

Abstract
Whereas moderately increased cellular oxidative stress is supportive for cancerous growth of cells, excessive levels of reactive oxygen species (ROS) is detrimental to their growth and survival. We demonstrated that high ROS levels, via increased oxidized glutathione (GSSG), induce isoform-specific S-glutathionylation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) at the residue Cys206, which is located near the entrance to the 6-phosphofructo-2-kinase catalytic pocket. Upon this ROS-dependent, reversible, covalent modification, a marked decrease in its catalytic ability to synthesize the Fructose-2,6-bisphosphate (Fru-2,6-P2), the key glycolysis allosteric activator, was observed. This event was coupled to a decrease in glycolytic flux and an increase in glucose metabolic flux into the pentose phosphate pathway (PPP). This shift, in turn, caused an increase in reduced glutathione (GSH) and, ultimately, resulted in ROS detoxification inside HeLa cells. The ability of PFKFB3 to control the Fru-2,6-P2 levels in an ROS-dependent manner allows the PFKFB3-expressing cancer cells to continue energy metabolism with a reduced risk of excessive oxidative stress and, thereby, to support their cell survival and proliferation. This study provides a new insight into the roles of PFKFB3 as switch that senses and controls redox homeostasis in cancer in addition to its role in cancer glycolysis.
Keywords: reactive oxygen species, Warburg effect, TCA cycle, glutathione, pentose phosphate pathway
Introduction
Oncogenic cell transformation involves switching to aberrant metabolic patterns and mitochondrial impairment [1]. It has been well documented that cancer cells are prone to autonomous metabolism that utilizes glycolysis and glutaminolysis as major catabolic pathways for production of energy and anabolic precursors required for survival and rapid growth [2]. As first theorized by Warburg, the majority of pyruvate from glycolysis is converted to lactate [3]. However, a small but significant fraction of pyruvate still goes into the TCA cycle after conversion to acetyl-CoA as α-ketoglutarate produced from glutaminolysis does [4]. Persistent running of the TCA cycle in impaired mitochondria causes a fast buildup of reactive oxygen species (ROS) [5],[6]. Consequently, the levels of ROS in cancer cells are higher than those of normal cells, despite the fact that normal cells rely more heavily on oxidative phosphorylation than cancer cells do [7,8]. The ROS level elevated such a way is known to support tumor progression [9]. Nonetheless, even in cancer cells, ROS production must still be regulated below the detrimental level, so as not to face the fatal effects associated with excessive oxidative stress [10].
One of the most abundant and regenerable cellular antioxidants employed for ROS detoxification inside cells is glutathione (γ-glutamyl-cysteine-glycine), which exists in two interchangeable redox states: reduced (GSH) and oxidized (glutathione disulfide: GSSG) [11,12]. The ROS protection by glutathione is achieved by the formation of mixed disulfides via a thiol exchange reaction (RS–SR + R′SH ↔ R′S–SR + RSH), in which a free electron is donated to ROS [12]. Through this mechanism, GSH can detoxify heavy metals, organic xenobiotics, and many other toxic metabolites which are produced as side products of cellular metabolism [13]. As such, the concentration ratio of GSH to GSSG within cells often serves as a sensitive indicator of oxidative stress [14]. In the presence of oxidative stress, a thiol exchange reaction of GSH also can be made with proteins through a procedure called S-glutathionylation, which describes the formation of a disulfide bridge between the thiol group of glutathione and an accessible free thiol on proteins [15,16]. S-glutathionylation is reversible and known to regulate signal transduction as well as localization and functionality of redox-sensitive proteins [17,18].
The regeneration of GSH from GSSG is catalyzed by the enzyme glutathione reductase (GR) at the expense of reduced nicotinamide adenine dinucleotide phosphate (NADPH). Over 60% of NADPH is produced during the initial steps of the pentose phosphate pathway (PPP) and the rest at the reactions following glutaminolysis [14]. The PPP diverges from glycolysis, using glucose-6-phosphate as the entry substrate. The NADPH-producing glucose-6-phosphate dehydrogenase (G6PDH) catalyzes the rate-limiting first step and the turnover rate is determined by substrate concentration. Consequently, glucose metabolic flux redirects to the PPP in situations where glycolysis is interrupted. The increased PPP flux causes an increase in the NADPH production and the ROS detoxification, as well as the increased production of precursors for anabolic metabolism [19]. This metabolic redirection is especially important for cancer cells, which have to perform persistent glycolysis and ROS regulation simultaneously [20–22]. Recent studies have suggested a possibility that glycolysis, in cancer cells, is interrupted for ROS regulation via promotion of the PPP through oxidative stress- and/or metabolic status-dependent mechanisms. Covalent modifications of the glycolysis rate-regulating enzymes such as pyruvate kinase M (PKM) and phosphofructokinase (PFK) have been suggested as the molecular models. Both S-glutathionylation of PKM and O-linked glycosylation of PFK with N-acetyl glucosamine caused decreases in glycolysis and increases in the PPP [23–25].
Among a family of bifunctional enzymes, PFKFB1–4, each of which controls glycolytic flux by controlling the levels of the key glycolytic stimulator, fructose-2,6-bisphosphate (Fru-2,6-P2) in a tissue-specific manner, PFKFB3 is cancer-predominant and responsible for markedly increased glycolysis within cancer cells [26–29]. We observed that excessive ROS induces S-glutathionylation of PFKFB3, causing a severe decrease in its 2-Kinase catalytic activity for the synthesis of Fru-2,6-P2. The ROS-dependent modulation of PFKFB3 2-Kinase activity resulted in a decrease in the Fru-2,6-P2 levels and an increase in the GSH/GSSG ratios at the cellular level, implicating the redirection of glucose metabolic flux from glycolysis to the PPP for regulation of the cellular oxidative stress homeostasis. The experimental results are presented and discussed in this report.
Results
Isoform-specific S-glutathionylation of PFKFB3
To test whether or not PFKFB isoforms are regulated by oxidative stress, their capacity for S-glutathionylation was tested using anti-glutathione antibody as probe. All four purified recombinant PFKFB isoforms were treated with a physiological level (3 mM) of total glutathione. But, the GSH to GSSG molar ratio was kept low at 1:1, which is extremely favorable for S-glutathionylation, in order to detect the presence of S-glutathionylated PFKFB isoforms, if any exist. The result of this test shows no other isoform, other than PFKFB3, was S-glutathionylated in an immunoblot analysis, even if the ratio was raised to a level 50-fold higher than physiological levels (Figure 1A). Moreover, S-glutathionylation of human PFKFB3 was induced in a dose-dependent manner (Figure 1B) and a significant extent of S-glutathionylation was also detected at a near physiological GSH to GSSG ratio (30:1) (lane 3 in Figure 1B).
Figure 1. Isoform-specific S-glutathionylation of PFKFB3 and its functional effect.

(a) Immunoblotting of S-glutathionylation of PFKFB isoforms after 25 min incubation with 3 mM 1:1 molar ratio of GSH to GSSG, using anti-glutathione antibody. SDS-PAGE separated input PFKFB isoforms (1–4) were visualized by Coomassie blue staining as the loading control. (b) Immunoblotting of a dose-dependent PFKFB3 S-glutathionylation after 25 min treatment with varying molar ratios of GSH to GSSG. Bottom: Immunoblotting of input PFKFB3 using anti-PFKFB3 antibody. (c) 6-phosphofructo-2-kinase activity (2-Kinase) of S-glutathionylated PFKFB3 after 25 min pre-treatment with varied molar ratio of GSH to GSSG. The percentage activities against the control (without preincubation) are represented (n=3 experiments).
When tested for the functional effect of S-glutathionylation on PFKFB3, the 2-Kinase activity for catalysis of Fru-2,6-P2 synthesis was also severely decreased in a dose-dependent manner, implicating a correlation between the extent of S-glutathionylation and the magnitude of activity loss (Figure 1C). Supporting the speculation that the observed decreases in the enzymatic activity are caused by reversible S-glutathionylation, the full enzyme activity was recovered upon treatment with reducing agents such as DTT or GSH. Moreover, a ~20% loss of the catalytic activity of PFKFB3 for F-2,6-P2 synthesis was observed at a near physiological ratio. This magnitude is comparable to or larger than losses of the catalytic activities of pyruvate kinase M2 by S-glutathionylation or phosphofructokinase by the attachment of O-linked N-acetyl glucosamine. A loss of the catalytic activity of either enzyme in such a magnitude was observed to be significant enough to divert glucose metabolic flux to the PPP [23,25].
The effect of S-glutathionylation on PFKFB3 structure/function relationships
To investigate the molecular basis of the functional effect of S-glutathionylation on PFKFB3, we determined the crystal structure of glutathionylated PFKFB3 to 2.2 Å resolution by a method of molecular replacement using the first PFKFB3 structure (PDB code: 2AXN) as a search model [30]. The involved crystallographic data is summarized in Table 1. The crystal structure revealed that this S-glutathionylation is made on Cys206 (Figure 2A, B, and C) and forms a hydrogen bond with Gln199. Among a total of nine cysteine residues in PFKFB, Cys206 and Cys440 are unique to PFKFB3, whereas Cys102, Cys155, Cys193, Cys252, Cys329, Cys388, and Cys426 are conserved among the isoforms. Some residual electron densities were also shown on Cys440 but not sufficient to build in S-glutathionylation with a certainty, when compared to those on Cys206 (Figure 2B). However, this observation suggests an additional S-glutathionylation site in PFKFB3.
Table 1.
Statistics of reflection data and structure refinements
| PFKFB3·SSG | |
|---|---|
| Space group | P6522 |
| Unit cell dimensions | |
| a = b (Å) | 102.50 |
| c (Å) | 260.61 |
| Resolution range (Å) | 47.7-2.2 |
| No. reflections used | 37,905 |
| Completeness (%) | 99.7 |
| Redundancy | 3.6 (3.6) |
| I/σ (I) | 7.9 |
| Rsym | 0.081 |
| Rcrys | 0.209 |
| Rfree | 0.257 |
| No. amino acids | 457 |
| No. protein atoms | 3676 |
| No. hetero atoms | 109 |
| No. water molecules | 160 |
| r.m.s.d. from ideal | |
| Bond lengths (Å) | 0.024 |
| Bond angles (deg.) | 2.22 |
| Dihedral angles (deg.) | 20.5 |
| Mean Bfactor | |
| Protein atoms (Å2) | 33.6 |
| Hetero atoms (Å2) | 47.5 |
| Water atoms (Å2) | 36.3 |
Rsym = Σh(Σj|Ih,j−<Ih>|/ΣIh,j), where h = set of Miller indices, j = set of observations of reflection h, and <Ih>=the mean intensity. Rcrys= Σh||Fo,h|−|Fc,h||/Σh|Fo,h|. Rfree was calculated using 10% of the complete data set excluded from refinement. The numbers in parentheses represent values from the highest resolution shell.
Figure 2. Position of S-glutathionylation in PFKFB3.

(a) A ribbon diagram of the PFKFB3 dimer showing site of S-glutathionylation. S-glutathionylation is shown in only one subunit for a clearer view. The two catalytic domains are shown in different colors: the 2-Kinase domains, gray and magenta; the 2-Phosphatase domains, light green and dark green. S-glutathionylated C206 is labeled as GS-C206 and the position of Fru-6-P in the 2-Kinase catalytic pocket is also shown. (b) The glutathione moiety (labeled as GS-) bound to C206 is shown with an |Fo|–|Fc| omit map at a 2.5σ level. (c) Glutathione (GS-) bound to Cys206 is located on the kinase surface surrounded by basic residues, Arg131, His135, and Lys205. Glutathione has a hydrogen bond with Gln199. The protein residues are shown in yellow, glutathione in green, and the Fru-6-P and ADP in magenta for comparison. (d) Amino acid sequence alignment among human PFKFB isoforms, PFKFB1, PFKFB2, Two splicing variants of PFKFB3, and PFKFB4 from the ClustalW program. Fully conserved, conserved, and similar residues at each position are symbolized by an asterisk (*), colon (:), and dot (.), respectively. The conserved cysteine residues are in bold type and yellow shading shows PFKFB3 isoform-specific S-glutathionylated cysteine residue. (e) Immunoblotting of WT-PFKFB3 and the C206A mutant after pre-treatment with 3 mM 1:1 molar ratio of GSH to GSSG. (f) The 2-Kinase activities of WT-PFKFB3 and the C206A mutant after 25 min pre-treatment with varied molar ratio of GSH to GSSG.
A sequence comparison between PFKFB isoforms was made and a part of the compared sequences is shown in Figure 2D, in which C206 is unique to PFKFB3 and commonly exists in both of the two known PFKFB3 splicing isomers, PFKFB3_1 and PFKFB3_2. In this figure, two other conserved cysteine residues, Cys193 and Cys252, are also shown, because they represent all other conserved cysteine residues, which are not subject to S-glutathionylation according to Figure 1. Compared to Cys206, which is located on the surface of the protein, Cys252 is buried deep inside and, thus, unable to contact with glutathione (data not shown). Other cysteine residues belonging to this buried class are Cys102, Cys155, Cys193, Cys252, Cys329, and Cys388. On the other hand, Cys193 and Cys426 are on the surface but do not have a space wide enough to accommodate a bulky group such as glutathione (data not shown). Our observation supports PFKFB3 isoform-specific S-glutathionylation as suggested by the immunoblots in Figure 1. This conclusion was further supported by inter-species sequence comparisons. When the amino acid sequences of the same PFKFB isoforms from other mammals including mouse, rat, and primates were compared, the PFKFB3-unique Cys206 was found to be conserved between all other mammals evaluated (data not shown).
S-glutathionylation at Cys206 did not induce any significant global conformational changes in PFKFB3, when compared to that of the native protein (data not shown). The S-glutathionylated cysteine residue is located within a loop, which is known to regulate the entry of substrate, Fru-6-P, into the 2-Kinase catalytic pocket. The neighboring basic residues, Arg131, His135, Arg208, and Lys205, are likely to attract the negatively charged glutathione to the site and the negative charges from glutathione seems to facilitate deprotonation of Cys206. Supporting this speculation, Lys205 and His135 are unique to PFKFB3 (data not shown). The covalent attachment of a glutathione to the substrate entry region is likely to doubly compromise the enzymatic activity as shown in Figure 2C. It is likely that the addition of a bulky glutathione near the Fru-6-P binding pocket limits the conformational flexibility of the loop, which is known to be necessary for Fru-6-P entry. Additionally, the negatively charged glutathione is likely to exert repulsion on Fru-6-P, which is also negatively charged. Compared to the dramatic changes seen in the 2-Kinase activity, the phosphatase activity of PFKFB3, responsible for Fru-2,6-P2 hydrolysis, remained essentially unchanged upon S-glutathionylation (data not shown), suggesting that the structure/function effect is limited to the 2-Kinase.
To confirm the site of regulation, Cys206 was mutated to alanine (C206A) for tests. Whereas the wild-type (WT) PFKFB3 is glutathionylated with a roughly 80% loss of kinase activity at 1:1 molar ratios of GSH to GSSG (Figure 2E), C206A abrogated S-glutathionylation with no loss of kinase activity under the same condition (Figure 2F). Taken together, these findings suggest that Cys206 is crucial for S-glutathionylation-dependent inactivation of PFKFB3 2-Kinase activity.
S-glutathionylation of PFKFB3 by cellular oxidative stress
To test whether S-glutathionylation of PFKFB3 is a cellular event, the effect of increased oxidative stress on cellular PFKFB3 was investigated using cultured HeLa cells. To increase cellular oxidative stress, we exogenously applied to the cells with hydrogen peroxide (H2O2) or 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU), both of which are routinely employed to induce oxidative stress in cell studies. Whereas H2O2 increases cellular oxidative stress through abnormal decomposition reactions, BCNU induces accumulation of cellular GSSG by inhibiting glutathione reductase [31].
As shown in Figure 3A, both H2O2 and BCNU enhanced the generation of oxidative stress in HeLa cells, when ROS production was analyzed employing chloromethyl-H2DCFDA (CM-H2DCFDA, Invitrogen)-based fluorescence flow cytometry. A moderate increase in ROS was observed after treating with relatively low concentrations of H2O2 or BCNU (30μM and 10μM, respectively), whereas levels of ROS become excessively high with higher concentrations (100μM and 80 μM, respectively).
Figure 3. PFKFB3 S-glutathionylation induced by oxidative stress inside cancer cells.
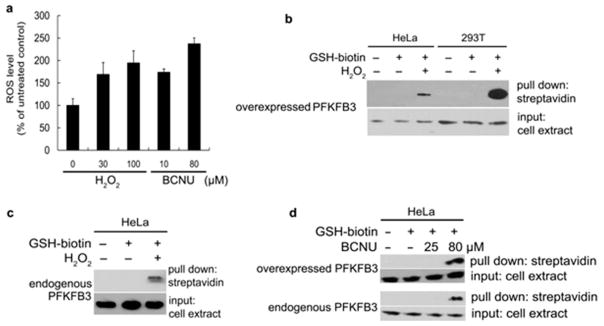
(a) ROS levels in HeLa cells after treatment with the varying amounts of H2O2 and BCNU. Error bars denote SEM. (b) Detection of ROS-induced S-glutathionylation of cellular PFKFB3. PFKFB3-overexpressing HeLa cells and 293T cells were treated with or without 1.5mM Bio-GEE and 100 μM H2O2. Then, biotin-GSS-protein conjugates were precipitated using streptavidin-agarose. Immunoblotting shows PFKFB3 levels before (input, bottom) and after precipitation (upper) (60 μg of total protein per lane). (c) Detection of ROS-induced S-glutathionylation of endogenous PFKFB3 in HeLa cells. With HeLa cells transfected with the empty vector, an experiment similar to (b) was performed. (d) Detection of ROS-induced S-glutathionylation of overexpressed (upper) and endogenous (lower) PFKFB3. HeLa cells were treated with the indicated doses of BCNU to induce S-glutathionylation and the experimental methods were the same as (b) and (c). Immunoblotting shows PFKFB3 levels before (input, bottom) and after precipitation (upper).
Detection of cellular S-glutathionylated PFKFB3 was performed using a pull-down assay followed by immunoblotting. The cells were preloaded with 1 mM cell-permeable biotinylated GSH ethyl ester (Bio-GEE), which is widely accepted for this type of studies, because it causes no significant changes in the cellular GSH concentrations or in the GSSG/GSH ratios with uncompromised GSH redox reactivity [45,46]. The induction of high ROS conditions was achieved by treating the cells with either H2O2 (100 μM) or BCNU (80μM). S-glutathionylation of cellular PFKFB3 was induced by increased cellular oxidative stress regardless whether the protein is endogenous or exogenously overexpressed. S-glutathionylation of both exogenously overexpressed PFKFB3 and endogenous PFKFB3 were detected in HeLa cells (Figure 3B,C, and D), whereas only overexpressed PFKFB3 was detected in 293T cells (Figure 3B), which has no endogenous PFKFB3. This pull down assay confirmed that PFKFB3 is S-glutathionylated upon an increase in cellular oxidative stress.
Consequences of PFKFB3 S-glutathionylation in an in-vitro model
S-glutathionylation of PFKFB3 greatly decreased its 2-Kinase activity, resulting in decreases in the Fru-2,6-P2 levels. This event will lead to decreases in glycolytic flux, because the rate-limiting step of glycolysis is catalyzed by phosphofructokinase (PFK) whose activity is under the tight control by Fru-2,6-P2 levels [32,33]. Consequently, the overall rate of glycolysis is controlled by Fru-2,6-P2 levels. However, unlike glycolysis, the overall rate of glycolysis-interconnecting pentose phosphate pathway (PPP) is controlled by abundance of the entry substrate, glucose-6-phosphate (Figure 4A). Because of this fundamental difference in the rate-controlling mechanisms between the two pathways, it is likely that the decreased glycolytic flux will reroute the metabolic flux to the PPP, resulting in the increased PPP activity.
Figure 4. A model for the carbohydrate flux upon PFKFB3 S-glutathionylation.
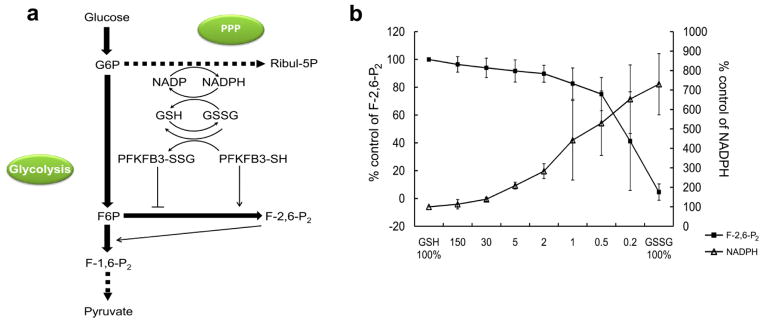
(a) This diagram is provided to shows a hypothetical role played by S-glutathionylation of PFKFB3 in a relationship between fructose-2,6-bisphosphate (Fru-2,6-BP), PFKFB3, and the PPP. It has been hypothesized that increased S-glutathionylation of PFKFB3 leads to decreases in the Fru-2,6-BP level and in the carbohydrate flux to glycolysis and, in turn, an increase in the flux to the PPP and the level of GSH. Solid/bold lines represent direct, one-step biochemical reactions, and indirect, multi-step reactions are represented by dotted lines. Inhibition and activation of enzymatic steps are indicated by solid/plain lines. G-6-P, glucose-6-phosphate; Ribul-5-P, ribulose-5-phosphate; Fru-6-P, fructose -6-phosphate; Fru-1,6-BP, fructose-1,6-bisphosphate; Fru-2,6-BP, fructose-2,6-bisphosphate; PFKFB3-SSG, S-glutathionylated PFKFB3; PFKFB3-SH, unglutathionylated PFKFB3. (b) Enzymatic assay for rerouting of carbohydrate metabolic flux in response to PFKFB3 S-glutathionylation. Changes in the formation of Fru-2,6-BP and NADPH from varied extents of S-glutathionylation of PFKFB3 was measured. The extent of S-glutathionylation was controlled with varied molar ratio of GSH to GSSG in a total concentration kept as 3 mM.
To test this possibility, an enzymatic assay for rerouting of carbohydrate metabolic flux between glycolysis and PPP in response to S-glutathionylation-induced PFKFB3 kinase inactivation was designed. In this assay, the competitive usage of Fru-6-P by PFKFB3 and Glu-6-P dehydrogenase was assessed at varying doses of the GSH/GSSG ratio by measuring Fru-2,6-P2 and NADPH (Figure 4B). The GSSG treatment of PFKFB3 decreased Fru-2,6-P2 levels and increased NADPH levels in a dose-dependent manner, implicating a decrease in glycolysis and an increase in the PPP, respectively (Figure 4B). Thus, the results from this experiment suggest that the S-glutathionylation of PFKFB3 is likely to reroute the glucose metabolic flux from glycolysis to the PPP inside cells.
Cellular metabolic effects of PFKFB3 S-glutathionylation
With cultured HeLa cells, we tested the cellular validity of our model. In this experiment, overexpression of WT-PFKFB3 and C206A was employed, to control the PFKFB3 abundance and the glycolytic rate. It was expected that the overexpression of both WT and C206A would increase overall glycolytic flux with similar magnitudes. However, when the oxidative stress is increased, WT, but not C206A, is S-glutathionylated, resulting in a sharp decrease in the Fru-2,6-P2 production. As a consequence, a bottleneck in the glycolytic pathway is created at the step for conversion of Fru-6-P to Fru-1,6-P2 and the upstream substrates including Fru-6-P are rerouted into the PPP, producing greatly increased NADPH. The magnitude of this redirecting must be dependent on the abundance of PFKFB3 whose activity can be modulated in a ROS-dependent manner.
It was confirmed that the glycolytic rates indicated by the secreted lactate levels coincide with the Fru-2,6-P2 levels in all test conditions (Figure 5A and B). Compared to the empty vector, overexpression of WT-PFKFB3 increased the concentrations of Fru-2,6-P2 and the release of lactate, however H2O2 (or BCNU)-induced oxidative stress abolished such responses. On the other hand, overexpression of C206A displayed a consistent increase in lactate and Fru-2,6-P2 levels both in the presence and absence of H2O2 or BCNU, compared with empty vector control and WT-PFKFB3. Thus, acute oxidative stress triggers a ROS-dependent S-glutathionylation of PFKFB3, resulting in decreased Fru-2,6-P2 levels and, subsequently, glycolysis.
Figure 5. PFKFB3 S-glutathionylation increases the carbohydrate flux to the PPP.
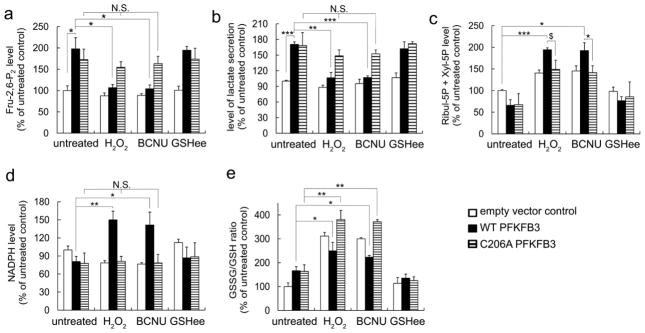
(a) Intracellular Fru-2,6-P2 concentration, (b) the secreted lactate levels (c), cellular concentration of Ribul-5P and Xyl-5P, (d) NADPH, and (e) GSH/GSSG ratio in HeLa cells transiently expressing empty vector pcDNA3.1 (Con), expression plasmids for WT- or C206A-PFKFB3 after treatment with 100 μM H2O2, 50 μM BCNU, or 1.5 mM GSHee to induce PFKFB3 S-glutathionylation. Absolute values were normalized to the total protein concentration and changes in metabolite levels are expressed as a percent of control (without oxidant treatment). T-test (*P < 0.05, **P < 0.01, ***P< 0.001, $P<0.06 (marginally significant); N.S., not significant) used for all the rest figures.
We then tested whether the decreased rates of glycolysis caused by PFKFB3 S-glutathionylation would coincide with increases in the PPP rates inside cells. When no treatment was made, a slight decrease in Ribul-5P+Xyl-5P levels and NADPH levels accompanied with an increase in the GSSG-to-GSH ratio in cells expressing WT and C206A (Figure 5C, D, and E, respectively). This difference might be caused by increases in glycolysis as a result of overexpressed PFKFB3. Treatment with oxidizing agents increased intracellular NADPH concentrations in cells expressing WT, but not in cells expressing C206A (Figure 5D). As expected, H2O2 or BCNU treatment moderately enhanced the GSSG-to-GSH ratio in cells expressing WT, while the treatment excessively increased GSSG levels in cells expressing C206A (Figure 5E). All together it was indicated that the PFKFB3 S-glutathionylation plays an important role in the PPP activation and that the magnitude of activation is significant enough to generate reducing equivalents under oxidative conditions.
Regulation of cellular oxidative stress by PFKFB3 S-glutathionylation
Finally, we investigated if the increased PPP, as a result of PFKFB3 S-glutathionylation, plays a significant role in controlling of cellular levels of ROS. When the cells were treated with increasing doses of H2O2, control cells with an empty vector showed consistent increases in ROS levels. However, cells expressing WT demonstrated ROS regulation through which ROS levels decrease after an initial burst, which was shown at lower doses of H2O2 (Figure 6A). This initial burst seems to be attributable to glycolysis stimulated by increases in PFKFB3 abundance and in the resulting Fru-2,6-P2 levels. Altogether, these results strongly suggest that PFKFB3 plays a role in maintaining the levels of ROS below the threshold toxic yet elevated enough to promote proliferation, oncogenic mutations, and stress responses [21, 34].
Figure 6. Regulation of cellular ROS by PFKFB3 S-glutathionylation.
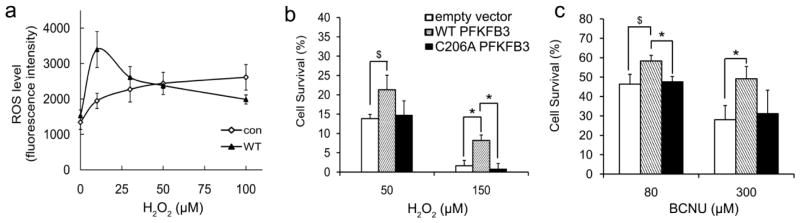
(a) Intracellular ROS levels for empty vector (Con) and WT-PFKFB3 expression after H2O2 treatment. The results are expressed as the mean DCFru-fluorescence. Cell survival after 30 hrs treatment with H2O2 (b) or BCNU (c) using HeLa cells expressing empty vector pcDNA3.1 (Con), expression plasmids for WT- or C206A-PFKFB3.
To test whether this mechanism is relevant to cancer cell survival under oxidative stress, a cell viability assay was performed. In response to ROS-inducing treatments by H2O2 or BCNU, increased cell survival was observed from WT-PFKFB3 overexpression whereas an insignificant difference was shown between C206A and the empty vector (Figure 6B and C). The magnitude of the increase in cell survival rates was greater, when the treating concentrations of H2O2 or BCNU were higher. This observation supports the argument that the magnitude of the PPP influx is dependent on the magnitude of glycolytic flux. The increased glycolytic flux by WT-PFKFB3 overexpression is represented as increased PPP products, decreased ROS levels, and/or increased cell survival, after rerouting by S-glutathionylation-dependent impairment of glycolysis. The overexpressed C206A is incapable of S-glutathionylation-dependent rerouting and, as a consequence, its effects on the PPP and ROS-neutralization are not dissimilar from those of the empty vector. In conjunction with the previous sections, these results together suggest that the S-glutathionylation of PFKFB3 in cancer cells promotes cellular resistance against ROS-associated cell death. The results also reveal how efficiently glucose metabolism and cellular ROS levels can be regulated by the S-glutathionylation of PFKFB3 in order to protect cancer cells from oxidative stress. Hence, PFKFB3 can alternatively stimulate glycolysis and the PPP, depending on the cellular oxidative stress levels.
Discussion
Aberrant metabolism and impairment of mitochondria are the hallmarks of cancer [35]. The cancer-specific aberrant metabolism is represented by anomalously stimulated glutaminolysis and glycolysis [2]. Persistent tumorigenic glycolysis and glutaminolysis followed by the TCA cycle in impaired mitochondria produces ROS above the norm [5]. The excess ROS causes detrimental effects on cell growth and survival, unless counteracted by a neutralization mechanism [6],[10]. The most ubiquitous biological reductant for detoxifying ROS inside cells is GSH, the reducing power of which is recovered by an NADPH-dependent glutathione reductase. Over 60% of cellular NADPH is produced by the PPP in mammals and, thus, the PPP has to be regularly stimulated in order to protect cells from ROS [36,37]. Because glycolysis and the PPP are interconnected and because the PPP rate is dependent on the availability of the entry substrate, modulation of glycolysis is accompanied by an increase in the PPP. A possibility has been suggested that the intermittent impairment of glycolysis may serve as a means for the regulation of oxidative stress in cancer cells. In this regard, the recent reports on the regulation of the two glycolysis-rate-limiting enzymes by posttranslational modifications suggest a molecular physiological model for the regulation of oxidative stress homeostasis in proliferating cells. It has been shown that ROS-dependent S-glutathionylation of pyruvate kinase M2 isoform (PKM2) results in inhibition of glycolysis and promotes the PPP [38]. Similarly, the O-linked glycosylation of phosphofructokinase (PFK) with N-acetyl glucosamine also inhibits glycolysis and promotes the PPP [39]. Although two different covalent modifications were suggested, both the studies revealed that the promoted PPP plays a crucial role in the regulation of the cellular ROS levels for cell survival and proliferation.
We hypothesized that PFKFB3, which is known as the cancer-specific glycolysis pacemaker protein, is also involved in ROS regulation in cancer cells. It has been speculated that inactivation of PFKFB3 decreases glycolysis via decreased Fru-2,6-P2 and may increase the PPP influx, resulting in regulation of ROS. To test this hypothesis, we investigated if the catalytic activity of PFKFB3 for Fru-2,6-P2 synthesis is modulated by increased cellular oxidative stress, if the modulated PFKFB3 activity induces an increase in the PPP via a decrease in glycolysis, if the increased PPP reduces cellular oxidative stress, and if the resulting reduction of cellular oxidative stress is significant enough to enhance viability of cancer cells.
A set of Western blots and subsequent enzymatic experiments revealed that PFKFB3 activity and, hence, its influence on glycolysis is modulated by isoform-specific S-glutathionylation. The isoform specificity of this oxidative stress-dependent covalent modification seems to be valid across all mammals according to sequence analysis. An X-ray crystallographic study revealed the site of S-glutathionylation and the associated mechanism of activity modulation. An in vitro assay experiment designed to investigate the interplay between glycolysis and the PPP showed increases in the PPP flux in response to S-glutathionylation-induced PFKFB3 inactivation. Moreover, the results suggested that Fru-6-P can be retrogradely routed to the PPP after conversion to G-6-P, when the glycolytic consumption of Fru-6-P by PFK is limited. The results provided a biochemical foundation that glucose metabolism can be alternatively routed between glycolysis and the PPP, depending on the rate of either pathway.
The validity of our model was tested at the cellular level with the cultured HeLa cells. When cellular oxidative stress was elevated by treating with H2O2 and BCNU, S-glutathionylation of both the endogenous and overexpressed PFKFB3 was detected, using a widely accepted affinity precipitation and immune blotting method. We showed that the cellular PFKFB3 activity regulates Fru-2,6-P2 levels and, thereby, glycolysis rates. Compared to the empty vector, the WT-PFKFB3 overexpression increased levels of both Fru-2,6-P2 and secreted lactate to a similar extent, implying an increase in overall glycolytic flux. Modulation of PFKFB3 activity by elevated ROS decreases glycolysis and increases the PPP via rerouting glycolytic metabolites. The magnitude of the rerouted flux must be dependent on the magnitude of the initial glycolytic flux. Consequently, the PPP increase by WT-PFKFB3 overexpression is more efficient than that by the empty vector. But, such an increased metabolic shift could not be elicited by C206A, although the mutant increased glycolysis to the same extent as WT-PFKFB3. This is because the mutant is insensitive to ROS-dependent activity modulation and, thus, might be unable to elicit ROS-dependent regulation of glycolysis and metabolic rerouting.
Subsequently, we investigated if the metabolic rerouting is related to changes in the abundance of cellular GSH. Metabolic parameters and the GSSG/GSH ratios of the cells treated with 4 mM cell-permeable GSH, ethyl ester GSH (GSHee), were not significantly different from those of the untreated cells after 30 hrs of incubation. This result suggests that the regulation of cellular oxidative stress homeostasis is to control the steady GSH/GSSG ratios rather than the abundance of glutathione. Lastly, it was revealed that increases in the PPP by PFKFB3 S-glutathionylation significantly reduce ROS levels and, thereby, enhance cell viability. And, that ROS neutralization is more efficient, when the glycolytic flux was stronger, as suggested by comparison of the WT-PFKFB3 overexpression and the empty vector.
These results altogether provide a new understanding of how the S-glutathionylation of PFKFB3 controls glucose metabolism, thereby regulating ROS levels in order to protect cancer cells against oxidative stress. The PFKFB3 S-glutathionylation plays an additional role for ROS regulation in cancer cells, which was originally suggested by PKM S-glutathionylation. Modulation of the PFKFB3 activity regulates the glycolysis rate by controlling the first glycolysis-committed step, conversion of Fru-6-P to Fru-1,6-P2, whereas modulation of PKM plays a similar function by controlling the last step, conversion of phosphoenol pyruvate to pyruvate. A question raised from this study is why the two opposite ends of glycolysis are both involved in fulfillment of the same physiological goal, ROS regulation.
Our speculation is that a decrease in glycolysis has an additional function to ROS regulation, as if an increase in the PPP is more than just for NADPH production. In cancer cells which are prone to autonomous metabolism, intermittent activation of the PPP is necessary for biosynthesis of nucleotides, amino acids, and lipids for rapid cell proliferation. For the same reasons, glycolysis also has to be periodically regulated such that cells can perform biosynthesis of amino acids and phospholipids using the glycolysis intermediate metabolites as precursors. Modulation of both PFKFB3 and PKM enables cells to secure large quantities of the glycolysis intermediate metabolites necessary for many anabolic processes. Otherwise, a harmonious biosynthesis using the PPP products would not be possible. In this regard, the functional involvement of PFKFB3 S-glutathionylation in ROS regulation would also suggest a possible role for PFKFB3 in cell cycle progression in cancer cells and a broader and more general role for ROS in the regulation of numerous biological processes, although further studies are necessary to elucidate the details of this mechanism. This new understanding will contribute to the development of appropriate therapeutic strategies for cancer prevention and treatment based on their redox profile.
Materials and Methods
Materials
The following materials were obtained from the sources indicated. Dimethyl sulfoxide (DMSO), oxidized L-glutathione (GSSG), reduced L-glutathione (GSH), 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) (Sigma Chemical Co., St. Louis, MO, USA); Qdot 605 ITK streptavidin conjugate kit 2 μM solution, dextran alexafluor 488, anti-V5 antibody, calceinacetoxymethyl ester, PLUS Reagent, lipofectamine LTX Reagent, biotinylated glutathione ethyl ester (BioGEE) (Invitrogen Corporation, Carlsbad, CA, USA); anti-glutathione monoclonal antibody (ViroGen Corporation); V5 antibody affinity purified agarose immobilized conjugate (Bethyl Laboratories); goat anti-mouse glutaredoxin-1/GLRX1 antibody (R&D Systems Inc.); glutaredoxin-1, glutaredoxin-1 antibody (Abcam).
Plasmid Constructs and Mutagenesis
Full-length cDNA constructs for human PFKFB3 mutants, C206A, were generated by site-directed mutagenesis using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene) and the commercially available vector pcDNA3.1/myc-HisA with human PFKFB3 as template, following the manufacturer’s recommendations. Briefly, the mutant strand synthesis reaction was carried out by an initial denaturation at 95°C for 1 min, followed by 18 thermal cycles at 95°C for 50 s, 60°C for 50 s, and 68°C for 9 min 24 s, with a final extension at 68°C for 7 min. To create the bacterial expression plasmids, C206A-PFKFB3 was then subcloned from pcDNA3.1/myc-HisA into the pET3a vector using NheI and BamHI restriction sites. All constructs were sequenced for verification.
Preparation and S-Glutathionylation of PFKFB isoforms and Western Blotting
The 6XHis-tagged human PFKFB1-4 wild-type isoforms and the PFKFB3 C206A mutant were expressed in Escherichia coli BL21(DE3) pLysS and purified using Ni-NTA affinity columns and, subsequently, Mono Q anion-exchange chromatography. The experimental details are described elsewhere [40].
To induce protein S-glutathionylation, purified PFKFB isoforms (0.3 mg/ml) in 20 mM Tris•HCl (pH 8.0), 10 mM NaPi, 5% glycerol, and 0.2 mM EDTA were incubated for 25 min with 3 mM glutathione (with the GSH/GSSG molar ratios of 1:1) in 50 mM Tris•HCl pH 7.4 at room temperature. The samples were then mixed without reducing agent or heating with Laemmli sample buffer containing 1 mM NEM for the alkylation of accessible thiols on PFKFB3. Samples were then resolved in non-reducing SDS-PAGE, and subsequently transferred to nitrocellulose membranes. The Membranes were then probed with anti-glutathione antibody (Virogen), followed by detection with horseradish peroxidase-conjugated secondary antibody (Novagen). Bands were visualized by enhanced chemiluminescence (SuperSignal west pico, Pierce) according to the manufacturer’s instructions. Membranes were subsequently re-probed with anti-PFKFB3 antibody (Abgent).
Measurement of 2-Kase/2-Pase activity of PFKFB3
The catalytic activity of PFKFB3 2-Kinase for Fru-2,6-P2 synthesis was performed as described previously[41]. The 2-Kinase reactions were performed first and the Fru-2,6-BP produced was measured by a conventional enzyme coupled assay. The reaction mixture contained 20 mM Tris•HCl (pH 8.5), 0.5 mM MgCl2, 0.1 mM ATP, and 10–20 μg PFKFB3. The reaction was initiated by adding 0.05 mM Fru-6-P and incubating at 25 °C for 10 min, and the concentration of Fru-2,6-P2 was determined every 5 min. Fru-2,6-P2 was measured spectrophotometrically at 340 nm using the potato pyrophosphate-dependent 6-phosphofructokinase (PPi-PF1K) activation assay.
To measure the catalytic activity of PFKFB3 2-Phosphatase for Fru-2,6-P2 hydrolysis, a spectrophotometeric coupled enzyme assay was carried out using phosphoglucose isomerase and Glu-6-P dehydrogenase as described previously. The reaction mixture (total volume 200μl) contained 100 mM Tris•HCl, pH 7.5, 0.2 mM EDTA, 100 μM NADP, 0.4 unit of Glu-6-P dehydrogenase, and 1 unit of phosphoglucose isomerase. PFKFB3 (0.2–0.4 mg) was used in the 2-Phosphatase activity assay. The reaction was initiated by the addition of 100 μM Fru-2,6-P2 and all experiment were run at 25 °C. NADPH formation was measured at 340 nm using a microplate reader (Model 3550-UV, Bio- Rad).
S-Glutathionylation of PFKFB3 Crystals and Diffraction Data
The purified PFKFB3 was kept, after concentrating to 8 mg/ml protein, in 20 mM Tris•HCl (pH 8.0), 10 mM NaPi, 5 mM β-mercaptoethanol, 5% glycerol. Crystals were prepared by the sitting-drop vapor-diffusion method with a 1:1 (v/v) mixture of the protein sample with a reservoir solution of 50 mM MES pH 7 and 7% (w/v) polyethylene glycol 4000 [40]. Crystals with a size of 0.2 mm × 0.1 mm × 0.2 mm grew in two to three weeks.
Crystals were soaked with a cryoprotectant solution containing different molar ratios of GSH/GSSG at constant total glutathione concentration of 5 mM. A soaked crystal was flash-cooled at 100 K using an Oxford cryo-device and kept at the same temperature during data collections. The diffraction data were collected at The Northeastern Collaborative Access Team (NE-CAT) Beamline at the Advanced Photon Source, Argonne National Laboratory, Argonne, IL. The X-ray source wavelength was 0.9792 Å. The data recorded on a ADSC Q315 detector were integrated, merged, and scaled using XDS [42]. Statistics of the diffraction data and structure refinement are summarized in Table 1. The crystals belong to P6522 space group with the cell dimensions similar to those of the native protein. The reduced data was formatted for the program suites of CCP4 [43] and 10% of the data was marked for free R-factor measurements in subsequent structure refinements.
Structure Determination and Refinement
The search model was built from the coordinates of the PFKFB3•ADP•EDTA complex structure (PDB accession code 2AXN) [30] by stripping all included ligand and solvent molecules. The initial model was determined using REFMAC within the CCP4 suit and processed through iterated cycles of manual model rebuilding and validation using the program COOT [43].
Bindings of ligands were confirmed, referring to the |Fo|–|Fc| omit maps that were generated, when Rcrys/Rfree reached 0.23/0.29 or below. Referring to these maps, GSH was incorporated into the corresponding complex models. As summarized in Supplementary Table 1, the final model of the glutathionylated PFKFB3 has Rcrys/Rfree of 0.209/0.257 using a total of 3,939 scatterers, including solvent molecules, against all available 37,905 reflections in the resolution range of 47.7–2.2 Å. The structure contains a total of 443 amino acid residues of the 520 residue full-length protein. As in the PFKFB3•ADP•EDTA complex, the C-terminus (residues 461–520) is mostly disordered. The pdb file of the refined structure with the structure factor was deposited with an accession code 4MA4.
In Vitro Metabolic Rerouting between Glycolysis and the PPP
To measure in vitro metabolic rerouting between glycolysis and the PPP upon PFKFB3 S-glutathionylation, a spectrophotometeric coupled enzyme assay was carried out using PFKFB3, phosphoglucose isomerase, and Glu-6-P dehydrogenase. The combined reactions of the first step of the PPP via phosphoglucose isomerase and Fru-2,6-P2 synthesis by PFKFB3 were initiated by addition of 0.2 mM Fru-6-P. Formation of Fru-2,6-P2 and NADPH were measured subsequently as representative products. The reaction mixture contained 50 mM glycylglycine (pH 7.5), 0.5 mM MgCl2, 0.2 mM ATP, 0.4 mM β-nicotinamide adenine dinucleotide phosphate (NADP), 0.2–0.6 unit of Glu-6-P dehydrogenase, 1 unit of phosphoglucose isomerase, and 8–20 μg PFKFB3. The reaction was initiated by adding 0.2 mM Fru-6-P and incubating at 25 °C for 10 min and collecting aliquots at every 5 minutes. The aliquots were transferred into 125 mM NaOH. The NADPH formation was directly measured at 340 nm and Fru-2,6-P2 produced was measured as described above.
Cell Culture, Transient Transfection, and Oxidant Treatments
293T and HeLa cells were obtained from ATCC and cultured in DMEM containing 2 mM glutamine, 10% fetal calf serum (FCS), 100 U/ml penicillin and 100 μg/ml streptomycin (all from GIBCO-BRL). All cells were cultured in a humidified incubator at 37°C, 5% CO2. Cells expressing PFKFB3 and its mutant were established by transfecting cells with pcDNA3.1/myc-HisA-PFKFB3 WT and mutant(s) using GeneJuice® transfection reagent (Novagen). Overexpression of PFKFB3 was confirmed by immunoblotting analysis. BCNU [N,N′-bis(2-chloroethyl)-N-nitro-sourea] and H2O2 (hydrogen peroxide) were from Sigma and used as described in the text.
Detection of Glutathionylation of PFKFB3 in 293T and HeLa cells
293T and HeLa cells expressing PFKFB3 were loaded with 1 mM biotinylated glutathione ethyl ester (BioGEE, Invitrogen) and incubated for 1 hr followed by an H2O2 treatment for 15 min or BCNU treatment for 3 hrs. The cells were lysed in a modified RIPA buffer (0.01 M sodium phosphate pH 7.2), 150 mM NaCl, 0.1mM EDTA, 1% Nonidet P-40, 1% sodium deoxycholate, and 0.1% SDS, 10 mM N-ethylmaleimide) supplemented with a protease inhibitor mixture (Roche). Glutathionylated PFKFB3 levels were measured after pull down with streptavidin-dynabeads (Invitrogen) according to the manufacturer’s protocol followed by reducing SDS-PAGE and Western blot analysis with anti-PFKFB3 antibody. For detection of endogenous PFKFB3 glutathionylation, cells were treated with 10 μM proteasome inhibitor MG132 for 8 hrs before BioGEE incubation. Then cells were prepared as described above.
Determination of Cellular Metabolites
HeLa cells were plated at a density of 1×107 in T 175 culture flasks in DMEM containing 10% FCS, 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. Cells expressing a specific PFKFB3 mutant were established by transient transfection the following day. After 36 hours of incubation, the media were replaced with fresh DMEM containing either H2O2, BCNU, or GSHee at the indicated concentrations and time. Media samples were collected for measuring the lactate secretion levels using a lactate oxidase-based assay kit (Sigma-Aldrich). Cell extracts were prepared with the lysis buffer containing 0.01 M sodium phosphate (pH 7.5), 150 mM NaCl, 5 mM EDTA, 1% Nonidet P-40, and 5% 5-sulfosalicylic acid (SSA). Before each assay, ice cold 2M KOH was added to the SSA- preserved sample for neutralization. The Fru-2,6-P2 level was determined in the collected cells after the treatment by the method described previously [41].
For the quantification of the PPP, levels of ribulose 5-phosphate (Ribul-5P) and xylulose-5-phosphate (Xyl-5P) were monitored. For this, the mixture of Ribul-5P, Xyl-5P, and glyceraldehyde-3-phosphate (G-3P) was quantitated first and the quantity of G-3P was subtracted. First, the quantification of mixture of the three was performed in a system containing 58 mM glycylglycine, 3.3 mM Xyl-5P, 0.002% (w/v) cocarboxylase, 15 mM magnesium chloride, 0.14 mM β-nicotinamide adenine dinucleotide, reduced form, 15 mM MgCl2, 20 units a-glycerophosphate dehydrogenase, 5 units triosephosphate isomerase, 0.1 unit ribulose 5-phosphate 3-epimerase, and 5 units transketolase as described previously[44]. Then, levels of G-3P have been measured under same condition but in the absence of ribulose 5-phosphate 3-epimerase and transketolase. Protein concentration was measured using QuantiPro BCA assay kit (Sigma-Aldrich) and metabolite concentrations were normalized to the total cellular protein concentration.
NADP/NADPH, GSH/GSSG and ROS measurements
Cells were prepared as described above in Metabolite determination. The cellular GSH/GSSG ratios and NADP/NADPH ratios were analyzed spectrophotometrically by using a GSH/GSSG measurement kit or NADPH assay kit (Abcam) according to the manufacturer’s instructions.
For ROS measurements, the medium was aspirated, pre-treated cells were washed 1x with PBS, harvested by trypsinization, and processed for flow cytometry. Cells were subsequently incubated with HBSS containing 10 μM chloromethyl-H2DCFDA (CM-H2DCFDA, Invitrogen) in DMSO for 30 min at 37°C, while still maintaining the H2O2 or BCNU treatment. The fluorescence of samples were analyzed by flow cytometry (FACSCalibur®, Becton Dickinson Immunocytometry), using the CellQuest (BD Bioscience) software.
Cell Viability assay
Cell viability assays were performed by trypan blue staining. Cells were plated at a density of 3.5×104 per well in a 24-well plate in DMEM containing 10% FCS. Cells expressing the PFKFB3 mutant were established by transient transfection the following day. After 20 hours of incubation, the media were replaced with fresh DMEM containing H2O2, BCNU, or GSHee at the indicated concentrations. After 30 hours of incubation with the vehicle or compounds, cells were trypsinized and cell viability was determined by the trypan blue exclusion assay using a hemacytometer.
Highlights.
A cancer-specific mechanism for ROS neutralization is necessary.
PFKFB3 activity is down-regulated by ROS-dependent S-glutathionylation.
S-glutathionylated PFKFB3 redirects metabolic flux to pentose phosphate pathway.
S-glutathionylation of PFKFB3 is involved in ROS neutralization in cancer.
Acknowledgments
The authors thank J. Kim (Louisiana State University Biological Sciences) for providing us with the HeLa cell line. This work was supported by the National Cancer Institute grant 1R01 CA124758-01 to Y.-H.L.
Abbreviations
- ROS
reactive oxygen species
- GSSG
oxidized glutathione
- GSH
reduced glutathione
- PFKFB
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase
- Fru-2, 6-P2
fructose-2,6-bisphosphate
- Fru-6P
fructose-6-phosphate
- G6PDH
glucose-6-phosphate dehydrogenase
- PKM
pyruvate kinase M
- PFK
phosphofructokinase
- PPP
the pentose phosphate pathway
- GSHee
ethyl ester GSH
- BCNU
1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU)
- Ribul 5-P
ribulose 5-phosphate
- Xyl-5-P
xylulose-5-phosphate
- G-3-P
glyceraldehydes-3-phosphate
Footnotes
Author Contributions
M.S. designed and performed experiments, analyzed data, and wrote the manuscript; Y.L. supervised and directed the project and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wechalekar K, Sharma B, Cook G. PET/CT in oncology--a major advance. Clin Radiol. 2005;60:1143–1155. doi: 10.1016/j.crad.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 2.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 4.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 5.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 6.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 7.Lawless MW, O’Byrne KJ, Gray SG. Targeting oxidative stress in cancer. Expert Opin Ther Targets. 2010;14:1225–1245. doi: 10.1517/14728222.2010.526933. [DOI] [PubMed] [Google Scholar]
- 8.Visconti R, Grieco D. New insights on oxidative stress in cancer. Curr Opin Drug Discov Devel. 2009;12:240–245. [PubMed] [Google Scholar]
- 9.Perera RM, Bardeesy N. Cancer: when antioxidants are bad. Nature. 2011;475:43–44. doi: 10.1038/475043a. [DOI] [PubMed] [Google Scholar]
- 10.Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006;25:695–705. doi: 10.1007/s10555-006-9037-8. [DOI] [PubMed] [Google Scholar]
- 11.Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 12.Lushchak VI. Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids. 2012;2012:736837. doi: 10.1155/2012/736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franco R, Schoneveld OJ, Pappa A, Panayiotidis MI. The central role of glutathione in the pathophysiology of human diseases. Arch Physiol Biochem. 2007;113:234–258. doi: 10.1080/13813450701661198. [DOI] [PubMed] [Google Scholar]
- 14.Pastore A, Federici G, Bertini E, Piemonte F. Analysis of glutathione: implication in redox and detoxification. Clin Chim Acta. 2003;333:19–39. doi: 10.1016/s0009-8981(03)00200-6. [DOI] [PubMed] [Google Scholar]
- 15.Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 16.Dalle-Donne I, Rossi R, Giustarini D, Colombo R, Milzani A. S-glutathionylation in protein redox regulation. Free Radic Biol Med. 2007;43:883–898. doi: 10.1016/j.freeradbiomed.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 17.Gallogly MM, Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol. 2007;7:381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 18.Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 20.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annu Rev Pharmacol Toxicol. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 21.Dreher D, Junod AF. Role of oxygen free radicals in cancer development. Eur J Cancer. 1996;32A:30–38. doi: 10.1016/0959-8049(95)00531-5. [DOI] [PubMed] [Google Scholar]
- 22.Toyokuni S, Okamoto K, Yodoi J, Hiai H. Persistent oxidative stress in cancer. FEBS Lett. 1995;358:1–3. doi: 10.1016/0014-5793(94)01368-b. [DOI] [PubMed] [Google Scholar]
- 23.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–U274. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 24.Sun QA, Chen XX, Ma JH, Peng HY, Wang F, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4129–4134. doi: 10.1073/pnas.1014769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yi W, Clark PM, Mason DE, Keenan MC, Hill C, et al. Phosphofructokinase 1 Glycosylation Regulates Cell Growth and Metabolism. Science. 2012;337:975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, et al. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J. 2004;381:561–579. doi: 10.1042/BJ20040752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Telang S, Yalcin A, Clem AL, Bucala R, Lane AN, et al. Ras transformation requires metabolic control by 6-phosphofructo-2-kinase. Oncogene. 2006;25:7225–7234. doi: 10.1038/sj.onc.1209709. [DOI] [PubMed] [Google Scholar]
- 28.Atsumi T, Chesney J, Metz C, Leng L, Donnelly S, et al. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002;62:5881–5887. [PubMed] [Google Scholar]
- 29.Calvo MN, Bartrons R, Castano E, Perales JC, Navarro-Sabate A, et al. PFKFB3 gene silencing decreases glycolysis, induces cell-cycle delay and inhibits anchorage-independent growth in HeLa cells. FEBS Lett. 2006;580:3308–3314. doi: 10.1016/j.febslet.2006.04.093. [DOI] [PubMed] [Google Scholar]
- 30.Kim S-G, Manes NP, El-Maghrabi MR, Lee Y-H. Crystal Structure of the Hypoxia-Inducible Form of 6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatase (PFKFB3): A Possible New Target for Cancer Therapy. J Biol Chem. 2006;281:2939–44. doi: 10.1074/jbc.M511019200. [DOI] [PubMed] [Google Scholar]
- 31.Forman HJ. Use and abuse of exogenous H2O2 in studies of signal transduction. Free Radic Biol Med. 2007;42:926–932. doi: 10.1016/j.freeradbiomed.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.El-Maghrabi MR, Noto F, Wu N, Manes N. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: suiting structure to need, in a family of tissue-specific enzymes. Curr Opin Clin Nutr Metab Care. 2001;4:411–418. doi: 10.1097/00075197-200109000-00012. [DOI] [PubMed] [Google Scholar]
- 33.Van Schaftingen E, Jett MF, Hue L, Hers HG. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc Natl Acad Sci U S A. 1981;78:3483–3486. doi: 10.1073/pnas.78.6.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benz CC, Yau C. Ageing, oxidative stress and cancer: paradigms in parallax. Nat Rev Cancer. 2008;8:875–879. doi: 10.1038/nrc2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, et al. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005;65:613–621. [PubMed] [Google Scholar]
- 36.Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011;30:546–555. doi: 10.1038/emboj.2010.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yi W, Clark PM, Mason DE, Keenan MC, Hill C, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012;337:975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seo M, Kim JD, Neau D, Sehgal I, Lee YH. Structure-Based Development of Small Molecule PFKFB3 Inhibitors: A Framework for Potential Cancer Therapeutic Agents Targeting the Warburg Effect. Plos One. 2011;6 doi: 10.1371/journal.pone.0024179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seo M, Kim JD, Neau D, Sehgal I, Lee YH. Structure-based development of small molecule PFKFB3 inhibitors: a framework for potential cancer therapeutic agents targeting the Warburg effect. PLoS One. 2011;6:e24179. doi: 10.1371/journal.pone.0024179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 44.Boada J, Roig T, Perez X, Gamez A, Bartrons R, et al. Cells overexpressing fructose-2,6-bisphosphatase showed enhanced pentose phosphate pathway flux and resistance to oxidative stress. FEBS Lett. 2000;480:261–264. doi: 10.1016/s0014-5793(00)01950-5. [DOI] [PubMed] [Google Scholar]
- 45.Meissner F, Molawi K, Zychlinsky A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat Immunol. 2008;9:866–72. doi: 10.1038/ni.1633. [DOI] [PubMed] [Google Scholar]
- 46.Sullivan DM, Levine RL, Finkel T. Detection and affinity purification of oxidant-sensitive proteins using biotinylated glutathione ethyl ester. Methods Enzymol. 2002;353:101–13. doi: 10.1016/s0076-6879(02)53040-8. [DOI] [PubMed] [Google Scholar]