

Abstract
Objective
One of the major risk factors for atherosclerosis is the plasma level of low density lipoprotein (LDL), which is a product of very low density lipoprotein (VLDL). Hepatic apolipoprotein B100 (apoB100) is the essential component that provides structural stability to VLDL particles. Newly translated apoB100 is partially lipidated in the endoplasmic reticulum (ER), forming nascent apoB100-VLDL particles. These particles are further modified to form fully mature VLDLs in the Golgi apparatus. Therefore, the transport of nascent VLDL from the ER to the Golgi represents a critical step during VLDL maturation and secretion, and in regulating serum LDL cholesterol levels. Our previous studies showed that apoB100 exits the ER in COPII vesicles, but the cohort of related factors that control trafficking is poorly defined.
Approach and Results
Expression levels of KLHL12, an adaptor protein known to assist COPII-dependent transport of procollagen, were manipulated by using a KLHL12-specific siRNA and a KLHL12 expression plasmid in the rat hepatoma cell line, McArdle-RH7777 (McA). KLHL12 knockdown decreased the secreted and intracellular pools of apoB100, an effect that was attenuated in the presence of an autophagy inhibitor. KLHL12 knockdown also significantly reduced secretion of the most lipidated apoB100-VLDL species and led to the accumulation of apoB100 in the ER. Consistent with these data, KLHL12 overexpression increased apoB100 recovery and apoB100-VLDL secretion. Images obtained from confocal microscopy revealed co-localization of apoB100 and KLHL12, further supporting a direct link between KLHL12 function and VLDL trafficking from the ER.
Conclusions
KLHL12 plays a critical role in facilitating the ER exit and secretion of apoB100-VLDL particles, suggesting that KLHL12 modulation would influence plasma lipid levels.
Keywords: Kelch-like protein 12, apolipoprotein B, very low density lipoprotein, COPII vesicles
Introduction
ApoB100 normally associates with very low density lipoprotein (VLDL) particles in mammalian liver. The trafficking of nascent apoB100-containing VLDL (apoB100-VLDL) from the ER to the Golgi is essential for the final maturation and secretion of these particles. We previously showed that nascent apoB100-VLDL particles require coat complex II (COPII) for ER-Golgi transport; however, the apoB100-VLDL transport vesicles had physical properties distinct from those containing other secretory proteins1. Notably, apoB100-VLDL transport vesicles were larger in diameter and denser than those transporting typical cargo proteins 1–5. It has been speculated that large cargo molecules, such as VLDL, require specialized factors to promote their packaging and exit from the ER 6. In fact, a recent study by Jin et al. (2012) 7 established that KLHL12 (Kelch-like protein 12), a substrate-specific adaptor protein in the Cullin3-based (CUL3) ubiquitin ligase complex, facilitates the transport of procollagen fibers from the ER to the Golgi apparatus. Like VLDL, procollagen is much larger than nearly every other secretory cargo and is unable to “fit” inside the confines of a typical COPII vesicle 7–10. To overcome this hurdle, KLHL12 binds to a COPII-component, Sec31, triggering its monoubiquitinylation, which leads to the assembly of larger vesicle coats 7. Here, we show that KLHL12 also facilitates the transport and secretion of apoB100-containing VLDL particles in a model of mammalian liver lipoprotein metabolism, rat hepatoma McArdle RH7777 (McA) cells.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results and Discussion
To determine whether KLHL12 plays a role in apoB100-VLDL trafficking and secretion, we first asked whether manipulating KLHL12 expression impacted apoB100 levels and the secretion of mature VLDL particles. KLHL12 in McA cells was depleted by transfecting cells with a KLHL12-specific siRNA, and the levels of secreted and intracellular apoB100 were measured under steady state radiolabeling conditions. A significant reduction of KLHL12 expression (confirmed by quantitative PCR and by Western blotting; online-only Data Supplement, Figs. I and II, respectively) in the presence of oleic acid/bovine serum albumin (OA/BSA) complexes resulted in >50% decreases in the levels of secreted and intracellular apoB100 (Fig. 1A and 1B). The effect of KLHL12 knockdown was specific to apoB100, as secretion of the constitutive hepatic protein albumin was unaltered (online-only Data Supplement Fig. III). This result provides further support that KLHL12 facilitates the transport of atypical, large COPII cargo. Data from density gradient fractionation showed that knockdown of KLHL12 had a profound effect on the secretion of the most lipidated species of apoB100, namely VLDL1 (Fig. 1C). To further support a role of KLHL12 during the ER-to-Golgi trafficking of apoB100-VLDL, apoB100 levels were measured in ER microsomes and Golgi membranes isolated from KLHL12 silenced McA cells. The ratio of apoB100 recovery from ER to that from the Golgi apparatus increased by 60% in KLHL12 depleted cells compared to that of the controls (Fig. 1D; the ER and Golgi markers distributions are shown in the online-only Data Supplement Fig. IV). This shift suggested a decrease in VLDL vesicular transport and a concomitant accumulation in the ER. Overall, then, the collective data strongly imply that the trafficking of apoB100 from the ER to Golgi, where VLDL fully matures 11–13, was compromised when KLHL12 expression was reduced.
Figure 1. Effects of KLHL12 knock-down and overexpression on apoB100 and apo100-lipoproteins in McArdle-RH7777 (McA) cells incubated with oleic acid.
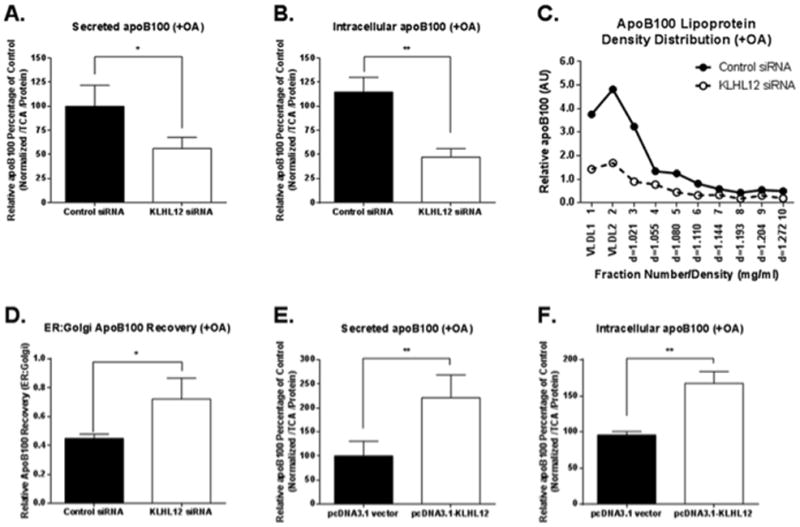
McA cells were transfected with control or KLHL12-specific siRNA. 48 h later, the cells were incubated with 0.6 mM OA/BSA complex for 1 h, and the incubation was continued for an additional 3 h with 35S-met/cys added to radiolabel apoB100 to steady state. Secreted (A) and intracellular (B) recoveries of radiolabeled apoB100 in the conditioned media and cell lysates were normalized against total TCA-precipitated counts and total protein amounts. (C) Density gradient analysis of secreted apoB100-associated lipoproteins. (D) McA cells were treated as in A and B. Cell homogenates were separated by sucrose density gradient centrifugation to obtain ER microsomes and Golgi membranes, from which apoB100 was immunoprecipitated and quantified. The ER:Golgi ratio of apoB100 recovery was calculated and is displayed. E and F: McA cells were transfected with either Flag-tagged pcDNA3.1-KLHL12 or empty pcDNA3.1 vector (as a control) for 48 h. Cells were then incubated with 0.6 mM OA/BSA complex for 1 h, and the incubation was continued for an additional 3 h with 35S-met/cys to radiolabel apoB100 to steady state. Secreted (D) and intracellular (E) recoveries of radiolabeled apoB100 in the conditioned media and cell lysates were normalized against total TCA-precipitated counts and total protein amounts. A,B,D,E,F: shown are the means, +/− SEM, n=3. *p < 0.05, and **p < 0.01; C: shown are the results representative of 3 independent experiments.
Opposite results were seen in McA cells overexpressing KLHL12 (Fig. 1E and 1F; the overexpression was confirmed by Western blotting as shown in the online-only Data Supplement Fig. V). In this case, the levels of secreted apoB100 were markedly increased, and there was a concomitant increase in the population of secreted VLDL1 (on-line Data Supplement Fig. VI). In contrast, there was little effect on albumin secretion (online-only Data Supplement Fig. VII), indicating the specificity of action of KLHL12 overexpression. These over-expression data suggest that the native level of KLHL12 is limiting for apoB100-VLDL trafficking in McA cells.
Though the focus of this report is on apoB100 (the sole form produced by human liver), rodent hepatic cells also produce apoB48. Limited analyses of the effects of KLHL12 knockdown and overexpression on apoB48 were also performed. Upon KLHL12 knockdown, there was also decreased recovery of apoB48, but to a lesser degree than for apoB100; in addition, there were minimal effects when KLHL12 was overexpressed (online-only Data Supplement Fig. VIII). These results most likely reflect the smaller proportion of apoB48 (~1/3 that of apoB100) that can be assembled into VLDL (e.g., 14), which would be expected to decrease the reliance on KLHL12 for the ER export of apoB48-containing lipoproteins.
In the experiments presented above, the McA cells were incubated with OA/BSA complexes. This treatment augments apoB100 lipidation and secretion, but has no effect on the preferential association of apoB100 with VLDL particles. In the absence of exogenous fatty acids, the low level of endogenous lipid synthesis can result in an increase in the amount of apoB100 destroyed by the ER-associated degradation (ERAD) pathway, but of the surviving molecules, most would be secreted in association with VLDL (reviewed in 15, 16). Under these conditions, KLHL12 again modulated apoB100 secretion (Fig. 2A and 2B), as the amount of apoB100 decreased upon KLHL12 knockdown (primarily in the VLDL-density fractions; data not shown) and increased upon KLHL12 over-expression (online-only Data Supplement Fig. IX).
Figure 2. KLHL12 regulates apoB100 metabolism when lipids are primarily endogenously synthesized and co-localizes with apoB100 in the secretory pathway.

McA cells were transfected with control or KLHL12-specific siRNA without OA/BSA complexes. 48 h later, cells were incubated with 35S-met/cys for 3 h to radiolabel apoB100 to steady state. Secreted (A) and intracellular (B) levels of radiolabeled apoB100 were decreased similar to that observed in Fig. 1. (C) McA cells were radio-labeled to steady state, and the incubation with the autophagy inhibitor 3-MA increased intracellular apoB100 recovery in KLHL12 siRNA-treated cells. (D) Confocal indirect immunofluorescence images from McA cells probed with antibodies to apoB100 (red), COPII (red), or albumin (red), or KLHL12 (green) as indicated. Arrows in the insets indicate co-localization (yellow) of apoB100, COPII, or albumin with KLHL12. Nuclei were stained with DAPI. Magnification is 63X. The inset is a ×10 zoom of the area of interest. (E) Quantification of KLHL12 co-localization with either albumin or apoB100. A,B,C: shown are the means, +/− SEM, n=3. *p < 0.05, and **p < 0.01. D: shown are the results representative of 3 independent experiments. E: same as A,B,C, but n > 10 cells analyzed for each protein.
Our laboratory and others have shown that apoB100 can be degraded by either the proteasome-mediated ER-associated degradation (ERAD) or by autophagy after ER exit (reviewed in 15). To define the fate of apoB100 after KLHL12 knockdown, we first focused on the role of autophagy, given that it is highly unlikely that an assembled apoB100-VLDL particle could “retro-translocate” from the ER lumen and gain access to the cytoplasmic proteasome, which must occur in ERAD. In contrast, autophagy is known to collect and destroy large substrates, even those that reside within cellular organelles (reviewed in 17). Consistent with this hypothesis, the autophagy inhibitor, 3-methyladenine (3-MA), significantly increased the level of intracellular apoB100 in KLHL12 knockdown cells (Fig. 2C).
Finally, we asked whether KLHL12 co-localizes with apoB100, which would provide additional support that this factor facilitates apoB100-VLDL secretion. By utilizing a monoclonal antibody specific for rat apoB100, we observed that KLHL12 co-localized ~ 8X more (p<0.01) with apoB100 than with albumin, and that KLHL12 co-localized with COPII-positive vesicles (Fig. 2D and E). Furthermore, the data also verified KLH12 association with the ER and not with the Golgi (online-only Data Supplement Fig. X). These co-localizations of KLHL12 are consistent with the sub-cellular distribution previously reported in the pro-collagen studies7.
In summary, our data demonstrate for the first time that KLHL12 plays a crucial role in delivering apoB100-VLDL particles to the Golgi apparatus for further maturation, an event that ultimately leads to the secretion of mature VLDL. Furthermore, KLHL12 appears to be a limiting factor that acts during ER-to-Golgi trafficking, at least in McA cells, since either increasing or decreasing KLHL12 levels had a significant effect on apoB100-VLDL secretion. We also find that autophagy is most likely responsible for the degradation of apoB100-containing particles that escape ERAD, but do not progress to the Golgi. The atypical physical properties of the ER-derived secretory vesicles we previously described1 may have reflected a difference in physical structure relative to those vesicles carrying smaller cargo, but also the presence of additional proteins needed to form them, such as KLHL12 (this study) and others2, 18. Our next goal is to confirm the function of KLHL12 in vivo, and to determine the molecular mechanism and relationships among KLHL12 and other factors required for apoB100-VLDL assembly and trafficking.
Supplementary Material
Significance.
It has been speculated that the transport of apoB-containing lipoproteins in secretory vesicles involves novel factors, given the size of VLDL and chylomicrons produced by the liver and small intestine, respectively. Recently it was reported that a factor called KLHL12 regulates the ER to Golgi trafficking of the large cargo pro-collagen by modifying the COPII vesicle component sec31. We now show that KLHL12 function is also required, and may even be limiting, for the secretion of apoB100-containing lipoproteins by hepatic cells. As for pro-collagen, the evidence supports a role for KLHL12 in the ER to Golgi trafficking step of secretion. The findings, then, add a new player to the repertoire of factors required in the complex process of apoB-containing lipoprotein production.
Acknowledgments
We thank Dr. Janet Sparks (University of Rochester) for providing the rat apoB100 specific monoclonal antibody and Dr. Randall Moon (University of Washington) for the gift of the KLHL12 expression plasmid.
Sources of Funding: This study was supported by National Institutes of Health grant HL58541 (to E.A.F. and J.L.B). Liang Guo received support from postdoctoral fellowship 13POST16810071 from American Heart Association.
Disclosures: None
Abbreviations
- KLHL12
Kelch-like protein 12
- VLDL
very low density lipoprotein
Non-standard abbreviations
- COPII
coat complex II
- CUL3
Cullin3
- DAPI
4',6-diamidino-2-phenylindole
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- KLHL12
Kelch-like protein 12
- MCA
McArdle RH-7777
- OA
oleic acid
- 35S-met/cys
35S-methionine and cysteine
- 3-MA
3-methyl adenine
- TCA
tri-chlor acetic acid
References
- 1.Gusarova V, Brodsky JL, Fisher EA. Apolipoprotein B100 exit from the endoplasmic reticulum (ER) is COPII-dependent, and its lipidation to very low density lipoprotein occurs post-ER. J Biol Chem. 2003;278:48051–48058. doi: 10.1074/jbc.M306898200. [DOI] [PubMed] [Google Scholar]
- 2.Rahim A, Nafi-valencia E, Siddiqi S, Basha R, Runyon CC, Siddiqi SA. Proteomic analysis of the very low density lipoprotein (VLDL) transport vesicles. J Proteomics. 2012;75:2225–2235. doi: 10.1016/j.jprot.2012.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuge O, Dascher C, Orci L, Rowe T, Amherdt M, Plutner H, Ravazzola M, Tanigawa G, Rothman JE, Balch WE. Sar1 promotes vesicle budding from the endoplasmic reticulum but not Golgi compartments. J Cell Biol. 1994;125:51–65. doi: 10.1083/jcb.125.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schekman R, Orci L. Coat proteins and vesicle budding. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- 5.Rothman JE, Wieland FT. Protein sorting by transport vesicles. Science. 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- 6.Schekman R, Mellman I. Does COPI go both ways? Cell. 1997;90:197–200. doi: 10.1016/s0092-8674(00)80326-8. [DOI] [PubMed] [Google Scholar]
- 7.Jin L, Pahuja KB, Wickliffe KE, Gorur A, Baumgartel C, Schekman R, Rape M. Ubiquitin-dependent regulation of COPII coat size and function. Nature. 2012;482:495–500. doi: 10.1038/nature10822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stagg SM, LaPointe P, Razvi A, Gurkan C, Potter CS, Carragher B, Balch WE. Structural basis for cargo regulation of COPII coat assembly. Cell. 2008;134:474–484. doi: 10.1016/j.cell.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fath S, Mancias JD, Bi X, Goldberg J. Structure and organization of coat proteins in the COPII cage. Cell. 2007;129:1325–1336. doi: 10.1016/j.cell.2007.05.036. [DOI] [PubMed] [Google Scholar]
- 10.Fromme JC, Schekman R. COPII-coated vesicles: Flexible enough for large cargo? Curr Opin Cell Biol. 2005;17:345–352. doi: 10.1016/j.ceb.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 11.Tran K, Thorne-Tjomsland G, DeLong CJ, Cui Z, Shan J, Burton L, Jamieson JC, Yao Z. Intracellular assembly of very low density lipoproteins containing apolipoprotein B100 in rat hepatoma mca-RH7777 cells. J Biol Chem. 2002;277:31187–31200. doi: 10.1074/jbc.M200249200. [DOI] [PubMed] [Google Scholar]
- 12.Olofsson SO, Boren J. Apolipoprotein B: A clinically important apolipoprotein which assembles atherogenic lipoproteins and promotes the development of atherosclerosis. J Intern Med. 2005;258:395–410. doi: 10.1111/j.1365-2796.2005.01556.x. [DOI] [PubMed] [Google Scholar]
- 13.Gusarova V, Seo J, Sullivan ML, Watkins SC, Brodsky JL, Fisher EA. Golgi-associated maturation of very low density lipoproteins involves conformational changes in apolipoprotein B, but is not dependent on apolipoprotein E. J Biol Chem. 2007;282:19453–19462. doi: 10.1074/jbc.M700475200. [DOI] [PubMed] [Google Scholar]
- 14.Fisher EA, Pan M, Chen X, Wu X, Wang H, Jamil H, Sparks JD, Williams KJ. The triple threat to nascent apolipoprotein B. Evidence for multiple, distinct degradative pathways. J Biol Chem. 2001;276:27855–27863. doi: 10.1074/jbc.M008885200. [DOI] [PubMed] [Google Scholar]
- 15.Brodsky JL, Fisher EA. The many intersecting pathways underlying apolipoprotein B secretion and degradation. Trends Endocrinol Metab. 2008;19:254–259. doi: 10.1016/j.tem.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ginsberg HN, Fisher EA. The ever-expanding role of degradation in the regulation of apolipoprotein B metabolism. J Lipid Res. 2009;50 (Suppl):S162–166. doi: 10.1194/jlr.R800090-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Z, Klionsky DJ. Eaten alive: A history of macroautophagy. Nature Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tiwari S, Siddiqi S, Siddiqi SA. Cideb protein is required for the biogenesis of very low density lipoprotein (VLDL) transport vesicles. J Biol Chem. 2013;288:5157–5165. doi: 10.1074/jbc.M112.434258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.