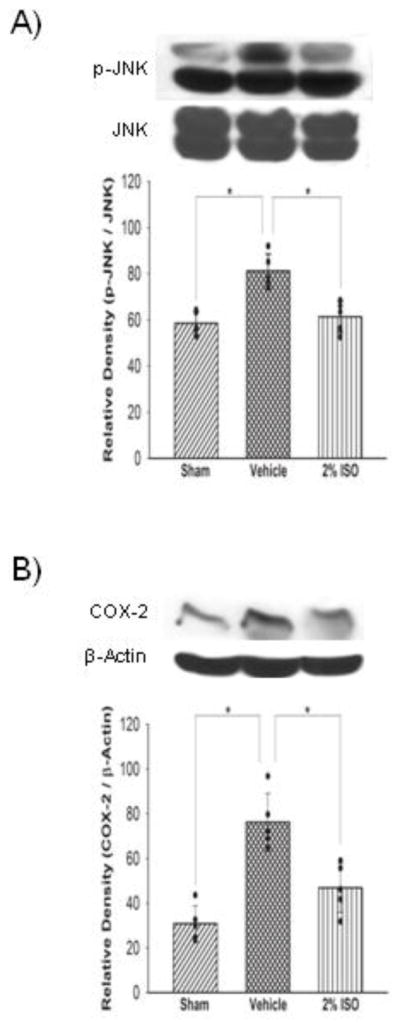
Representative Western blots and quantitative analysis of p-JNK (A) and COX-2 (B) expressions in the left cerebral hemisphere at 24 hours after SAH. The protein band density values are calculated as a ratio of that of JNK or β-actin. Vehicle, SAH+vehicle-air group; 2% ISO, SAH+2% isoflurane group; n=5 per group; values, mean±SD; *P<0.05, ANOVA.
DMS and VPC23019 block anti-inflammatory effects of isoflurane
No sham-operated mice died. Mortality rate was not significantly different among the DMSO+SAH+2% isoflurane (23.5 %, 4 of 17 mice), DMS+SAH+2% isoflurane (35%, 7 of 20) and VPC23019+SAH+2% isoflurane (35%, 7 of 20) groups. SAH grade also did not show significant differences among the groups (P=0.701). Both DMS and VPC23019 treatments significantly aggravated neurological scores (P<0.001, P=0.001, respectively) and brain edema in the left cerebral hemisphere (P<0.035, P=0.05, respectively) compared with the vehicle-treated SAH+2% isoflurane group as shown in our previous studies (Fig. S2) (Altay et al., 2012a, 2012b). Both DMS and VPC23019 treatments also increased ICAM-1 and IL-1β expressions in the left cerebral hemisphere compared with the vehicle-treated SAH+2% isoflurane group (P=0.004, P=0.045/P=0.003, P=0.038, respectively; Figure 5).
Figure 5.
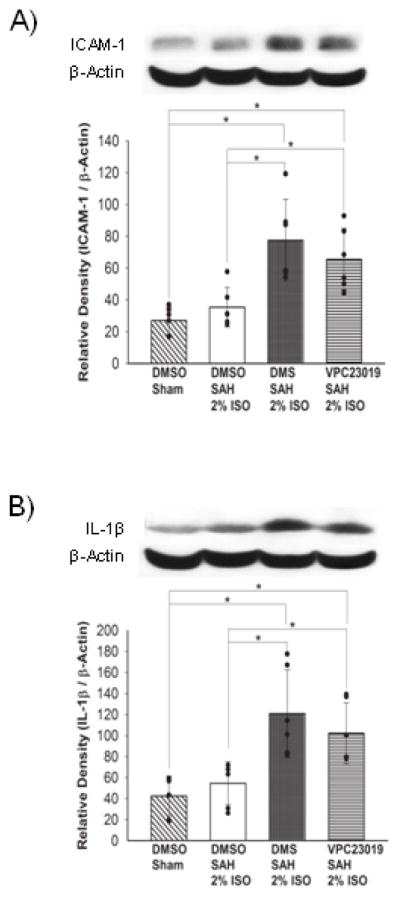
Representative Western blots and quantitative analysis of ICAM-1 (A) and IL-1β (B) expressions in the left cerebral hemisphere at 24 hours after SAH. The protein band density values are calculated as a ratio of that of β-actin. 2% ISO, 2% isoflurane group; n=6 per group; values, mean±SD; *P<0.05, ANOVA.
Discussion
In the present study, we showed consistent results when compared with previous publications (Altay et al., 2012a, 2012b) that 2% isoflurane post-treatment improved neurological score, brain edema, and BBB permeability after SAH in mice. In addition, we demonstrated that many inflammatory mediators increased in the brain after SAH at 24 hours, and that isoflurane prevented this inflammatory reaction.
The presence of a blood clot in the subarachnoid space and local accumulation of luminal platelet aggregates in parenchymal microvessels may initiate post-SAH inflammation (Sehba et al., 2004; Friedrich et al., 2010). BBB dysfunction is also well known to occur after SAH and may contribute to a “vicious circle” of the disease process: that is, it may allow greater influx of blood-borne cells and substances into brain parenchyma and activate the inflammatory response through a complex series of cellular and molecular events, thus amplifying inflammation and leading to further parenchymal damage (Doczi, 1985; Zhang et al., 2012). Experimental models using the magnetic resonance imaging techniques have shown a fall of the apparent diffusion gradient as early as 2 minutes after SAH, demonstrating a global ischemic insult and edema (Busch et al., 1998).
Vascular injury is associated with activation of the coagulation cascade and release of thrombin, which activates cytokine/chemokine production (Szaba and Smiley, 2002; Chen-Roetling et al., 2012). Monocyte-endothelial interactions are key initiating events for vascular inflammation (Bolick et al., 2005; Hammond et al., 2012), and increase endothelial permeability (Mehta and Malik, 2006). In the injured brain, macrophages are generally recruited from the blood to sites of inflammation by chemokine gradients (Fathali et al., 2013). In addition microglia function as the resident brain macrophage and microglia activation is considered the hallmark of brain inflammation (Dheen et al., 2007; Simard et al., 2012). Activated microglia cells are identified by the marker iba-1 (Schäbitz et al., 2008). Activated monocytes release cytokines such as TNF-α and IL-1β that mediate a variety of pathological responses including neuroinflammation (Aggarwal, 2002). MPO, a member of the haeme peroxidase-COX superfamily, is abundantly expressed in neutrophils (Klebanoff, 2005) and is considered to play a role in the pathogenesis of endothelial dysfunction and brain inflammation (Eiserich et al., 2002; Baldus et al., 2001). Adhesion molecules, such as P-selectin and ICAM-1 that belong to the immunoglobulin superfamily are required for leukocyte migration (Springer, 1990). Leukocyte-astrocyte interaction, via ICAM-1 stimulation, might contribute to cerebral TNF-α production by astrocytes, and thus, participate in an amplification of the inflammatory response (Etienne-Manneville et al., 1999). Moreover, TNF-α can affect astrocytes by overexpression of ICAM-1, leading to a further enhancement of leukocyte adhesion and activation (Etienne-Manneville et al., 1999). COX-2 an inducible isoform thought to mediate inflammatory events in response to inflammatory stimuli, such as IL-1β and TNF-α, is a critical factor in the cytotoxicity associated with inflammation (Iadecola et al., 2001). It has been also shown that the level of inflammatory cytokines, including IL-1β and TNF-α were related to the severity of SAH and brain damage (Mathiesen et al., 1997). P-selectin as an early mediator of BBB injury plays an essential role in the initial recruitment of leukocytes (Del Conde et al., 2005): it is expressed on stimulated endothelial cells, and mediates the rolling and loose tethering of leukocytes on the luminal surface of endothelia (Springer, 1990). vWF is a large adhesive glycoprotein that serves as a tool to assess the extent of endothelial cell activation (van Mourik et al., 1999). TNF-α may stimulate the vWF release in plasma and on the surface of endothelial cells to induce platelet aggregation and adhesion on the vascular endothelium, describing a potential linkage between inflammation and thrombosis (Bernardo et al., 2004) which may aggravate secondary ischemic brain damage (Balduini et al., 2004). JNK is a member of the mitogen-activated protein kinase group, activated by cytokines (Manning and Davis, 2003). JNK signaling has been shown to be involved in SAH-induced BBB disruption (Yatsushige et al., 2007). Besides, it has been reported that IL-1β activation causes BBB disruption partly via the JNK pathway in EBI after SAH in mice (Sozen et al., 2009). Activation of JNK may promote the binding and recruitment of neutrophils to the vascular endothelium as well as neutrophil adhesion-dependent oxidant production (Kapitonov et al., 2009).
Murine macrophages express only S1P1 and S1P2 receptors (Hughes et al., 2008). FTY720, a S1P receptor modulator, reduced macrophage infiltration into the peripheral nerves in experimental autoimmuneneuritis, which was accompanied by an increase in S1P1/5 and a decrease in S1P3/4 receptor subtypes (Zhang et al. 2008). Others have demonstrated that FTY720 reduced leukocyte infiltration into the central nervous system in experimental autoimmune encephalomyelitis in vivo (Fujino et al., 2003). Besides that, S1P1 mediates inhibitory effects on motility and trans-endothelial migration attributable to the coupling of S1P2 to the G12/13/Rho pathway (Sugimoto et al., 2003). It has been reported that S1P may inhibit actin filament reorganization in leading edges of neutrophil pseudopodia by acting through the cell surface receptor, thus resulting in the inhibition of motility and trans-endothelial migration (Kawa et al. 1997).
This study has revealed that isoflurane treatment after SAH decreases brain expression levels of neutrophils marker MPO and early mediator of initial recruitment of leukocytes P-selectin, which indirectly demonstrates reduced amount of leukocytes in injury sites. More specifically, the results suggest that, isoflurane can attenuate leukocyte motility, trans-endothelial migration and initiation of leukocyte-platelet-endothelial cell and neuroglial cell interactions by activating the SphK-related pathway. Our previous studies demonstrated that 2% isoflurane increased SphK1 in post-SAH brain (Altay et al., 2012a, 2012b). SphK1 generates S1P, which binds to the receptor on macrophages, vascular endothelium and neuroglial cells, and it has been reported that nanomolar concentrations of S1P activates a potent anti-inflammatory signaling cascade to prevent initiation of inflammation in in-vitro and in-vivo studies (Taussef et al. 2008; Hughes et al., 2008; Kawa et al. 1997; Bolick et al. 2005; Whetzel et al. 2006). Consistently, SphK inhibitor DMS and S1P1/3 receptor inhibitor VPC23019 used in the present study abolished anti-inflammatory effects of isoflurane.
This study has some limitations. First, this study demonstrated that isoflurane decreased brain expression levels of marker of activated microglia cells/macrophages iba-1, suggesting reduced monocyte activation in injury sites. As activated monocytes release cytokines that mediate neuroinflammation (Aggarwal, 2002), microglia/macrophages may be a main source of the changes in pro-inflammatory markers in this study. However, lack of the immunostaining data does not allow us to exclude other possibilities. Secondly, we repeatedly reported that SAH does not change brain SphK levels, but 2% isoflurane increases SphK1 in post-SAH brain (Altay et al., 2012a, 2012b). As well, this study showed that SphK inhibitor DMS abolished anti-inflammatory effects of isoflurane, indirectly suggesting isoflurane’s anti-inflammatory effects through SphK upregulation. The direct demonstration, however, would support the sphingosine-related mechanisms more surely. Lastly, reduced inflammatory reaction does not necessarily mean the causative role of inflammation in post-SAH bran injury, and may merely be a result of reduced brain injury. Sozen et al (Sozen, 2009) reported that an anti-proinflammatory agent administered at 1 hour pot-SAH suppressed brain inflammation and then prevented EBI in the endovascular perforation model of SAH in mice used in this study, demonstrating the causative role of inflammation on post-SAH EBI and supporting the anti-inflammatory effects of isoflurane in this study. However, it would be helpful in defining the causative role to detail the time course of inflammatory mediator expression and neuronal injury in this model. Anyway, the identification of anesthetics with properties that help to attenuate post-SAH brain injury is clinically highly relevant, because the first step to treat aneurysmal SAH is aneurysmal obliteration under general anesthesia. This study showed that isoflurane is the good candidate. Further studies are warranted to elaborate the protocol of isoflurane treatment.
Conclusion
This study investigated the mechanisms of isoflurane on brain inflammation. From the results obtained in this study, we speculate that one possibility is that isoflurane may prevent inflammation directly by activating the SphK-related/S1P1 pathway inhibiting leukocyte-platelet-endothelial cell interactions. On the other hand, we cannot exclude the possibility that isoflurane’s anti-inflammatory action may turn out to be a secondary effect in such a scenario, isoflurane may prevent EBI [neuronal apoptosis and BBB disruption (Altay et al., 2012a, 2012b)] firstly, and as a result, inflammation may be suppressed. However, even in this case, isoflurane still may have some direct anti-inflammatory effects, because SAH by itself may cause brain inflammation.
Supplementary Material
Highlights.
The presence of a blood clot in the subarachnoid space and local accumulation of luminal platelet aggregates in parenchymal microvessels may initiate post-SAH inflammation.
Inflammatory mediators increased in brain after SAH at 24 hours.
2% Isofluranemay attenuate leukocyte motility and trans-endothelial migration.
2% isoflurane may suppress post-SAH brain inflammation possibly via the sphingosine-related pathway.
Acknowledgments
This study is partially supported by NIH NS060936 to JT and NS053407 to JHZ.
Abbreviations
- SAH
Subarachnoid hemorrhage
- EBI
Early brain injury
- SphK
Sphingosine kinase
- S1P
Sphingosine 1-phosphate
- S1P1/3
Sphingosine-1-phosphate receptor-1/3
- BWC
Brain water content
- iba-1
Ionized calcium binding adaptor molecule-1
- MPO
Myeloperoxidase
- TNF-α
Tumor necrosis factor-alpha
- IL-1β
Interleukin-1beta
- ICAM-1
Intercellular adhesion molecule-1
- COX-2
Cyclooxygenase-2
- p-JNK
Phosphorylated c-Jun N-terminal kinase
- vWF
von Willebrand factor
- DMS
N, N-dimethylsphingosine
Footnotes
The authors report no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aggarwal BB, Shishodia S, Ashikawa K, Bharti AC. The role of TNF and its family members in inflammation and cancer: lessons from gene deletion. Curr Drug Targets Inflamm Allergy. 2002;1:327–341. doi: 10.2174/1568010023344571. [DOI] [PubMed] [Google Scholar]
- Altay O, Hasegawa Y, Sherchan P, Suzuki H, Khatibi NH, Tang J, et al. Isoflurane delays the development of early brain injury after subarachnoid hemorrhage through sphingosine-related pathway activation in mice. Crit Care Med. 2012a;40:1908–1913. doi: 10.1097/CCM.0b013e3182474bc1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altay O, Suzuki H, Hasegawa Y, Caner B, Krafft PR, Fujii M, et al. Isoflurane attenuates blood-brain barrier disruption in ipsilateral hemisphere after subarachnoid hemorrhage in mice. Stroke. 2012b;43:2513–2516. doi: 10.1161/STROKEAHA.112.661728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antkowiak B. How do general anaesthetics work? Naturwissenschaften. 2001;88:201–213. doi: 10.1007/s001140100230. [DOI] [PubMed] [Google Scholar]
- Balduini W, Carloni S, Mazzoni E, Cimino M. New therapeutic strategies in perinatal stroke. Curr Drug Targets CNS Neurol Disord. 2004;3:315–323. doi: 10.2174/1568007043337247. [DOI] [PubMed] [Google Scholar]
- Baldus S, Eiserich JP, Mani A, Castro L, Figueroa M, Chumley P, et al. Endothelial transcytosis of myeloperoxidase confers specificity to vascular ECM proteins as targets of tyrosine nitration. J Clin Invest. 2001;108:1759–1770. doi: 10.1172/JCI12617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood. 2004;104:100–106. doi: 10.1182/blood-2004-01-0107. [DOI] [PubMed] [Google Scholar]
- Bolick DT, Srinivasan S, Kim KW, Hatley ME, Clemens JJ, Whetzel A, et al. Sphingosine-1-phosphate prevents tumor necrosis factor-{alpha}-mediated monocyte adhesion to aortic endothelium in mice. Arterioscler Thromb Vasc Biol. 2005;25:976–981. doi: 10.1161/01.ATV.0000162171.30089.f6. [DOI] [PubMed] [Google Scholar]
- Broderick JP, Brott TG, Duldner JE, Tomsick T, Leach A. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke. 1994;25:1342–1347. doi: 10.1161/01.str.25.7.1342. [DOI] [PubMed] [Google Scholar]
- Busch E, Beaulieu C, de Crespigny A, Moseley ME. Diffusion MR imaging during acute subarachnoid hemorrhage in rats. Stroke. 1998;29:2155–2161. doi: 10.1161/01.str.29.10.2155. [DOI] [PubMed] [Google Scholar]
- Chen-Roetling J, Sinanan J, Regan RF. Effect of iron chelators on methemoglobin and thrombin preconditioning. Transl Stroke Res. 2012;3:452–459. doi: 10.1007/s12975-012-0195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med. 2005;201:871–879. doi: 10.1084/jem.20041497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dheen ST, Kaur C, Ling EA. Microglial activation and its implications in the brain diseases. Curr Med Chem. 2007;14:1189–1197. doi: 10.2174/092986707780597961. [DOI] [PubMed] [Google Scholar]
- Doczi T. The pathogenetic and prognostic significance of blood–brain barrier damage at the acute stage of aneurysmal subarachnoid haemorrhage. Clinical and experimental studies. Acta Neurochir (Wien) 1995;77:110–132. doi: 10.1007/BF01476215. [DOI] [PubMed] [Google Scholar]
- Eiserich JP, Baldus S, Brennan ML, Ma W, Zhang C, Tousson A, et al. Myeloperoxidase: a leukocyte derived vascular NO oxidase. Science. 2002;296:2391–2394. doi: 10.1126/science.1106830. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Chaverot N, Strosberg AD, Couraud PO. ICAM-1-coupled signaling pathways in astrocytes converge to cyclic AMP response element-binding protein phosphorylation and TNF-alpha secretion. J Immunol. 1999;163:668–674. [PubMed] [Google Scholar]
- Fathali N, Ostrowski RP, Hasegawa Y, Lekic T, Tang J, Zhang JH. Splenic immune cells in experimental neonatal hypoxia-ischemia. Transl Stroke Res. 2013;4:208–219. doi: 10.1007/s12975-012-0239-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich V, Flores R, Muller A, Sehba FA. Escape of intraluminal platelets into brain parenchyma after subarachnoid hemorrhage. Neuroscience. 2010;165:968–975. doi: 10.1016/j.neuroscience.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii M, Yan J, Rolland WB, Soejima Y, Caner B, Zhang JH. Early brain injury, an evolving frontier in subarachnoid hemorrhage research. Transl Stroke Res. 2013;4:432–446. doi: 10.1007/s12975-013-0257-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino M, Funeshima N, Kitazawa Y, Kimura H, Amemiya H, Suzuki S, et al. Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J Pharmacol Exp Ther. 2003;305:70–77. doi: 10.1124/jpet.102.045658. [DOI] [PubMed] [Google Scholar]
- Hammond MD, Ai Y, Sansing LH. Gr1+ Macrophages and dendritic cells dominate the inflammatory infiltrate 12 hours after experimental intracerebral hemorrhage. Transl Stroke Res. 2012;3:s125–s131. doi: 10.1007/s12975-012-0174-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt L, Badaut J, Thevenet J, Granziera C, Regli L, Maurer F, et al. D-JNKI1, a cell-penetrating c-Jun-N-terminal kinase inhibitor, protects against cell death in severe cerebral ischemia. Stroke. 2004;35:1738–1743. doi: 10.1161/01.STR.0000131480.03994.b1. [DOI] [PubMed] [Google Scholar]
- Huang J, van Gelder JM. The probability of sudden death from rupture of intracranial aneurysms: A meta-analysis. Neurosurgery. 2002;51:1101–1105. doi: 10.1097/00006123-200211000-00001. [DOI] [PubMed] [Google Scholar]
- Hughes JE, Srinivasan S, Lynch KR, Proia RL, Ferdek P, Hedrick CC. Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circ Res. 2008;102:950–958. doi: 10.1161/CIRCRESAHA.107.170779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E, et al. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc Natl Acad Sci. 2001;98:1294–1299. doi: 10.1073/pnas.98.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov D, Allegood JC, Mitchell C, Hait NC, Almenara JA, Adams JK, et al. Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 2009;69:6915–6923. doi: 10.1158/0008-5472.CAN-09-0664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawa S, Kimura S, Hakomori S, Igarashi Y. Inhibition of chemotactic motility and trans-endothelial migration of human neutrophils by S1P. FEBS Lett. 1997;420:196–200. doi: 10.1016/s0014-5793(97)01516-0. [DOI] [PubMed] [Google Scholar]
- Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- Manaenko A, Chen H, Kammer J, Zhang JH, Tang J. Comparison Evans Blue injection routes: intravenous versus intraperitoneal, for measurement of blood-brain barrier in a mice hemorrhage model. J Neurosci Methods. 2011;195:206–210. doi: 10.1016/j.jneumeth.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AM, Davis RJ. Targeting jnk for therapeutic benefit: From junk to gold? Nat Rev Drug Discov. 2003;2:554–565. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- Mathiesen T, Edner G, Ulfarsson E, Andersson B. Cerebrospinal fluid interleukin-1 receptor antagonist and tumor necrosis factor-alpha following subarachnoid hemorrhage. J Neurosurg. 1997;87:215–220. doi: 10.3171/jns.1997.87.2.0215. [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- Plachinta RV, Hayes JK, Cerilli LA, Rich GF. Isoflurane pretreatment inhibits lipopolysaccharide-induced inflammation in rats. Anesthesiology. 2003;98:89–95. doi: 10.1097/00000542-200301000-00017. [DOI] [PubMed] [Google Scholar]
- Schäbitz WR, Krüger C, Pitzer C, Weber D, Laage R, Gassler N, et al. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF) J Cereb Blood Flow Metab. 2008;28:29–43. doi: 10.1038/sj.jcbfm.9600496. [DOI] [PubMed] [Google Scholar]
- Sehba FA, Bederson JB. Mechanisms of acute brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28:381–398. doi: 10.1179/016164106X114991. [DOI] [PubMed] [Google Scholar]
- Sehba FA, Mostafa G, Knopman J, Friedrich V, Jr, Bederson JB. Acute alterations in microvascular basal lamina after subarachnoid hemorrhage. J Neurosurg. 2004;101:633–640. doi: 10.3171/jns.2004.101.4.0633. [DOI] [PubMed] [Google Scholar]
- Simard JM, Tosun C, Ivanova S, Kurland DB, Hong C, Radecki L, et al. Heparin reduces neuroinflammation and transsynaptic neuronal apoptosis in a model of subarachnoid hemorrhage. Transl Stroke Res. 2012;3(Suppl 1):155–165. doi: 10.1007/s12975-012-0166-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozen T, Tsuchiyama R, Hasegawa Y, Suzuki H, Jadhav V, Nishizawa S, et al. Role of interleukin-1beta in early brain injury after subarachnoid hemorrhage in mice. Stroke. 2009;40:2519–2525. doi: 10.1161/STROKEAHA.109.549592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer TA. Adhesion receptors of the immune system. Nature. 1990;346:425–434. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- Sugimoto N, Takuwa N, Okamoto H, Sakurada S, Takuwa Y. Inhibitory and stimulatory regulation of Rac and cell motility by the G12/13-Rho and Gi pathways integrated downstream of a single G protein-coupled sphingosine-1-phosphate receptor isoform. Mol Cell Biol. 2003;23:1534–1545. doi: 10.1128/MCB.23.5.1534-1545.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaba FM, Smiley ST. Roles for thrombin and fibrin(ogen) in cytokine/chemokine production and macrophage adhesion in vivo. Blood. 2002;99:1053–1059. doi: 10.1182/blood.v99.3.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauseef M, Kini V, Knezevic N, Brannan M, Ramchandaran R, Fyrst H, et al. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ Res. 2008;103:1164–1172. doi: 10.1161/01.RES.0000338501.84810.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Mourik JA, Boertjes R, Huisveld IA, Fijnvandraat K, Pajkrt D, van Genderen PJ, et al. von Willebrand factor propeptide in vascular disorders: A tool to distinguish between acute and chronic endothelial cell perturbation. Blood. 1999;94:179–185. [PubMed] [Google Scholar]
- Whetzel AM, Bolick DT, Srinivasan S, Macdonald TL, Morris MA, Ley K, et al. Sphingosine-1 phosphate prevents monocyte/endothelial interactions in type 1 diabetic nod mice through activation of the s1p1 receptor. Circ Res. 2006;99:731–739. doi: 10.1161/01.RES.0000244088.33375.52. [DOI] [PubMed] [Google Scholar]
- Yatsushige H, Ostrowski RP, Tsubokawa T, Colohan A, Zhang JH. Role of c-Jun N-terminal kinase in early brain injury after subarachnoid hemorrhage. J Neurosci Res. 2007;85:1436–1448. doi: 10.1002/jnr.21281. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Badaut J, Tang J, Obenaus A, Hartman R, Pearce WJ. The vascular neural network--a new paradigm in stroke pathophysiology. Nat Rev Neurol. 2012;8:711–716. doi: 10.1038/nrneurol.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhang ZY, Fauser U, Schluesener HJ. FTY720 ameliorates experimental autoimmune neuritis by inhibition of lymphocyte and monocyte infiltration into peripheral nerves. Exp Neurol. 2008;210:681–690. doi: 10.1016/j.expneurol.2007.12.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.