

Abstract
Botulinum neurotoxin A (BoNT/A) is the most potent toxin known. Unfortunately, it is also a potential bioweapon in terrorism, which is without an approved therapeutic treatment once cellular intoxication takes place. Previously, we reported how hydroxamic acid prodrug carbamates increased cellular uptake, which translated to successful inhibition of this neurotoxin. Building upon this research, we detail BoNT/A protease molecular modeling studies accompanied by the construction of small library of hydroxamic acids based on 2,4-dichlorocinnamic hydroxamic acid scaffold and their carbamate prodrug derivatization along with the evaluation of these molecules in both enzymatic and cellular models.
Keywords: Botulinum neurotoxin, SNARE, SNAP-25, protease inhibitor, zinc-dependent metalloprotease, hydroxamic acid, carbamate prodrug
1. Introduction
Botulinum neurotoxin, a protein produced by Clostridium botulinum, is the most potent toxin known to date.1 Although the toxin has been historically recognized in food poisoning, its application to medical maladies such as chronic pain and migraines; as well as cosmetic improvements including facial wrinkles, provides a clinical significance that has led to its commercial production.2,3 This, however, also implicates the increased potential use of this toxin as a bioweapon in terrorism.4 Indeed, Botulinum neurotoxin has been classified as a category A agent, the highest class for potential misuse as a biothreat by the Center for Disease Control and Prevention.
Botulinum neurotoxin consists of 100 kDa heavy chain and 50 kDa light chain, which are appended to each other by a disulfide linkage.5 In brief, the etiology of this neurotoxin consists of the heavy chain acting to deliver the light chain into the cytosol by translocation through target receptors on nerve terminals, and depending on the serotype, the light chain cleaves one of the proteins necessary for SNARE complex formation.6 As a result, the release of acetylcholine into neuromuscular junction ceases, which can lead to fatal flaccid paralysis. The serotypes A, B, and E are known to cause Botulism in humans. Among the SNARE components, type A and E target SNAP-25 whereas type B cleaves VAMP.7,8 In addition to the aforementioned human intoxication, its potency (LD50 value of 0.001 µg per kg)9 and the length of duration (up to several months) sets Botulinum neurotoxin A apart from the other serotypes and renders it to be a focal point of many laboratories including our own.10
While there are known treatments such as vaccination and antitoxin treatment for serotype A, no therapeutic is currently available once BoNT/A cellular intoxication takes place. Because the BoNT protease is responsible for the pathology seen with all neurotoxins, it is incumbent to develop small molecules to counteract its mechanism of action. To date, several potent light chain protease inhibitors have been reported based on in vitro enzyme assays, including peptidic and non-peptidic molecules.11,12 However, the development of efficacious inhibitors that can function intracellularly still remains a major hurdle to overcome in the scientific community.
Hydroxamic acids developed in our laboratory as BoNT/A light chain protease inhibitors have suffered from a lack of cellular protection, presumably due to their poor cellular uptake and toxicity.11 However, we recently discovered that these issues could be resolved and ultimately circumvented through protection of the hydroxamic acid moiety as a carbamate (Scheme 1).13 Thus, the N-Benzyl carbamate of hydroxamic acid A became cell-permeable and now demonstrated inhibition of BoNT/A with the EC50 value of 20 µM in human induced pluripotent stem cells (hiPSCs).14
Scheme 1.

O-carbamation of the hydroxamic acid moiety as a prodrug approach.
With this prodrug approach in mind, we next turned our focus to additional druggable scaffolds (Scheme 2). Previously our laboratory disclosed cinnamic hydroxamic acid 1 as a potent hit from hydroxamic acid library screening,15,16 and a congener of this molecule, hydroxyethyl hydroxamate 2 as an inhibitor with an additional element of chemical functionality.17 To further diversify this line of research, we devised two other scaffolds: amides (3) and ethers (4) as a means to further increase hydrophobic interactions within the active site of enzyme. Herein, we communicate the synthesis/molecular modeling of amides (3), ethers (4), and β-(2,4-dichlorophenyl)-hydroxamic acid O-carbamates along with their inhibitory activity as determined through enzymatic as well as cellular assays.
Scheme 2.

Application of a prodrug approach to β-(2,4-dichlorophenyl)-hydroxamic acids.
2. Results and Discussion
2.1 Computational studies
The program Autocorrelator was utilized to construct computational models predicting and ranking potency of derived structures against BoNT LC/A using 2 as a starting point for synthetically accessible targets, amides 3 and ethers 4.18 In brief, a model was developed through Autocorrelator using the 2IMB co-crystal structure of BoNT LC/A with a computational pipeline consisting of Omega v2.4.6 and Fred v2.2.5 (R2 = 0.757).16,19,20 Autocorrelator was also used to derive a new scoring function, produced by the LARS methodology in R, shown in Equation 121,22:
| (Eq.1) |
Thus, computational modeling provided us a series of structures as potential inhibitors with predicted IC50 values (Figure 1). From this computational exercise, aryl groups or planar structures seemed to be important for engaging active site interactions. Two docking solutions are shown in Figure 2, and based on these modeling parameters, compounds such as 3a and 3c displayed optimized steric and hydrogen-bonding interactions within the active site. The size of the substituents also affected both the orientation and hydrogen-bonding partners of our docked poses. Thus, larger substituents forced molecules to lose interactions with Thr11, Arg56, and Thr6, although this should have only a modest impact on potency, as predicted molecules were able to still maintain the key hydroxamate-zinc interaction. Hence, among the compounds shown in Figure 1, we selected 3a–c, 3f, and 4a due to the synthetic accessibility. Lastly, as many of the structures shown in Figure 1 were synthetically challenging, close congeners (3d, 3e, 4b–d, Figure 3) were prepared for examination.
Figure 1.

Predicted BoNT/A protease inhibitors and their IC50 values.
Figure 2.

The docking solutions of 3a and 3c within the 2IMB co-crystal using the automated pipeline derived by Autocorrelator.
Figure 3.

Selected synthetic targets as possible protease inhibitors.
2.2 Synthesis of hydroxamic acids
Synthesis of the target amide molecules was conducted using a similar strategy to our previous report (Scheme 3).17 Thus, dichlorobenzaldehyde was condensed with dimethyl malonate in the presence of piperidine to provide the α, β-unsaturated diester 5. Dimethyl malonate underwent a Michael addition with 5 under basic conditions, affording tetraester 6, which was then subjected to decarboxylation to furnish diacid 7. This diacid was mono-esterified via the initial formation of an anhydride with TFAA and subsequent nucleophilic opening with MeOH. The resulting acid 8 was converted to various amides 9 under the conventional coupling conditions. Finally, each amide was reacted with hydroxylamine to provide the desired hydroxamic acid 3.23
Scheme 3.

Synthesis of amides 3.
In terms of a similar set of ethers, we initially envisioned the alkylation of alcohol B or C as a convergent approach (Scheme 4). However, either route was found to be untenable, likely due to the instability of starting materials under basic conditions. Thus, although slightly more cumbersome, an alternative strategy was undertaken. Hence, diacid 7 was reduced to diol 10, which was then mono-alkylated with several electrophiles to provide alcohols 11. Each alcohol was subjected to three transformations in one-pot: 1) oxidation of the alcohol to an acid by Jones’ reagent; 2) formation of methyl ester using diazomethane; 3) conversion of the ester to hydroxamic acid 4. We note that using this sequence all attempts to isolate the methyl ester were not successful.
Scheme 4.

Synthesis of ethers 4.
2.3 Screening of selected hydroxamic acids against the BoNT/A light chain 1–425
With desired hydroxamic acids in hand, their inhibitory activity was evaluated in vitro using an established FRET-based assay (Table 1).24 The compounds were tested against truncated Botlulinum neurotoxin A light chain (1–425 residues) in the presence of SNAPtide, a 13mer SNAP-25 pseudosubstrate containing a FITC fluorophore and a DABCYL quencher. Based on the results of the SNAPtide assay, the aryl moiety seems to be important. This was true with either the amide or ether appendage, whereas simple alkyl chains did not contribute to the inhibition seen. Based on these findings, the most potent amide and ether homologue of 2 were further evaluated in terms of Ki using our 66mer assay, where a cleaved product from 66 residues found within SNAP-25 (141–206 residues) was quantified by LCMS analysis.25 As anticipated, inhibitors 3a and 4a showed competitive inhibition with Ki values of 1.0 and 2.1 µM respectively (Table 1 and Figure 4). Importantly, using Equation 1, all of our molecules prepared and tested (3a–4d), have their IC50 correctly predicted within a log of our experimental results.
Table 1.
In vitro evaluation of amides 3 and ethers 4.
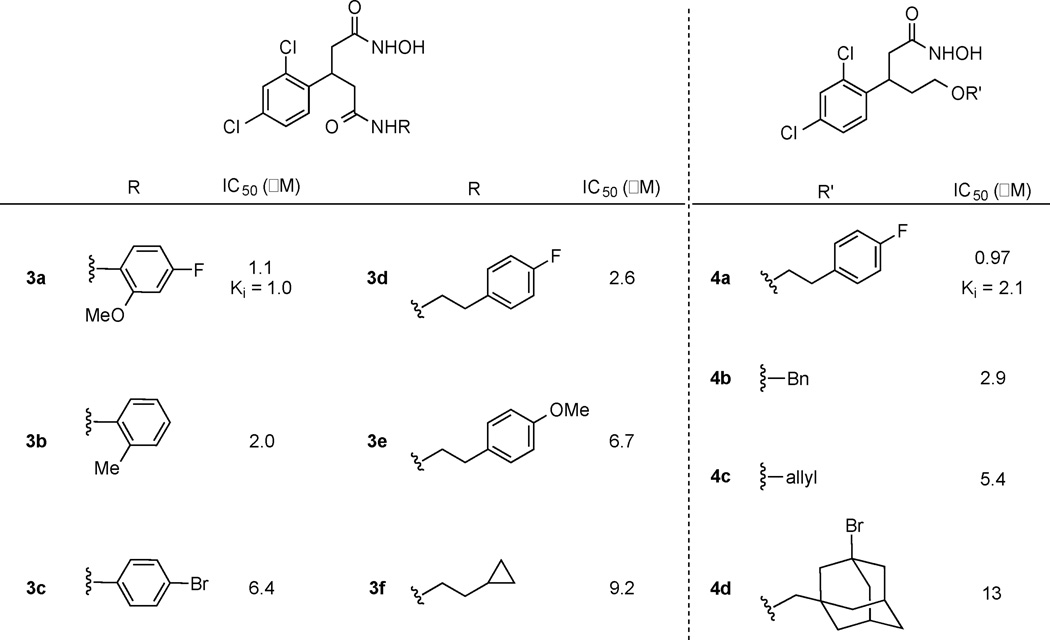 |
Figure 4.

Kinetic analysis of inhibitor 3a.
2.4 Synthesis of BoNT/A protease prodrugs
With in vitro assessment accomplished, several of the hydroxamic acids (1,2,3a–3d, 4a–b, 4d) were converted to the corresponding benzylcarbamates (1’,2’, 3a’–3d’, 4a’-b’, 4d’) as prodrugs via the formation of a carbonate intermediate and subsequent nucleophilic addition of benzylamine (Scheme 5).26 This protocol achieves a selective O-carbamation (vs N-carbamation) as shown previously.14
Scheme 5.

Generic scheme showing synthetic access to the carbamate prodrugs.
2.5 Testing of selected protease inhibitors in a BoNT/A cell based assay
Selected hydroxamic acids and corresponding prodrugs were tested in a hiPSC-derived neuron assay. The importance of this cell line and assay is that it expresses the necessary receptors and substrates for BoNT/A intoxication. The intoxicated cells were incubated with each compound for 8 hours, and upon cell lysis, the SNAP-25 cleavage was analyzed by Western blot. The initial screening was conducted at one or two concentrations (typically 0.1 mM and 1.0 mM). Unfortunately this assay revealed that most of the hydroxamic acids were not effective due to cytotoxicity. Interestingly, 2, was an exception, as it did not show cytotoxicity, although no inhibition was observed at 200 µM. Next, the carbamate prodrugs were examined in this cellular assay (Table 2). Carbamate prodrugs 1’ and 2’ were found to be ineffective inhibitors due to cytotoxicity and potency respectively, whereas amide prodrugs 3a’–3d’ showed reasonable protection of SNAP-25 from the neurotoxin without apparent cytotoxicity. The EC50 values were further determined for these compounds with 3c’ being the most potent, 198 µM (Table 2). Disappointingly, prodrug ethers (4a’–4b’, 4d’) did not improve their cellular activity. This could partially be due to a lack of solubility, which may also contribute to cytotoxicity seen.
Table 2.
Prodrug screening using hiPSC-derived neurons
| Prodrug | Inhibition |
|---|---|
| 1' | cytotoxic at 150 µMa |
| 2' | partial inhibition at 300 µM |
| 3a' | EC50 = 374 µM |
| 3b' | EC50 = 209 µM |
| 3c' | EC50 = 198 µM |
| 3d' | EC50 = 572 µM |
| 4a' | cytotoxic at 1 mMa |
| 4b' | cytotoxic at 1 mMa |
| 4d' | EC50 > 1 mM |
No inhibition was observed up to the cytotoxic concentration examined.
3. Conclusions
In summary, we have applied molecular modeling and a prodrug strategy to our 2,4-dichlorocinnamic hydroxamic acid scaffold, thus preparing a series of amide and ether O-carbamate hydroxamic acids. The computational modeling studies granted insights into the BoNT/A protease’s active site, which translated to a more effective approach, generating molecules for our screening efforts. In general, these molecules as hydroxamic acids were toxic to the cells, whereas their prodrug O-carbamates showed modest inhibitory activity without major cellular toxicity. While we were pleased to see our prodrug approach was translatable to alternate scaffolds, in vitro cellular potency was marginal. We surmise this could be due to the inefficient release of the hydroxamic acid warhead from the corresponding prodrug, presumably enzyme-assisted within the cell. Future research will entail exploration of the enzyme responsible for carbamate hydrolysis as well as an alternative Zn chelator to avoid the inherent toxicity of hydroxamic acids.
4. Experimental section
4.1 Chemistry
Tetramethyl 2-(2,4-dichlorophenyl)propane-1,1,3,3-tetracarboxylate (6)
To a stirred solution of dimethylmalonate (26.2 mmol, 3.46g, 1.20 equiv) in NaOMe/MeOH solution (0.5 M, 26.2 mmol, 52 mL), dimethyl 2-(2,4-dichlorobenzylidene)malonate (21.9 mmol, 6.32 g) was added dropwise at ambient temperature. After 1 h, the reaction mixture was cooled to 0 °C and quenched by the addition of AcOH (87.4 mmol, 5.24 g, 4.0 equiv). Upon evaporation of volatiles, reaction mixture was re-dissolved in dichloromethane and H2O. The partitioned organic layer was dried over MgSO4, and concentrated in vacuo. The crude mixture was subjected to column chromatography (Hexanes/EtOAc) to afford the titled compound as clear oil (9.02 g, 98%).
1H NMR (600 MHz, CDCl3) δ 7.38 (s, 2H), 7.19 (d, J = 8.5 Hz, 1H), 4.79 (s, 1H), 4.37 − 4.12 (m, 2H), 3.70 (s, 6H), 3.57 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 168.1, 167.8, 135.9, 134.5, 134.2, 130.0, 129.9, 127.4, 54.0, 53.0, 52.8; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C17H19Cl2O8: 421.0451, found 421.0450.
3-(2,4-Dichlorophenyl)pentanedioic acid (7)
Tetraester 6 (10.4 mmol, 4.40 g) was dissolved in aq. HCl (37%, 30 mL) and heated to reflux overnight. The white solid was collected by filtration to provide the titled compound as white solid (2.56g, 88%).
1H NMR (600 MHz, MeOD-d4) δ 7.42 (s, 1H), 7.36 (d, J = 8.0 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 4.06 (p, J = 6.9 Hz, 1H), 2.70 (m, 4H); 13C NMR (151 MHz, MeOD-d4) δ 174.9, 140.6, 135.8, 133.9, 130.4, 130.3, 128.4, 39.4, 35.4; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C11H11Cl2O4: 277.0029, found 277.0033.
3-(2,4-Dichlorophenyl)-5-methoxy-5-oxopentanoic acid (8)
To a solution of diacid 7 (3.00 mmol, 831 mg, 1.0 equiv) in chloroform (20 mL) at 0 °C, TFAA (6.00 mmol, 1.26 g, 2.0 equiv) was added dropwise. The reaction was stirred at ambient temperature for 2h and concentrated in vacuo. The resulting acid anhydride was dissolved in MeOH (40 mL) at 0 °C. TEA (12.0 mmol, 1.21 g, 4.0 equiv) was added to this solution, and the reaction mixture was stirred at ambient temperature overnight. Upon evaporation of solvent, the crude mixture was re-dissolved in EtOAc, washed with aq. HCl (1 M), brine, dried over MgSO4, and concentrated to afford the titled product (900 mg, quant.) as yellow oil, which was used to next reaction without further purification.
1H NMR (600 MHz, CDCl3) δ 7.39 (s, 1H), 7.24 − 7.15 (m, 2H), 4.07 (m, 1H), 3.61 (s, 3H), 2.76 (m, 4H); 13C NMR (151 MHz, CDCl3) δ 177.1, 171.7, 138.1, 134.6, 133.4, 130.0, 128.9, 127.5, 52.0, 38.4, 38.2, 34.0; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C12H13Cl2O4: 290.0185, found 290.0189.
3-(2,4-Dichlorophenyl)pentane-1,5-diol (10)
To a solution of diacid 7 (4.00 mmol, 1.11 g, 1.0 equiv) in THF (40 mL), BH3 (THF solution of dimethyl sulfide complex, 2 M, 12.0 mmol, 6.00 mL, 3.0 equiv) was added at −78 °C. The reaction mixture was warmed to ambient temperature and stirred for 3 h, which was quenched by the addition of aq. NaHCO3 at −78 °C. This crude mixture was extracted with EtOAc, washed with brine, dried over MgSO4, and concentrated in vacuo. The resulting crude product was purified by column chromatography (MeOH/dichloromethane) to afford the titled compound (918 mg, 92%) as clear oil.
1H NMR (600 MHz, CDCl3) δ 7.37 (s, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 3.61 − 3.41 (m, 5H), 1.97 (m, 2H), 1.90 − 1.70 (m, 4H); 13C NMR (151 MHz, CDCl3) δ 140.7, 135.0, 132.5, 129.5, 129.1, 127.8, 60.6, 38.8, 33.9; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C11H15Cl2O2: 249.0444, found 249.0448.
General procedure for mono-alkylation of diol 10
To a solution of diol 10 (1.0 equiv) in THF (0.1 M) was added NaH (2.0 equiv) at 0 °C. After stirring for 0.5 h, electrophile (1.0 equiv) was added, and the solution was stirred at ambient temperature overnight. Upon evaporation of solvent, crude mixture was directly subjected to column chromatography (Hexanes/EtOAc) to afford mono-alkylated product as clear oil.
For the synthesis of 11c, TBAI (1.0 equiv) was added together with electrophile
For the synthesis of 11d, 18-Crown-6 (1.0 equiv) was added together with electrophile.
3-(2,4-Dichlorophenyl)-5-(4-fluorophenethoxy)pentan-1-ol (11a)
43.8 mg, 49% 1H NMR (600 MHz, CDCl3) δ 7.36 (s, 1H), 7.21 (d, J = 8.4 Hz, 1H), 7.13 (d, J = 7.4 Hz, 3H), 6.96 (t, J = 8.2 Hz, 2H), 3.60 − 3.38 (m, 6H), 3.31 (q, J = 6.5, 5.9 Hz, 1H), 3.23 (q, J = 8.0, 7.5 Hz, 1H), 2.79 (t, J = 6.9 Hz, 2H), 1.96 (tq, J = 13.9, 6.7 Hz, 2H), 1.82 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 161.6 (JC-F = 243 Hz), 140.7, 135.1, 134.8 (JC-F = 3.02 Hz), 132.4, 130.4 (JC-F = 7.55 Hz), 129.4, 129.1, 127.6, 115.2 (JC-F = 21.1 Hz), 72.0, 68.7, 60.8, 39.0, 35.9, 35.6, 34.4; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C19H22Cl2FO2: 371.0975, found 371.0973.
4-fluorophenethyl trifluoromethanesulfonate
To a solution of 4-fluorophenethyl alcohol (1.00 mmol, 140 mg, 1.0 equiv) in chloroform (5 mL) were added Tf2O (1.50 mmol, 423 mg, 1.5 equiv) and 2,6-lutidine (1.70 mmol, 182 mg, 1.7 equiv) at 0 °C. After stirring for 1 h, the reaction was concentrated in vacuo, re-dissolved in Hexanes/EtOAc (2:1), washed with aq. citric acid (10%), aq. NaHCO3, brine, dried over MgSO4, and again concentrated. Due to the instability of titled compound on SiO2, the obtained crude product (244 mg, 90%, reddish oil) was used for the synthesis of 11a without further purification.
1H NMR (600 MHz, CDCl3) δ 7.23 – 7.13 (m, 2H), 7.04 (t, J = 8.2 Hz, 2H), 4.67 (t, J = 6.8 Hz, 2H), 3.11 (t, J = 6.8 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 162.3 (JC-F = 246 Hz), 130.67, 130.67 (JC-F = 7.55 Hz), 118.7 (q, JC-F = 320 Hz), 115.8 (JC-F = 21.1 Hz), 77.1, 35.1; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C9H9F4O3S: 273.0209, found none.
5-(Benzyloxy)-3-(2,4-dichlorophenyl)pentan-1-ol (11b)
47.4 mg, 47% 1H NMR (600 MHz, CDCl3) δ 7.39 − 7.29 (m, 3H), 7.27 (s, 2H), 7.24 − 7.13 (m, 2H), 4.40 (t, J = 8.2 Hz, 2H), 3.50 (m, 4H), 3.37 (m, 1H), 3.30 (m, 1H), 1.93 (m, 5H); 13C NMR (151 MHz, CDCl3) δ 140.7, 138.4, 135.1, 132.4, 129.5, 128.5, 128.4, 127.8, 127.7, 127.6, 73.2, 68.2, 60.8, 39.1, 35.9, 34.5; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C18H21Cl2O2: 339.0913, found 339.0908.
5-(allyloxy)-3-(2,4-dichlorophenyl)pentan-1-ol (11c)
44.2 mg, 51% 1H NMR (600 MHz, CDCl3) δ 7.37 (s, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.18 (d, J = 8.3 Hz, 1H), 5.85 (ddt, J = 15.8, 10.3, 5.6 Hz, 1H), 5.21 (d, J = 17.2 Hz, 1H), 5.13 (d, J = 10.4 Hz, 1H), 3.87 (d, J = 4.5 Hz, 2H), 3.57 − 3.43 (m, 3H), 3.31 (q, J = 7.1 Hz, 1H), 3.24 (q, J = 8.9, 7.9 Hz, 1H), 1.99 (m, 2H), 1.85 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 140.7, 135.1, 134.8, 132.4, 129.5, 129.1, 127.6, 117.1, 72.1, 68.1, 60.8, 39.1, 36.0, 34.4; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C14H19Cl2O2: 289.0757, found 289.0754.
((1r,3r)-3-bromoadamantan-1-yl)methyl trifluoromethanesulfonate
To a solution of alcohol (1.10 mmol, 270 mg, 1.0 equiv) in dichloromethane (10 mL) were added Tf2O (1.32 mmol, 372 mg, 1.2 equiv) and 2,6-lutidine (1.43 mmol, 153 mg, 1.3 equiv) at 0 °C. After stirring for 3 h, the reaction was concentrated in vacuo, re-dissolved in Et2O, washed with aq. citric acid (10%), aq. NaHCO3, brine, dried over MgSO4, and again concentrated. The obtained crude product (377 mg, 91%, pinkish oil) was used for the synthesis of 11d without further purification. The alcohol, starting material, was prepared from the corresponding acid according to the literature procedure.27
1H NMR (500 MHz, CDCl3) δ 4.11 (s, 2H), 2.35 (d, J = 11.7 Hz, 2H), 2.30 − 2.21 (m, 4H), 2.18 (s, 2H), 1.78 − 1.71 (m, 1H), 1.69 − 1.62 (m, 1H), 1.59 (m, 4H); 13C NMR (126 MHz, CDCl3) δ 118.8 (q, JC-F = 320 Hz), 84.2, 62.7, 49.6, 48.2, 38.5, 36.7, 34.7, 31.5; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C12H17BrF3O3S: 375.9956, found none.
5-(((1r,3r)-3-bromoadamantan-1-yl)methoxy)-3-(2,4-dichlorophenyl)pentan-1-ol (11d)
98.4 mg, 51% 1H NMR (600 MHz, CDCl3) δ 7.37 (s, 1H), 7.23 (d, J = 8.3 Hz, 1H), 7.18 (d, J = 8.3 Hz, 1H), 3.51 (m, 3H), 3.24 (m, 2H), 2.91 (s, 2H), 2.36 – 2.21 (m, 4H), 2.12 (m, 4H), 2.03 – 1.91 (m, 2H), 1.91 – 1.75 (m, 2H), 1.67 (m, 1H), 1.60 (m, 2H), 1.47 (m, 4H); 13C NMR (151 MHz, CDCl3) δ 140.9, 135.0, 132.3, 129.5, 129.1, 127.6, 80.6, 69.4, 66.4, 60.8, 51.3, 48.9, 39.5, 39.3, 37.9, 35.9, 35.3, 34.6, 32.3; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C22H30BrCl2O2: 475.0801, found 475.0807.
General procedure for amide coupling
EDC coupling
To a mixture of acid (1.0 equiv), amine (1.2 equiv), HOBt (2.0 equiv), NMM (2.0 equiv) in DCM (0.1 M) was added EDC (2.0 equiv) at 0 °C. The reaction was warmed to ambient temperature and stirred overnight. Upon evaporation, crude mixture was re-dissolved in EtOAc, washed with aq. HCl (1 M), aq. NaHCO3, brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by column chromatography (Hexanes/EtOAc) to afford the amide product as white solid.
Coupling via acid chloride formation
To a solution of acid 8 (1.0 equiv), DMF (1 drop) in chloroform (0.1 M) at 0 °C was added (COCl)2 (4.0 equiv). The resulting solution was warmed to ambient temperature and stirred for 1 h. Upon evaporation, this acid chloride was dissolved in chloroform (0.1 M). The solution was cooled to 0 °C, followed by the addition of TEA (1.2 equiv), DMAP (1 crystal) and aniline (1.0 equiv), and stirred at ambient temperature overnight. Upon evaporation, the crude mixture was dissolved in EtOAc/H2O. The partitioned organic layer was washed with brine, dried over MgSO4, and concentrated. The crude product was purified by column chromatography (Hexanes/EtOAc) to provide amide product as white solid.
Methyl 3-(2,4-dichlorophenyl)-5-((4-fluoro-2-methoxyphenyl)amino)-5-oxopentanoate (9a)
The aniline was prepared from the corresponding nitro compound according to the literature procedure.28 55.0 mg, 66% 1H NMR (600 MHz, CDCl3) δ 8.20 (t, J = 6.8 Hz, 1H), 7.56 (s, 1H), 7.39 (s, 1H), 7.21 (q, J = 8.2 Hz, 2H), 6.60 (m, 2H), 4.18 (q, J = 6.5 Hz, 1H), 3.83 (s, 3H), 3.60 (s, 3H), 2.90 (m, 1H), 2.84 − 2.75 (m, 3H); 13C NMR (151 MHz, CDCl3) δ 172.0, 168.4, 159.3 (JC-F = 243 Hz), 149.1 (JC-F = 9.06 Hz), 138.4, 134.6, 133.3, 130.0, 129.1, 127.5, 123.5 (JC-F = 3.02 Hz), 120.9 (JC-F = 9.06 Hz), 106.8 (JC-F = 22.7 Hz), 98.8 (JC-F = 27.2Hz), 56.1, 51.9, 41.8, 38.1, 35.0; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C19H19Cl2FNO4: 414.0670, found 414.0678.
Methyl 3-(2,4-dichlorophenyl)-5-oxo-5-(o-tolylamino)pentanoate (9b)
114 mg, 84% 1H NMR (600 MHz, CDCl3) δ 7.71 (d, J = 8.0 Hz, 1H), 7.41 (s, 1H), 7.23 (m, 2H), 7.17 (m, 2H), 7.07 (m, 2H), 4.21 – 4.11 (m, 1H), 3.62 (s, 3H), 2.86 (m, 4H), 2.14 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 172.2, 168.8, 138.3, 135.5, 134.5, 133.5, 130.6, 130.0, 129.2, 129.1, 127.6, 126.9, 125.5, 123.2, 52.0, 41.5, 38.1, 35.4, 17.7; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C19H20Cl2NO3: 380.0815, found 380.0811.
Methyl 5-((4-bromophenyl)amino)-3-(2,4-dichlorophenyl)-5-oxopentanoate (9c)
159 mg, 71% 1H NMR (600 MHz, CDCl3) δ 7.51 – 7.30 (m, 6H), 7.24 – 7.16 (m, 2H), 4.19 – 4.09 (m, 1H), 3.62 (s, 3H), 2.92 – 2.69 (m, 4H); 13C NMR (151 MHz, CDCl3) δ 172.4, 168.7, 138.2, 136.8, 134.4, 133.5, 132.1, 130.0, 129.1, 127.6, 121.5, 117.1, 52.1, 41.6, 38.1, 35.0; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C18H17BrCl2NO3: 443.9763, found 443.9758.
Methyl 3-(2,4-dichlorophenyl)-5-((4-fluorophenethyl)amino)-5-oxopentanoate (9d)
56.7 mg, 80% 1H NMR (600 MHz, CDCl3) δ 7.37 (s, 1H), 7.19 (d, J = 8.4 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 7.06 (t, J = 6.2 Hz, 2H), 6.97 (t, J = 8.1 Hz, 2H), 5.46 (s, 1H), 4.03 (p, J = 6.9 Hz, 1H), 3.59 (s, 3H), 3.43 (dh, J = 14.1, 6.4 Hz, 2H), 2.80 (dd, J = 16.1, 6.3 Hz, 1H), 2.70 (q, J = 7.3 Hz, 3H), 2.53 (h, J = 7.6 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 172.1, 170.3, 161.8 (JC-F = 246 Hz), 138.5, 134.5, 134.4 (JC-F = 3.02 Hz), 133.3, 130.2 (JC-F = 7.55 Hz), 129.9, 129.1, 127.5, 115.6 (JC-F = 21.1 Hz), 51.9, 40.8, 40.7, 38.1, 35.1, 34.9; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C20H21Cl2FNO3: 412.0877, found 412.0875.
Methyl 3-(2,4-dichlorophenyl)-5-((4-methoxyphenethyl)amino)-5-oxopentanoate (9e)
58.8 mg, 81% 1H NMR (600 MHz, CDCl3) δ 7.37 (s, 1H), 7.19 (d, J = 8.4 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 7.01 (d, J = 1.5 Hz, 2H), 6.83 (d, J = 7.8 Hz, 2H), 5.45 (s, 1H), 4.03 (p, J = 7.4 Hz, 1H), 3.79 (s, 3H), 3.59 (s, 3H), 3.49 – 3.36 (m, 2H), 2.81 (dd, J = 15.9, 6.6 Hz, 1H), 2.75 – 2.62 (m, 3H), 2.52 (s, 2H); 13C NMR (151 MHz, CDCl3) δ 172.1, 170.3, 158.4, 138.5, 134.5, 133.2, 130.7, 129.9, 129.7, 129.1, 127.5, 114.2, 55.4, 51.9, 40.8, 40.8, 38.1, 35.1, 34.7; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C21H24Cl2NO4: 424.1077, found 424.1080.
Methyl 5-((2-cyclopropylethyl)amino)-3-(2,4-dichlorophenyl)-5-oxopentanoate (9f)
60.6 mg, 85% 1H NMR (600 MHz, CDCl3) δ 7.38 (s, 1H), 7.19 (s, 2H), 5.56 (s, 1H), 4.07 (pd, J = 7.2, 1.9 Hz, 1H), 3.60 (s, 3H), 3.25 (q, J = 6.5 Hz, 2H), 2.86 (dd, J = 15.9, 6.7 Hz, 1H), 2.75 (dd, J = 15.9, 7.5 Hz, 1H), 2.57 (h, J = 7.4 Hz, 2H), 1.30 (q, J = 6.9 Hz, 2H), 0.58 – 0.49 (m, 1H), 0.42 (d, J = 7.8 Hz, 2H), 0.01 (d, J = 4.9 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 172.1, 170.2, 138.5, 134.5, 133.3, 129.9, 129.2, 127.5, 51.9, 40.9, 39.9, 38.1, 35.2, 34.4, 8.6, 4.3, 4.3; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C17H22Cl2NO3: 358.0971, found 358.0976.
General procedure for the synthesis of hydroxamic acid23
To a solution of methyl ester 9 (1.0 equiv) and KCN (3.0 equiv) in THF/MeOH (1 mL/1 mL) was added aq. NH2OH (50%, 0.5 mL). This solution was stirred overnight at ambient temperature. Upon evaporation of solvent, crude mixture was purified by preparative TLC (MeOH/dichloromethane) to afford hydroxamic acid 3.
3-(2,4-Dichlorophenyl)-N1-(4-fluoro-2-methoxyphenyl)-N5-hydroxypentanediamide (3a)
39.4 mg, 45% 1H NMR (600 MHz, DMSO-d6) δ 10.39 (s, 1H), 9.12 (s, 1H), 8.71 (s, 1H), 7.68 (t, J = 7.7 Hz, 1H), 7.53 (s, 1H), 7.41 (q, J = 8.9 Hz, 2H), 6.93 (d, J = 10.8 Hz, 1H), 6.68 (t, J = 9.0 Hz, 1H), 4.03 (p, J = 7.9 Hz, 1H), 3.79 (d, J = 2.0 Hz, 3H), 2.72 (d, J = 7.5 Hz, 2H), 2.40 (dd, J = 14.9, 7.7 Hz, 1H), 2.32 (dd, J = 14.8, 6.8 Hz, 1H); 13C NMR (151 MHz, DMSO-d6) δ 169.0, 166.8, 159.1 (JC-F = 242 Hz), 151.5, 139.9, 139.2, 134.1, 131.4, 128.7, 128.0, 127.2, 124.9, 105.8 (JC-F = 21.1 Hz), 99.6 (JC-F = 27.2 Hz), 56.1, 40.4, 36.7, 34.4; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C18H18Cl2N2O4: 415.0622, found 415.0616.
3-(2,4-Dichlorophenyl)-N1-hydroxy-N5-(o-tolyl)pentanediamide (3b)
The titled compound was purified by preparative HPLC (acetonitrile/water) due to its limited solubility in organic media.
80.4 mg, 70% 1H NMR (600 MHz, DMSO-d6) δ 10.31 (s, 1H), 9.16 (s, 1H), 8.62 (s, 1H), 7.40 (s, 1H), 7.32 – 7.24 (m, 2H), 7.07 (d, J = 7.6 Hz, 1H), 7.01 (d, J = 7.2 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 6.90 (t, J = 7.2 Hz, 1H), 3.97 – 3.85 (m, 1H), 2.56 (m, 2H), 2.32 – 2.17 (m, 2H), 1.86 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 168.9, 167.0, 139.8, 136.2, 134.2, 131.9, 131.6, 130.3, 129.9, 128.9, 127.3, 126.0, 125.4, 125.3, 40.3, 37.1, 34.6, 17.7; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C18H19Cl2N2O4: 381.0767, found 381.0763.
N1-(4-Bromophenyl)-3-(2,4-dichlorophenyl)-N5-hydroxypentanediamide (3c)
The titled compound was purified by recrystallization from MeOH.
36 mg, 80% 1H NMR (600 MHz, DMSO-d6) δ 10.39 (s, 1H), 10.06 (s, 1H), 8.71 (s, 1H), 7.53 (s, 1H), 7.50 – 7.41 (m, 4H), 7.38 (s, 2H), 4.13 – 3.96 (m, 1H), 2.70 (d, J = 7.1 Hz, 2H), 2.40 (dd, J = 14.4, 8.1 Hz, 1H), 2.34 (dd, J = 14.5, 6.9 Hz, 1H); 13C NMR (151 MHz, DMSO-d6) δ 169.1, 166.7, 139.7, 138.3, 134.0, 131.5, 129.6, 128.8, 127.2, 124.9, 121.0, 114.6, 40.7, 36.6, 34.1; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C17H16BrCl2N2O3: 444.9716, found 444.9717.
3-(2,4-Dichlorophenyl)-N1-(4-fluorophenethyl)-N5-hydroxypentanediamide (3d)
28.1 mg, 60% 1H NMR (600 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.69 (s, 1H), 7.88 (s, 1H), 7.52 (s, 1H), 7.39 – 7.26 (m, 2H), 7.12 (t, J = 6.1 Hz, 2H), 7.06 (t, J = 8.2 Hz, 2H), 3.93 (p, J = 7.7 Hz, 1H), 3.16 (d, J = 5.9 Hz, 2H), 2.57 (t, J = 6.9 Hz, 2H), 2.37 (m, 3H), 2.24 (dd, J = 14.5, 6.6 Hz, 1H); 13C NMR (151 MHz, DMSO-d6) δ 169.7, 166.8, 160.8 (JC-F = 240 Hz), 139.9, 135.6 (JC-F = 3.02 Hz), 134.0, 131.3, 130.3 (JC-F = 7.55 Hz), 129.6, 128.7, 127.1, 114.9 (JC-F = 21.1 Hz), 40.1, 40.0, 36.6, 34.2, 34.1; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C19H20Cl2FN2O3: 413.0829, found 413.0823.
3-(2,4-Dichlorophenyl)-N1-hydroxy-N5-(4-methoxyphenethyl)pentanediamide (3e)
22.7 mg, 46% 1H NMR (600 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.69 (s, 1H), 7.87 (s, 1H), 7.53 (s, 1H), 7.39 – 7.28 (m, 2H), 7.01 (d, J = 7.4 Hz, 2H), 6.81 (d, J = 7.3 Hz, 2H), 4.00 – 3.90 (m, 1H), 3.71 (s, 3H), 3.13 (d, J = 6.0 Hz, 2H), 2.55 – 2.47 (m, 2H), 2.45 – 2.30 (m, 3H), 2.25 (dd, J = 14.3, 5.6 Hz, 1H); 13C NMR (151 MHz, DMSO-d6) δ 169.6, 166.8, 157.6, 139.9, 134.0, 131.3, 131.3, 129.6, 129.5, 128.7, 127.1, 113.7, 55.0, 40.3, 40.01, 36.6, 34.19, 34.17; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C20H23Cl2N2O4: 425.1029, found 425.1023.
N1-(2-Cyclopropylethyl)-3-(2,4-dichlorophenyl)-N5-hydroxypentanediamide (3f)
14.3 mg, 40% 1H NMR (600 MHz, DMSO-d6) δ 10.37 (s, 1H), 8.68 (s, 1H), 7.79 (s, 1H), 7.51 (s, 1H), 7.39 – 7.29 (m, 2H), 4.03 – 3.87 (m, 1H), 2.99 (s, 2H), 2.37 (m, 3H), 2.28 (dd, J = 14.8, 6.8 Hz, 1H), 1.16 (d, J = 6.8 Hz, 2H), 0.51 (s, 1H), 0.32 (d, J = 7.3 Hz, 2H), −0.04 (s, 2H); 13C NMR (151 MHz, DMSO-d6) δ 169.5, 166.9, 139.9, 134.0, 131.3, 129.6, 128.7, 127.1, 40.1, 38.6, 36.6, 34.3, 34.0, 8.4, 4.1; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C16H21Cl2N2O3: 359.0924, found 359.0926.
General procedure to synthesize hydroxamic acid from alcohol 11
To a solution of alcohol 11 (1.0 equiv) in acetone (0.1 M) was added Jones’ reagent (2.0 M solution, 2.0 equiv) at 0 °C.29 The reaction was stirred at ambient temperature for 1 h, and quenched by the addition of IPA (a few drops). The organic layer was washed with water, brine, dried over MgSO4, and concentrated in vacuo. The resulting acid was dissolved in THF (0.1 M), and diazomethane (excess) in Et2O was added at 0 °C until the yellow color stays in the solution. After stirring overnight, upon evaporation, crude methyl ester was subjected to the conditions above to form hydroxamic acid as reddish oil.
3-(2,4-dichlorophenyl)-5-(4-fluorophenethoxy)-N-hydroxypentanamide (4a)
20.0 mg, 42% 1H NMR (600 MHz, DMSO-d6) δ 10.35 (s, 1H), 8.70 (s, 1H), 7.53 (d, J = 2.3 Hz, 1H), 7.38 (dd, J = 8.4, 2.2 Hz, 1H), 7.32 (d, J = 8.5 Hz, 1H), 7.21 (dd, J = 8.5, 5.7 Hz, 2H), 7.07 (t, J = 8.9 Hz, 2H), 3.66 (p, J = 7.6 Hz, 1H), 3.49 – 3.39 (m, 2H), 3.23 (dt, J = 12.5, 6.3 Hz, 1H), 3.21 – 3.13 (m, 1H), 2.72 (t, J = 6.8 Hz, 2H), 2.29 (qd, J = 14.5, 7.6 Hz, 2H), 1.84 (dq, J = 13.0, 6.6 Hz, 1H), 1.75 (dt, J = 14.8, 7.3 Hz, 1H); 13C NMR (151 MHz, DMSO) δ 167.0, 160.8 (JC-F = 242 Hz), 140.5, 135.2 (JC-F = 3.02 Hz), 134.1, 131.3, 130.5 (JC-F = 9.06 Hz), 129.8, 128.7, 127.4, 114.8 (JC-F = 21.1 Hz), 70.9, 67.7, 40.4, 37.6, 34.6, 34.5; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C19H21Cl2FNO3: 400.0877, found 400.0895.
5-(benzyloxy)-3-(2,4-dichlorophenyl)-N-hydroxypentanamide (4b)
17 mg, 36% 1H NMR (600 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.69 (s, 1H), 7.52 (d, J = 1.8 Hz, 1H), 7.38 – 7.32 (m, 2H), 7.30 (t, J = 7.3 Hz, 2H), 7.23 (m, 3H), 4.38 – 4.29 (m, 2H), 3.72 (p, J = 7.7 Hz, 1H), 3.28 (m, 1H), 3.23 (m, 1H), 2.29 (hept, J = 7.6 Hz, 2H), 1.96 – 1.86 (m, 1H), 1.86 – 1.77 (m, 1H); 13C NMR (151 MHz, DMSO) δ 167.0, 140.4, 138.4, 134.2, 131.3, 129.8, 128.7, 128.1, 127.42, 127.35, 127.3, 71.8, 67.3, 40.4, 37.7, 34.4; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C18H20Cl2NO3: 368.0815, found 368.0813.
5-(allyloxy)-3-(2,4-dichlorophenyl)-N-hydroxypentanamide (4c)
11.2 mg, 27% 1H NMR (600 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.69 (s, 1H), 7.53 (s, 1H), 7.37 (d, J = 13.4 Hz, 2H), 5.80 (td, J = 10.9, 5.3 Hz, 1H), 5.16 (d, J = 17.2 Hz, 1H), 5.07 (d, J = 10.3 Hz, 1H), 3.81 (s, 2H), 3.75 – 3.61 (m, 1H), 3.20 (dt, J = 28.2, 8.2 Hz, 2H), 2.38 – 2.20 (m, 2H), 1.87 (dd, J = 13.7, 6.8 Hz, 1H), 1.78 (p, J = 7.1 Hz, 1H); 13C NMR (151 MHz, DMSO) δ 167.0, 140.5, 135.2, 134.1, 131.3, 129.7, 128.7, 127.4, 116.1, 70.8, 67.1, 37.7, 34.5, 34.2; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C14H18Cl2NO3: 318.0658, found 318.0658.
5-((3-bromoadamantan-1-yl)methoxy)-3-(2,4-dichlorophenyl)-N-hydroxypentanamide (4d)
53.5 mg, 55%1H NMR (600 MHz, DMSO-d6) δ 10.35 (s, 1H), 8.69 (s, 1H), 7.50 (s, 1H), 7.35 (q, J = 8.6 Hz, 2H), 3.68 (s, 1H), 3.17 (m, 2H), 2.92 – 2.77 (m, 2H), 2.23 (d, J = 12.2 Hz, 4H), 2.16 (d, J = 11.2 Hz, 2H), 2.02 (m, 4H), 1.84 (s, 1H), 1.77 (s, 1H), 1.62 (d, J = 12.4 Hz, 1H), 1.52 (d, J = 12.0 Hz, 1H), 1.40 (d, J = 11.9 Hz, 2H), 1.34 (s, 2H); 13C NMR (151 MHz, DMSO-d6) δ 167.0, 140.6, 134.1, 131.2, 129.8, 128.7, 127.3, 79.4, 68.6, 67.9, 50.8, 48.4, 39.0, 38.0, 37.0, 34.5, 34.3, 31.7, 29.1; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C22H29BrCl2NO3: 504.0702, found 504.0697.
General procedure to synthesize carbamate prodrug23,26
To a solution of hydroxamic acid (1.0 equiv) in acetonitrile (0.05 M) was added CDI (1.0 equiv) at 0 °C. After 0.5 h, BnNH2 was added to the solution, which was stirred overnight at ambient temperature. Upon evaporation, crude mixture was subjected to column chromatography (Hexanes/EtOAc) to provide carbamate prodrug.
Hydroxamic acid 1 and 2 were synthesized based on the literature procedure.17,30
(E)-N-((benzylcarbamoyl)oxy)-3-(2,4-dichlorophenyl)acrylamide (1’)
200 mg, 71% 1H NMR (500 MHz, D2O) δ 11.89 (s, 1H), 8.39 (s, 1H), 7.86 – 7.71 (m, 3H), 7.51 (dd, J = 8.5, 2.0 Hz, 1H), 7.40 – 7.22 (m, 5H), 6.63 (d, J = 15.8 Hz, 1H), 4.26 (d, J = 6.1 Hz, 2H); 13C NMR (151 MHz, D2O) δ 162.7, 155.2, 139.0, 135.0, 134.6, 134.3, 131.3, 129.5, 129.2, 128.4, 128.1, 127.1, 127.0, 121.5, 44.2; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C17H15Cl2N2O3: 365.0454, found 365.0450.
N-((benzylcarbamoyl)oxy)-3-(2,4-dichlorophenyl)-5-hydroxypentanamide (2’)
33 mg, 34% 1H NMR (400 MHz, DMSO-d6) δ 11.42 (s, 1H), 8.19 (t, J = 6.1 Hz, 1H), 7.53 (d, J = 1.5 Hz, 1H), 7.37 (s, 2H), 7.36 – 7.29 (m, 2H), 7.25 (d, J = 8.0 Hz, 3H), 4.42 (t, J = 5.0 Hz, 1H), 4.21 (d, J = 6.1 Hz, 2H), 3.65 (s, 1H), 3.25 (s, 2H), 2.43 (d, J = 7.8 Hz, 2H), 1.78 (d, J = 33.4 Hz, 2H); 13C NMR (151 MHz, D2O) δ 168.1, 155.2, 140.5, 139.0, 134.0, 131.2, 129.8, 128.7, 128.3, 127.4, 127.03, 126.98, 58.4, 44.1, 37.4, 37.2, 33.9; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C19H21Cl2N2O4: 411.0873, found 411.0865.
N1-((benzylcarbamoyl)oxy)-3-(2,4-dichlorophenyl)-N5-(4-fluoro-2-methoxyphenyl)pentanediamide (3a’)
7.1 mg, 43% 1H NMR (600 MHz, CDCl3) δ 10.20 (s, 1H), 8.07 (dd, J = 8.9, 6.3 Hz, 1H), 7.61 (s, 1H), 7.41 (s, 1H), 7.34 (t, J = 7.5 Hz, 2H), 7.29 (dt, J = 6.6, 3.3 Hz, 3H), 7.15 (s, 2H), 6.64 (td, J = 8.4, 2.4 Hz, 1H), 6.58 (dd, J = 10.1, 2.6 Hz, 1H), 5.65 (s, 1H), 4.41 (d, J = 5.9 Hz, 2H), 4.12 (m, 1H), 3.76 (s, 3H), 3.05 (m, 1H), 2.96 – 2.82 (m, 2H), 2.67 – 2.48 (m, 1H); 13C NMR (151 MHz, CDCl3) δ 171.3, 169.9, 159.9 (JC-F = 246 Hz), 155.4, 149.8, 138.0, 137.3, 134.1, 133.5, 129.8, 129.4, 128.9, 128.0, 127.7, 127.5, 122.7, 122.3, 107.0 (JC-F = 21.1 Hz), 99.0 (JC-F = 27.2 Hz), 56.1, 45.7, 39.9, 35.8, 35.6; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C26H25Cl2FN3O5: 548.1150, found 548.1138.
N1-((benzylcarbamoyl)oxy)-3-(2,4-dichlorophenyl)-N5-(o-tolyl)pentanediamide (3b’)
16.4 mg, 83% 1H NMR (600 MHz, DMSO-d6) δ 11.46 (s, 1H), 9.29 (s, 1H), 8.23 (s, 1H), 7.55 (s, 1H), 7.46 (d, J = 7.7 Hz, 1H), 7.41 (d, J = 8.3 Hz, 1H), 7.33 (m, 2H), 7.23 (m, 4H), 7.14 (d, J = 7.1 Hz, 1H), 7.09 (t, J = 7.3 Hz, 1H), 7.03 (t, J = 6.7 Hz, 1H), 4.22 (s, 2H), 4.06 (s, 1H), 2.78 (d, J = 8.3 Hz, 2H), 2.50 (m, 2H), 1.99 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 168.7, 167.7, 155.2, 139.5, 139.0, 136.1, 134.0, 131.8, 131.6, 130.2, 129.7, 128.8, 128.5, 128.3, 127.0, 127.0, 125.8, 125.2, 125.2, 44.1, 40.1, 36.7, 34.3, 17.6; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C26H26Cl2N3O4: 514.1295, found 514.1294.
N1-((benzylcarbamoyl)oxy)-N5-(4-bromophenyl)-3-(2,4-dichlorophenyl)pentanediamide (3c’)
18.6 mg, 87% 1H NMR (600 MHz, DMSO-d6) δ 11.47 (s, 1H), 10.07 (s, 1H), 8.22 (s, 1H), 7.54 (s, 1H), 7.48 – 7.35 (m, 6H), 7.32 (t, J = 7.2 Hz, 2H), 7.24 (d, J = 6.5 Hz, 3H), 4.21 (s, 2H), 4.04 (ddt, J = 15.1, 9.0, 4.5 Hz, 1H), 2.77 (s, 2H), 2.50 (s, 2H); 13C NMR (151 MHz, DMSO-d6) δ 169.0, 167.7, 155.2, 139.5, 139.0, 138.3, 134.0, 131.5, 131.5, 129.6, 128.8, 128.3, 127.3, 127.0, 127.0, 121.0, 114.7, 44.1, 40.3, 36.5, 33.9; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C25H23BrCl2N3O4: 578.0243, found 578.0240.
N1-((benzylcarbamoyl)oxy)-3-(2,4-dichlorophenyl)-N5-(4-fluorophenethyl)pentanediamide (3d’)
9.4 mg, 77% 1H NMR (600 MHz, CDCl3) δ 10.22 (s, 1H), 7.40 – 7.27 (m, 5H), 7.16 (d, J = 6.6 Hz, 2H), 7.08 – 7.00 (m, 2H), 6.97 (d, J = 8.8 Hz, 2H), 5.68 (d, J = 10.7 Hz, 2H), 4.40 (d, J = 6.1 Hz, 2H), 3.94 (t, J = 7.3 Hz, 1H), 3.41 (dt, J = 17.7, 6.8 Hz, 2H), 2.91 – 2.59 (m, 5H), 2.59 – 2.41 (m, 1H); 13C NMR (151 MHz, CDCl3) δ 171.8, 169.9, 161.8 (JC-F = 245 Hz), 155.3, 138.2, 137.3, 134.1, 134.1, 133.5, 130.2 (JC-F = 7.55 Hz), 129.8, 129.3, 128.9, 128.0, 127.6, 127.4, 115.6 (JC-F = 21.1 Hz), 45.7, 40.7, 38.9, 35.9, 35.5, 34.7; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C27H27Cl2FN3O4: 546.1357, found 546.1361.
N-((benzylcarbamoyl)oxy)-3-(2,4-dichlorophenyl)-5-(4-fluorophenethoxy)pentanamide(4a’)
9.4 mg, 77% 1H NMR (600 MHz, CDCl3) δ 9.20 (s, 1H), 7.39 – 7.26 (m, 6H), 7.16 (m, 3H), 7.03 (d, J = 8.4 Hz, 1H), 6.97 (t, J = 8.6 Hz, 2H), 5.55 (s, 1H), 4.39 (d, J = 5.6 Hz, 2H), 3.76 (m, 1H), 3.65 – 3.51 (m, 2H), 3.34 (s, 2H), 2.83 (t, J = 6.4 Hz, 2H), 2.61 (dd, J = 14.1, 8.1 Hz, 1H), 2.49 (dd, J = 13.2, 4.7 Hz, 1H), 2.06 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 170.0, 161.7 (JC-F = 245 Hz), 155.0, 139.0, 137.3, 134.7, 134.6, 132.9, 130.3 (JC-F = 7.55 Hz), 129.7, 129.4, 128.9, 128.0, 127.7, 127.3, 115.3 (JC-F = 21.1 Hz), 72.0, 68.9, 45.6, 37.9, 35.8, 35.5, 32.8; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C27H28Cl2FN2O4: 533.1405, found 533.1399.
N-((benzylcarbamoyl)oxy)-5-(benzyloxy)-3-(2,4-dichlorophenyl)pentanamide (4b’)
9.5 mg, 46% 1H NMR (600 MHz, CDCl3) δ 9.36 (s, 1H), 7.36 – 7.30 (m, 5H), 7.28 (d, J = 8.9 Hz, 6H), 7.14 (dd, J = 8.4, 2.2 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 5.68 (s, 1H), 4.49 – 4.39 (m, 2H), 4.36 (d, J = 5.9 Hz, 2H), 3.84 (p, J = 6.3 Hz, 1H), 3.40 (m, 2H), 2.64 (dd, J = 14.3, 8.0 Hz, 1H), 2.52 (m, 1H), 2.07 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 169.8, 155.2, 139.0, 138.0, 137.4, 134.7, 132.9, 129.7, 129.4, 128.9, 128.6, 128.0, 127.90, 127.88, 127.6, 127.4, 73.3, 68.0, 45.5, 37.9, 35.6, 33.3; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C26H27Cl2N2O4: 501.1342, found 501.1339.
N-((benzylcarbamoyl)oxy)-5-(((1r,3r)-3-bromoadamantan-1-yl)methoxy)-3-(2,4-dichlorophenyl)pentanamide (4d’)
5.0 mg, 50% 1H NMR (600 MHz, CDCl3) δ 9.05 (s, 1H), 7.35 (dd, J = 16.2, 9.0 Hz, 3H), 7.29 (dd, J = 13.0, 7.2 Hz, 3H), 7.20 (q, J = 8.2 Hz, 2H), 5.46 (s, 1H), 4.40 (d, J = 4.8 Hz, 2H), 3.87 – 3.77 (m, 1H), 3.40 – 3.23 (m, 2H), 2.95 (q, J = 9.0 Hz, 2H), 2.68 – 2.52 (m, 2H), 2.30 (d, J = 11.8 Hz, 2H), 2.25 (d, J = 11.8 Hz, 2H), 2.12 (m, 4H), 1.68 (d, J = 12.7 Hz, 1H), 1.65 – 1.42 (m, 7H); 13C NMR (151 MHz, CDCl3) δ 172.0, 155.7, 139.8, 137.9, 135.3, 133.6, 130.5, 130.2, 129.7, 128.7, 128.4, 128.2, 81.5, 70.2, 61.3, 52.0, 49.5, 46.4, 40.1, 39.2, 38.6, 35.9, 34.1, 33.0, 30.6; HRMS (ESI-TOF) m/e calc’d for [M+H]+ C30H36BrCl2N2O4: 637.1230, found 637.1232.
4.2 Biology
4.3.1 SNAPtide assay and 66mer assay
Recombinant Botulinum neurotoxin light chain A (1–425) was prepared as previously described.31 SNAPtide assay was conducted according to the literature procedure with the enzyme concentration of 37 nM.31 66mer assay was conduted according to the literature procedure with the enzyme concentration of 0.5 nM. Prism 5 was used for Ki determination.25
4.3.2 Cell assay
Pure Botulinum neurotoxin (BoNT) A was prepared from C. botulinum strains Hall A hyper as previously described.32 The toxin was dissolved in phosphate buffered saline, pH 7.4 and 40 % glycerol, and stored at −20°C until use. Activity of the BoNT/A preparation was determined by the mouse bioassay,33,34 and specific toxicity was about 1.25 × 108 mouse LD50 Units/mg. The inhibitors were dissolved in 100 % DMSO to 100 mM and stored at 4°C.
The hiPSC derived neurons and culture medium were purchased from Cellular Dynamics International (Madison, WI), and cultured in 96-well plates as described for 5 days prior to the assay.35 For the inhibition assay, 200 LD50 Units of BoNT/A1 was added to the cells in 50 µl stimulation medium (modified neurobasal containing 2.2 mM CaCl2 and 56 mM KCl (Invitrogen) and supplemented with B27 and glutamax), and the cells were incubated at 37°C in a humidified 5 % CO2 atmosphere for 7.5 min. The toxin was removed and cells were washed 3 times in 200 µl of culture medium (provided by Cellular Dynamics), and the inhibitors were added in culture medium, 1 % DMSO at the indicated concentrations. Cells were incubated for 8 h at 37°C, 5 % CO2 to allow for SNAP-25 cleavage, and the inhibitor mixtures were aspirated and cells lysed in 50 µl of 1 × LDS lysis buffer (Invitrogen). The samples were analyzed by Western blot using a monoclonal anti-SNAP-25 antibody (Synaptic Systems, Germany) as described previously,36,37 except that Phosphaglo chemiluminescent reagent (KPL) was used and the bands visualized on a Fotoanalyst FX (Fotodyne) equipped with a CCD camera and GraphQuant software (Fotodyne) for densitometry. GrahPad Prism 6 software was used for graph creation and data analysis.
Acknowledgements
The authors gratefully acknowledge support of this project by the National Institute of Allergy and Infectious Diseases, National Institute of Health and the Department of Health and Human Services under contract number AI080671. The authors thank Regina C. M. Whitemarsh for harvesting cells. B.B. thanks the Spanish Ministry of Science and Innovation for her FPU fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Burnett JC, Henchal EA, Schmaljohn AL, Bavari S. Nat. Rev. Drug Discov. 2005;4:281. doi: 10.1038/nrd1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hackett R, Kam PC. Med. Chem. 2007;3:333. doi: 10.2174/157340607781024438. [DOI] [PubMed] [Google Scholar]
- 3.Truong DD, Jost WH. Parkinsonism Relat. Disord. 2006;12:331. doi: 10.1016/j.parkreldis.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 4.Josko D. Clin. Lab. Sci. 2004;17:30. [PubMed] [Google Scholar]
- 5.Montecucco C, Schiavo G. Q. Rev. Biophys. 1995;28:423. doi: 10.1017/s0033583500003292. [DOI] [PubMed] [Google Scholar]
- 6.Simpson LL. Annu. Rev. Pharmacol. Toxicol. 2004;44:167. doi: 10.1146/annurev.pharmtox.44.101802.121554. [DOI] [PubMed] [Google Scholar]
- 7.Gill DM. Microbiol. Rev. 1982;46:86. doi: 10.1128/mr.46.1.86-94.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blasi J, Chapman ER, Link E, Binz T, Yamasaki S, Decamilli P, Sudhof TC, Niemann H, Jahn R. Nature. 1993;365:160. doi: 10.1038/365160a0. [DOI] [PubMed] [Google Scholar]
- 9.Schantz EJ, Johnson EA. Microbiol. Rev. 1992;56:80. doi: 10.1128/mr.56.1.80-99.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Willis B, Eubanks LM, Dickerson TJ, Janda KD. Angew. Chem. Int. Ed. Engl. 2008;47:8360. doi: 10.1002/anie.200705531. [DOI] [PubMed] [Google Scholar]
- 11.Šilhár P, Silvaggi NR, Pellett S, Capkova K, Johnson EA, Allen KN, Janda KD. Bioorg. Med. Chem. 2013;21:1344. doi: 10.1016/j.bmc.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuniga JE, Schmidt JJ, Fenn T, Burnett JC, Arac D, Gussio R, Stafford RG, Badie SS, Bavari S, Brunger AT. Structure. 2008;16:1588. doi: 10.1016/j.str.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlimme S, Hauser AT, Carafa V, Heinke R, Kannan S, Stolfa DA, Cellamare S, Carotti A, Altucci L, Jung M, Sippl W. Chemmedchem. 2011;6:1193. doi: 10.1002/cmdc.201100007. [DOI] [PubMed] [Google Scholar]
- 14.Šilhár P, Eubanks L, Seki H, Pellett S, Javor S, Tepp W, Johnson E, Janda KD. J. Med. Chem. 2013 doi: 10.1021/jm400873n. Just Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boldt GE, Kennedy JP, Hixon MS, McAllister LA, Barbieri JT, Tzipori S, Janda KD. J. Comb. Chem. 2006;8:513. doi: 10.1021/cc060010h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silvaggi NR, Boldt GE, Hixon MS, Kennedy JP, Tzipori S, Janda KD, Allen KN. Chem. Biol. 2007;14:533. doi: 10.1016/j.chembiol.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 17.Stowe GN, Silhar P, Hixon MS, Silvaggi NR, Allen KN, Moe ST, Jacobson AR, Barbieri JT, Janda KD. Org. Lett. 2010;12:756. doi: 10.1021/ol902820z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lardy MA, LeBrun L, Bullard D, Kissinger C, Gobbi A. J. Chem. Inf. Model. 2012;52:1328. doi: 10.1021/ci200558e. [DOI] [PubMed] [Google Scholar]
- 19. [accessed Sep 2011];The OEChem Toolkit, OMEGA, ROCS, FRED, and SZYBKI are distributed by OpenEye Scientific Software, Santa Fe, NM. http://www.eyesopen.com.
- 20.McGann MR, Almond HR, Nicholls A, Grant JA, Brown FK. Biopolymers. 2003;68:76. doi: 10.1002/bip.10207. [DOI] [PubMed] [Google Scholar]
- 21.R: A Language and Environment for Statistical Computing, version 2.0.1. Vienna, Austria: R Development Core Team; 2004. [Google Scholar]
- 22.Efron BHT, Johnstone I, Tibshirani R. Ann. Statist. 2004;32:407. [Google Scholar]
- 23.Ho CY, Strobel E, Ralbovsky J, Galemmo RA. J. Org. Chem. 2005;70:4873. doi: 10.1021/jo050036f. [DOI] [PubMed] [Google Scholar]
- 24.Shine NR. 6504,006 B1. U.S. Patent. 2003
- 25.Capkova K, Hixon MS, McAllister LA, Janda KD. Chem. Commun. 2008:3525. doi: 10.1039/b808305c. [DOI] [PubMed] [Google Scholar]
- 26.Dubé P, Nathel NFF, Vetelino M, Couturier M, Aboussafy CLe, Pichette S, Jorgensen ML, Hardink M. Org. Lett. 2009;11:5622. doi: 10.1021/ol9023387. [DOI] [PubMed] [Google Scholar]
- 27.Yusuff N, Dore M, Joud C, Visser M, Springer C, Xie X, Herlihy K, Porter D, Toure BB. ACS Med. Chem. Lett. 2012;3:579. doi: 10.1021/ml300095a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nitabaru T, Nojiri A, Kobayashi M, Kumagai N, Shibasaki M. J. Am. Chem. Soc. 2009;131:13860. doi: 10.1021/ja905885z. [DOI] [PubMed] [Google Scholar]
- 29.Eisenbraun EJ. Org. Synth. 1965;45:28. [Google Scholar]
- 30.Boldt GE, Kennedy JP, Janda KD. Org. Lett. 2006;8:1729. doi: 10.1021/ol0603211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eubanks LM, Hixon MS, Jin W, Hong S, Clancy CM, Tepp WH, Baldwin MR, Malizio CJ, Goodnough MC, Barbieri JT, Johnson EA, Boger DL, Dickerson TJ, Janda KD. Proc. Natl. Acad. Sci. U.S.A. 2007;104:2602. doi: 10.1073/pnas.0611213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malizio CJ, Goodnough MC, Johnson EA. Methods Mol. Biol. 2000;145:27. doi: 10.1385/1-59259-052-7:27. [DOI] [PubMed] [Google Scholar]
- 33.Hatheway CL. In: Laboratory Diagnosis of Infectious Diseases - Principles and Practice. Balows AHJWJOM, Turano A, editors. Vol. 1. New York: Springer-Verlag; 1988. p. 111. [Google Scholar]
- 34.Schantz EJ, Kautter DA. J. Assoc. Off. Ana. Chem. 1978;61:96. [Google Scholar]
- 35.Whitemarsh RC, Strathman MJ, Chase LG, Stankewicz C, Tepp WH, Johnson EA, Pellett S. Toxicol. Sci. 2012;126:426. doi: 10.1093/toxsci/kfr354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pellett S, Tepp WH, Clancy CM, Borodic GE, Johnson EA. FEBS Lett. 2007;581:4803. doi: 10.1016/j.febslet.2007.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pellett S, Tepp WH, Toth SI, Johnson EA. J. Pharmacol. Toxicol. Methods. 2010;61:304. doi: 10.1016/j.vascn.2010.01.003. [DOI] [PubMed] [Google Scholar]