


Abstract
One of the most striking examples of small RNA regulation of gene expression is the process of RNA editing in the mitochondria of trypanosomes. In these parasites, RNA editing involves extensive uridylate insertions and deletions within most of the mitochondrial messenger RNAs (mRNAs). Over 1200 small guide RNAs (gRNAs) are predicted to be responsible for directing the sequence changes that create start and stop codons, correct frameshifts and for many of the mRNAs generate most of the open reading frame. In addition, alternative editing creates the opportunity for unprecedented protein diversity. In Trypanosoma brucei, the vast majority of gRNAs are transcribed from minicircles, which are approximately one kilobase in size, and encode between three and four gRNAs. The large number (5000–10 000) and their concatenated structure make them difficult to sequence. To identify the complete set of gRNAs necessary for mRNA editing in T. brucei, we used Illumina deep sequencing of purified gRNAs from the procyclic stage. We report a near complete set of gRNAs needed to direct the editing of the mRNAs.
INTRODUCTION
In Trypanosoma brucei, expression of the mitochondrial genome involves one of the most striking examples of small RNA directed regulation, RNA editing (1,2). In these parasites, hundreds of small RNAs direct the insertion and deletion of uridylate (U) residues needed to generate translatable mRNAs. The RNA editing process is developmentally regulated and alternative editing has been detected, creating the opportunity for unprecedented protein diversity (3,4). The small guide RNAs (gRNAs) are key components of RNA editing and are all encoded in the mitochondrial genome. This genome consists of several thousand, interlocked, circular DNA molecules organized into a disk-like structure called the kinetoplast or kDNA (5). Each cell has one mitochondrion and one kDNA network made up of maxicircles and minicircles. The kDNA maxicircle (∼22 kb) encodes 18 proteins, 2 ribosomal subunits and 2 gRNAs, and is present in ∼50 copies within the network. All other gRNAs are encoded on the minicircles that make up the bulk of the mitochondrial genome. Each network can contain from 5000–10 000 minicircles composed of ∼250 different minicircle sequence classes (6). With each minicircle encoding ∼3–5 gRNAs, this component of the genome has the capacity to encode over 1200 different gRNAs (7–9).
Despite the importance of the gRNAs to the editing process, a full complement has only been described in one laboratory strain of Leishmania tarentolae (10). Editing in this strain is limited, and five of the normally pan-edited genes are not productively edited. In T. brucei, a number of studies using conventional sequencing methods have been done in the attempt to identify gRNAs (11–13). However, despite these attempts, large numbers of the gRNAs needed for the extensive editing of the protein coding genes were still unidentified. We report here the characterization of the gRNA transcriptome of the procyclic stage of T. brucei using deep sequencing of purified mitochondrial gRNAs. Within this transcriptome, we have identified the full complement of gRNAs needed to direct the editing of ATPase 6 (A6), cytochrome oxidase III (COIII), C-rich region 4 (CR4), cytochrome b (CYb) and ribosomal protein subunit 12 (RSP-12). In contrast, a full complement of gRNAs were not identified for C-rich region 3 (CR3), maxicircle unidentified reading frame II (Murf II), NADH dehydrogenase (ND) subunits 3, 7, 8 and 9. Most striking was the large variation in transcript copy number observed for the identified gRNAs.
MATERIALS AND METHODS
Parasites, isolation of mitochondria and RNA extraction
T. brucei clone IsTar from stock EATRO 164 was grown in SDM79 and harvested at a cell density of 1–3 × 107 cells/ml. Harvested trypanosomes were washed in sodium buffered glucose (SBG), resuspended in DTE buffer and disrupted using a sterile Dounce homogenizer as previously described (14). Cell lysate was then treated with RNAse-free DNAse I (5 µ/ml) and incubated on ice for 45 min. The reaction was stopped by addition of an equal volume of STE (250 mM Sucrose, 20 mM Tris (pH 7.9), 2 mM EDTA) and cells/organelles collected by centrifugation (16 000 g, 10 min). Mitochondrial vesicles were then collected using a series of differential spins. Briefly, initial pellets were resuspended in STE and cleared of large particles and cell debris using two low-speed spins (1500 g, 10 min). Mitochondrial vesicles were then collected by centrifugation at 16 000g (10 min). The enriched mitochondrial pellet was then lysed using an acidic phenol–CHCl3 extraction in the presence of 4 M guanidinium isothiocyanate and 2% (w/v) sodium-N-lauroyl sarcosinate (15). The mitochondrial RNA (mtRNA) was precipitated, washed and resuspended in RNAse-free water. Alternatively, collected parasites were immediately lysed using the acidic phenol–CHCl3 protocol and total RNA collected.
Library preparation and Illumina sequencing
Approximately 100 µg of mitochondrial or total RNA was treated with DNAse RQ1 (Promega) and then size-fractionated by denaturing 10% (w/v) polyacrylamide electrophoresis (8 M urea). A gRNA marker lane was generated by 5′ capping 10 µg of mtRNA using 32P αGTP and Vaccinia capping enzyme (BioLabs) according to manufacturer’s directions. RNAs in the gRNA size range (∼40–80 nt) were excised from the gel, passively eluted and ethanol precipitated. To preserve strand information, we used a modified Illumina ‘Small RNA’ sample preparation protocol. The gRNAs have a 5′ tri-phosphate and a 3′ hydroxyl group. This allows the direct ligation of the RNA 3′ adaptor. However, addition of the RNA 5′ adapter required phosphatase treatment followed by polynucleotide kinase (PNK) to add a single phosphate. Ligation of the 5′ adapter was followed by RT-PCR amplification and gel purification of the gRNA library. The gRNA library, as determined by an Agilent Bioanalyzer, had a narrow distribution, centered at ∼135 bp, consistent with the estimated size of the gRNAs (plus adapters). Each library (gRNAs isolated from mtRNA and gRNAs isolated from total RNA) was sequenced on a Illumina GAIIx (single read 75 base run). Approximately 30 million raw reads were obtained from each of the gRNA libraries. After removal of the Illumina adapter sequences, quality-based trimming was done with prinseq (stand alone lite version, http://prinseq.sourceforge.net/). Reads with two or more N's or an overall mean Q-score < 25 were discarded. The 3′ end was further trimmed of low quality bases (mean Q-score < 20 over a 5 base window); any reads < 20 nt after trimming were discarded. Only a small fraction of reads were discarded at this step. Input raw reads were further processed using the following criteria: (i) remove redundant reads. The number of redundant reads is kept for each unique sequence; (ii) remove reads without at least four consecutive Ts.
RESULTS
Identification of gRNAs
To identify gRNAs that direct the editing process of the known mRNAs, we aligned each transcript read to the conventionally edited mRNAs based on known base-pairing mechanisms. A legal alignment between gRNA and the edited mRNA mainly contains canonical Watson–Crick DNA base pairs and the G-U base pair. We note that a small number of other types of base pairs may also exist in the alignment; however, these were not allowed in our initial screen. In addition, we allowed no gaps in the alignment, allowing us to formulate the gRNA-mRNA alignment problem as an extended longest common substring (LCS) problem. The LCS problem outputs the LCS between two input sequences. To use LCS to identify the gRNA-mRNA alignments, we defined match and mismatch base pairs as follows: (i) match: canonical Watson–Crick and G-U base pairs; and (ii) mismatch: any other type of base pair. Based on this definition, we formulated the extended LCS problem as follows: given two sequences x and y, find the LCS between x and y with at most T mismatches. The problem was solved using dynamic programming. The sub-problem is denoted using function LCS(i, j, τ), representing the length of the LCS ending at position i and j in two input sequences x and y with exact τ mismatches (τ ≤ T). Thus, the length of the LCS between x and y with τ mismatches is max1≤i≤|x|,1≤j≤|y| [LCS(i, j, τ)]. When T = 0, the extended LCS problem is reduced to the original LCS problem.
The following recursive functions are used to solve LCS(i, j, τ).
xi is the ith character of x. yj is the jth character of y.
Recursive functions:
if xi and yj form a match
LCS(i, j, τ) = LCS(i-1, j-1, τ) + 1
else
LCS(i, j, τ) = LCS(i-1,j-1, τ-1) + 1 for 1 ≤ τ ≤ T
We need to compute LCS(i, j, τ) for 1 ≤ i ≤ |x|, 1 ≤ j ≤ |y| and 0 ≤ τ ≤ T. The initialization is LCS(0, 0, τ) = 0. Once we record the maximum of LCS(i, j, τ) for all indexes in sequence x and y, we can easily recover the LCS itself. During our analyses, we did allow for at most three mismatches (i.e. T = 3). Thus, we have the alignments between gRNAs and edited mRNAs with 0 mismatches to 3 mismatches. However, the transcript reads that can be aligned with edited mRNAs with less number of mismatches have higher probability to be real gRNAs. Thus, when the edited sites in an mRNA can be aligned with gRNAs with τ mismatches, we did not use gRNA alignments containing τ+1 mismatches. Matched gRNAs were then scored as follows, two points for canonical Watson–Crick base pairs and one point for G-U base pairs. gRNAs with scores >45 were identified as guiding a specific region based on the identified mRNA fully edited sequence (numbered from the 5′ end). Using this criterion with the 0 mismatch data set, we found that all of the identified gRNAs had characteristics indicating that they were matched to the correct position. The matched gRNAs were sorted based on their guiding positions, and the populations analyzed and sorted into sub-populations (sequence variants that guide the same or nearly the same region). Using these data, we were able to generate two additional data files: (i) a best align file containing the highest scoring gRNA for any specific region and (ii) a coverage profile, containing the number of gRNAs that cover any specific nucleotide within the fully edited mRNA. Alignments of the identified gRNAs with the fully edited mRNA sequences indicated that we had identified a near full set of gRNA required for the editing of the mRNAs. Full coverage was obtained for A6, COIII, CR4, CYb and RSP-12. Full complements of gRNAs were not identified for CR3, Murf II or the ND subunits (ND3, ND7, ND8 and ND9). The identified gRNAs and the full gRNA-mRNA alignments for one fully edited mRNA, ATPase 6, are shown in Table 1 and in Figure 1. This mRNA is extensively edited and the data presented illustrate several key points. The identified gRNAs and the alignments for all other transedited mRNAs can be found in the supplemental data. gRNAs are designated by their guiding position on the fully edited mRNA. Both nucleotides and deletion sites in the fully edited mRNA were defined by a number, starting from the 5′ end (+1 = 0).
Table 1.
The major gRNA classes involved in the editing of ATPase 6
| mRNA 5′ | mRNA 3′ | Copy no. | ATPase 6: Major gRNA classes |
|---|---|---|---|
| 24 | 72 | 6a | ATATAC AACGCAACCAGAGTAAATCATGAAGGGAAAGTGAAGGCATATTTGTTTT T15 |
| 29 | 72 | 1630 | ATATAC AACGCAACCAGAGTAAATCATGAAGGGAAAGTGAAGGCATATTT T11 |
| ✰31 | 75 | 2044 | AT ATAAACGTAACTGAAATGAATCACGAGAGAAAGATAAAGATATAT AT12 |
| ✰31 | 75 | 143 | AT ATAAACGTAACTGAAATGAATCGCGAGAGAAAGATAAAGATATAT ATTTTTGT15 |
| ✰62 | 102 | 1435 | ATACA ATCATACACAGTAGTACATATATAGTGATAGACGTGATTAA T11 |
| ✰84 | 127 | 4a | ATAT AAATACACAGTAGAATATGATCTAGGTTATGTATGATGATATAT T14 |
| ✰86 | 127 | 2158 | ATAT AAATACACAGTAGAATATGATCTAGGTTATGTATGATGATAT T10 |
| ✰105 | 152 | 54a | AC ATCAAAAATCGACATTAGATAATTGAGGTATGTGATAGAGTATAATTT T5GT5 |
| ✰113 | 152 | 743 | ATAC ATCAAAAATCAACGTTAGACAGTTAAGATATGTGATAGAA GATAAT12 |
| ✰135 | 183 | 1a | ATATAAATCAAACAAACAGAATAGTAGAAAGTCAGAGATTGATGTTAA T11 |
| ✰138 | 183 | 430 | AT ATACAAATCAAACAGACAGAGTAATAGAAGGTTGAAGATTGATAT AGT11 |
| 144 | 177 | 210 | ATATC ATCAAACAAACAGAATAATAGAGAATCAGAGGT GAATGTTAAGT15 |
| ✰158 | 208 | 14a | ATAT ACAAACACAAACTGACGAATAGATACAGATTAAGTGAATGAAATAAT T11 |
| 164 | 208 | 54 | ATAT ATAAACACAAATCAACGAATAGATATAAGTCAGATAGATGG TGTATTAT12A11 |
| ✰176 | 210 | 36 | AT AAACAAACACAAATCAGTAGACGAGTACAAGT GAGATGGACGTATAGAT7 |
| ✰165 | 208 | 24 | ATAAT ACAAACACAAACTGATAGACGAATACGAGTTAGATGGACG TAT6 |
| ✰189 | 243 | 3a | ATATAAATTAAACAGCATAAACTGTAGCAGTGAAGATAGATGTGAATTAATA T14 |
| ✰192 | 243 | 172 | ATATAAATTAAACAACATAGATTACAGTGATAGAAGTAAATGTGAATTA T4 |
| ✰218 | 248 | 147 | ATC AGACTATGTGAGTTAGATGACGTGAATTATA CTGTATAT12 |
| ✰224 | 269 | 864a | ACATAA TAATACAATAATACGAGATTAGACTATGTGAATTAAATGATATGA T11G |
| ✰226 | 269 | 808 | ACATAA TAATACAATAATACGAGATTAGACTATGTGAATTAAATGATAT T8GT4 |
| ✰249 | 299 | 4a | AAAT AAACAACAAATATGAGTTCGAATAAGTGATATAATGGTATAAAATT T11 |
| ✰248 | 292 | 25 157 | ATATA AAATACAAATTCGAGTAGGTAGTACAATGATATGAGATTA T13 |
| ✰252 | 292 | 134 | ATATA AAATACAAATTCGAGTAGGTAGTACAATGATATAGA TTATTAAT7 |
| ✰255 | 292 | 244 | ATATA AAATACAAATTCGAGTAGGTAGTACAATGATAT TATTATTAAT15 |
| 253 | 298 | 5405 | ATATAT AACAACAAATATAGATTCAAGTAAGTGATGTAGTAATATGA T11 |
| ✰266 | 313 | 586 | AAAAAA AAAAAAACAATACAAGATGACAGGTATAAGTTTGGATGAGTAAT T12G |
| 300 | 346 | 263a | ATAT AAACAAAACAGAAATAGAAATGCAATATACGATAAGAAAATGGTATA T12 |
| ✰301 | 345 | 647 | ATAT AACAAAACAAAAGTAGAAGTGCAGTATATGATAGAAAAATGATGT CAAAT11 |
| ✰301 | 335 | 125 | ATAT ACAAAACAT AAATAAAAGTGCAGTATATGATAAAGAGATAATAT T11 |
| ✰331 | 375 | 24 736 | ATAT AATTATTAAACAAGAGAAAGTCACGTAAAAGGTAGAATGAAGATA TTTTTCT6 |
| ✰331 | 375 | 712 | ATAT AATTATTAAACAAGAGAAAGTCACGTAAAAAGTAGAATGAAGATA TTAT5 |
| ✰332 | 378 | 8776 | AT ATAAATTATTAAACAGAAAGAGATCATGTAGAAAGTGAGATAGAAAT T12CT |
| 331 | 371 | 3561 | ATATAA ATTAAACAAAAAGAAATCACGTAGAAGACAGAATAGAGATA T12G |
| 331 | 374 | 302 | ATAT ATTATTAAACAAAGAGAAATCATATAAGAGACAGAATGAGAATA T9AT5 |
| ✰332 | 378 | 387 | AT ATAAATTATTAAACAGAAAAGAGTCATATAGAAAATAAGATAGAAAT T12 |
| ✰332 | 378 | 144 | AT ATAAATTATTAAACAGAAAGAGATCATGTAGAAAGTGAGATAAAAAT T3 |
| ✰349 | 389 | 41 | ATATAA ATCACCAACTAATAAGTTATTGAATGAGAGAAAGTTATATA T12 |
| ✰360 | 407 | 181 | ATATAT ACATCCATAAAATTATCATCAGTTAATAGATTGTTAAATGAAAA T4 |
| 387 | 435 | 1428 | ATATAT AACACAACAAGAAACGAATGAGAGAAGTATCTATGAGATTATT T9CGT3CTTCT |
| ✰387 | 435 | 1049 | ATATAT AACACAACAAGAGACGAATAGAAAAGATATCTGTGAAATTATT T10ATT |
| ✰387 | 437 | 934a | ATATAT AAAACACAATAGAAAACGGATAAGAGAGATATTCATAGAGTTATT T9GTTT |
| 413 | 461 | 3a | ATAT ATACAACAAAGAAAGACACTCTAGAAGATACAGTGAGAGATGAGTAA T11 |
| ✰424 | 464 | 25 624 | ATAT ATGACACAACGAGGGAAGATACTCTAAAGGACACAGTGAAA T12 |
| ✰427 | 467 | 2307 | ATAT ATAACGACACAATAGAGAAAGATGCTCTGAGAGATGTAATA T12G |
| ✰421 | 460 | 1864 | ATAAAT TACAACAAAGAAAGATACTCTAGAAAGCACAGTGAGAAAT T8CT7 |
| 424 | 457 | 368 | AAATTAACGACA AACAAAGAGAAATACTCTGAGAAATATGATGAAA T12 |
| ✰455 | 491 | 368 | ATATATAATTAC AAACAAACGCAGAGATGTCGGTAAATAATGATATAAT T11 |
| ✰455 | 497 | 22a | ATAT ATTACAAAACAGACGTAAAGATGTCGATGAATGGTGGTATAAT T14 |
| ✰487 | 528 | 1a | ATAC ACATCAACAATAGAAGATGGGATGATAATAGATTGTGAGATA T27 |
| ✰487 | 526 | 8723 | ATACAA ATCAACAATAGAAGATGGGATGATAATAGATTGTGAGATA T16 |
| ✰521 | 567 | 232 | AA AAAAAAAAAAAACAAAAATAGAATAAAGAAAGTCAGAGAATGTTAAT T5 |
| ✰546 | 593 | 15a | AATAAATCGATAACAAAGAACACTGTAAAAAAAGAGAATGAGAGTAAA TATAT4 |
| ✰549 | 593 | 2587 | AC AATAAATCAATAACAGAGAATATCATAGAGAGGAAAGATAGAAAT T12GTTTGTACTT |
| ✰549 | 592 | 181 | ATAT ATAAATCAATGACAAGAAGCACTGTAGAAAAAGAGAGTGAAAAT TTTTAT8 |
| ✰557 | 593 | 69 619 | AATAAATCGATAACAAAGAACACTGTAAAAGAGAGAA TGAGAGTAAATAT9 |
| 568 | 611 | 670 | ATACT AAACACAAAAATGAATAAAATAAGTCAGTGATAGAAGATATTAT T12 |
| 583 | 629 | 2a | AT AAATAATAAACAGAAACAGAGCATAGAAGTAAGTAGAGTGAATTAAT T11 |
| 589 | 629 | 854 | AT AAATAATAAACAGAAACGGAATACGAGAATAAGTAAAGTGA TTTAAT13 |
| 612 | 657 | 3a | AT ATAAATCCAACAAGTATAAGAACATATAGAATAGTAGGTGAAAATA T6A |
| 613 | 654 | 618 | ATATAT AATCCAACAGATATAAGAGCATGTAAAATAGTAAGTGAAAAT T10AT |
| ✰613 | 657 | 183 | AT ATAAATCCAACAAGTATAAGAACATATAGAATAGTAGGTGAAAAT T7CT4 |
| ✰638 | 689 | 5a | ATAT ATAAATAACTGTAGTATGGTGGTAGATGAGTTTGATAGATATAAA T9 |
| ✰640 | 689 | 39 063 | ATAT ATAAATAACTGTAGTATGGTGGTAGATGAGTTTGATAGATATA T12 |
| ✰647 | 689 | 678 | ATAT ATAAATAACTGTAGTATGGTGGTAGATGAGTTTGAT T11 |
| ✰640 | 689 | 131 | ATAT ATAAATAACTGTAGTATGGCGGTAGATGAGTTTGATAGATATA T11 |
| ✰654 | 689 | 234 | ATAT ATAAATAACTGTAGTATGGTGGTAGATGA TTTTGATAGATATAT12 |
| ✰672 | 716 | 119a | ACACA ATCAACTGCAGAATTATATTACAGAGAGTGAGTAATTGTAA AAT12 |
| ✰680 | 714 | 7581 | AAAATA CAACTGCAAGATCGTGTTATAGAGGATAAGTGATT TAAT13 |
| ✰680 | 714 | 105 | AAATA CAACTGCAAGATCGTGTTGTAGAGGATAAGTGATT TAAT11 |
| ✰680 | 719 | 1291 | ATATAA ATTATCAACTGTGAGATTATATTACAAGGAATAAGTGATT T11AT |
| ✰686 | 728 | 12a | ATATATT AAAATCCATTATCGATTGTAGAGTTATGTTATAGAGAATAA TAT21 |
| ✰698 | 728 | 740 | ATT AAAATCCATTATCGATTGTAGAGTTATGT GATAGAGAATAAT11 |
| ✰715 | 760 | 2a | AT ATATAAAACTAAACAAATAGCAAAGACAGTGAGAGATTCGTTAT AAAT13 |
| ✰715 | 755 | 2272 | ATATATAT AAACTAAACAAATAGCAGAGACAGTGAGAGATTCGTTAT AAT13 |
| ✰720 | 767 | 4588 | AT AAATCAAATACAGAACTGAATAGACGATAAAGATAGTGAGAAATTT T10G |
| ✰720 | 767 | 165 | AT AAATCAAATACAGAACTAGATGAACAATAGAGATAGTGAGAAATTT TTTTTCT6 |
| ✰728 | 765 | 920 | AT ATCAAATACAAAACTGAGCAGATGACAGAGATAGTAAA TGATTTAT11G |
| ✰747 | 789 | 13 | ATAAAT ACAACAATATAATAACTGTCGAAGGTTGAATATGAGATTAAAT T11 |
| 770 | 822 | 1 | GGA CTATAACTCCGATAACGAATCAGATTTTGACAGTGATATGATAATTATT TCCCT3CTTCTC |
| ✰774b | 822 | 8663 | ATA CTATAACTCCAATGACGAAATCAGTTTTACAGTGATATGATAA T14 |
The gRNA is identified by its complementarity to the 5′ (column 1) and 3’ (column 2) number of the fully edited mRNA (+1 = 0). The gRNAs were sorted based on both mRNA regions covered and on guiding sequence class. Sequence variations observed in both the 5′ non-complementary region and the 3′ U-tail were ignored in assigning sequence classes. Transcript copy numbers (column 3) were determined by adding all gRNAs of the same sequence class. Major sequence classes were defined as containing greater than 100 transcript copies. The exact sequence shown is of the most abundant transcript in each sequence class. In the case of rare gRNA transcripts, the identified gRNAs are shown regardless of copy number. The asterisk indicates the highest scoring gRNAs identified for each population. Starred (✰) gRNAs indicate novel gRNAs not found in the KISS database (http://splicer.unibe.ch/kiss). A total of 84% of the gRNAs identified in this study are ‘novel gRNAs’, not previously identified.
aIdentified highest scoring gRNA (‘Best align’).
bmRNA 5′ border based on alternative sequence.
Figure 1.

The gRNA-mRNA sequence alignment for fully edited ATPase 6. The cDNA sequence of the most abundant gRNA in its sequence class is shown aligned beneath the fully edited mRNA. Lowercase u’s indicate uridines added by editing, asterisks indicate encoded uridines deleted during editing. Nucleotides and deletion sites in the fully edited mRNA were numbered starting from the 5′ end (+1 = 0). gRNAs are colored (on-line version only) based on transcript abundance as follows: Blue < 100; Green < 1000; Purple < 10 000; Orange < 100 000; Red > 100 000; Black = not quantified. Watson-Crick (|) and G:U base pairs (:) are indicated. Mismatches are indicated by the number sign (#) and shown in a contrasting color. The potential mRNA sequence generated by a more abundant alternative A6 initiating gRNA is also shown in Figure 1. Although this gRNA would introduce a number of sequence changes (indicated in red online) downstream of the stop codon (underlined), the generated anchor sequence for the next gRNA is maintained.
Overall characteristics of the gRNA populations
Analyses of the gRNA populations for the extensively edited (pan-edited) mRNAs indicate that editing involved a large number of gRNA populations for full editing. For example, sequence analyses identified 31 distinct gRNA populations involved in the editing of A6 and 40 involved in the editing of COIII (Table 1, Supplementary Table S3). In addition, most of the major populations (population defined as guiding the same or near same region of the mRNA) contained multiple sequence classes. In our initial sorting of the gRNA populations, it was often difficult to initially assign gRNAs to a specific population group, as the gRNA sequence classes have significant border variations at both the 5′ and 3′ ends. Variation at the 5′ end was often due to truncation of the sequence, suggestive of a distinct 5′–3′ exonuclease activity (Figure 2). Variation in the U-tail addition site was also often observed. For example, gA6 (224–269) and gA6 (226–269) differ only by the presence of a GA that may be due to differences in polyU site selection (Table 1). Similarly, although there are 14 major sequence classes that guide the COIII 699–753 region, they can be sorted into two distinct populations (Table 2). gCOIII (699–748) and gCOIII (701–748) have near identical guiding regions, differing by a single A residue that again may be due to differences in polyU site selection. The other main population guides the editing of a region within the mRNA that is shifted downstream by only 5 nt (706–753). In this population, there are six major sequence groups. Within each group, the different sequence classes are defined by differences at the 3′ polyU site. The different groups, however, are defined by distinct differences in the sequence of the guiding region. Although groups 2 and 3 differ from group 1 by single nucleotide change (bold and underlined), other groups show multiple differences in gene sequence, all R to R or Y to Y changes, allowing multiple gRNAs to guide the generation of the same mRNA sequence.
Figure 2.

Example of sequential 5′ end truncations suggestive of a 5′–3′ exonuclease activity. The gA6 (248–292) sequence class was large (∼25 000 transcripts, containing a large number of transcripts with both 5′ truncations and sequence variations in the U-tail (length of U-tail and U-tail punctuated with other nucleotides). The transcript numbers reported are for the specific sequence shown (i.e. 6788 cDNAs with a T-13 tail).
Table 2.
Major gRNA classes for the COIII 699–753 region
| mRNA 5′ | mRNA 3′ | Copy number | Major gRNA sequence classes for the COIII 699–753 region |
|---|---|---|---|
| 699 | 748 | 977 | ATATA TAATAAATCCAATGAAGATAAAGTAGAGTCAGAGATATTATGATTT TTTTTTTTTT |
| 701 | 748 | 2726 | ATA TAATAAATCCAATGAAGATAAAGTAGAGTCAGAGATATTATGAT ATTTTTTTTTTTTT |
| 7061 | 753 | 31 331 | ATATAT AAATGTAATAGATCTGATGAAAGTGAGGTAGAATTGAGAATATT TTTTTTTTTT |
| 7071 | 753 | 8037 | ATATAT AAATGTAATAGATCTGATGAAAGTGAGGTAGAATTGAGAATAT ATTTTTTTTTTTTTT |
| 7131 | 753 | 595 | ATATAT AAATGTAATAGATCTGATGAAAGTGAGGTAGAATTGAGAAT TTTTTTTTTAACCC |
| 7151 | 753 | 182 | ATATAT AAATGTAATAGATCTGATGAAAGTGAGGTAGAATTGAGA TTTATTTTTTTT |
| 7192 | 753 | 131 | ATATAT AAATGTAATAGATCTGATGAAAGTGAGGTAGAATT TAGAATATATTTTTTTT |
| 7073 | 753 | 206 | ATATAT AAATGTAATAGATCTGATAAAAGTGAGGTAGAATTGAGAATAT ATTTTTTATTTTTT |
| 7064 | 753 | 6744 | ATATAT AAATGTAATAGATCCAATGAAGGTAAGATAGAACTGAGAATATT TTTGTTTTT |
| 7074 | 753 | 3791 | ATATAT AAATGTAATAGATCCAATGAAGGTAAGATAGAACTGAGAATAT AATTATTTTTTT |
| 7134 | 753 | 154 | ATATAT AAATGTAATAGATCCAATGAAGGTAAGATAGAACTGAGAAT TTTTTTTGTTTTTT |
| 7065 | 753 | 1214 | ATATAT AAATGTAATAGATTCAATGAAGGTAAGATAGAACTGAGAATATT TTTTCTTTT |
| 7075 | 753 | 1124 | ATATAT AAATGTAATAGATTCAATGAAGGTAAGATAGAACTGAGAATAT AATTTTTTTTTTTTTT |
| 7076 | 752 | 848 | ATATAT AATGTAATAAATCTAATAGAGATAAGATAGAACTGAGGATAT ATTTTTTTTTTTT |
Fourteen major sequence classes that fall into two distinct populations were identified that could guide the editing of the 699–753 region. The two populations are shifted by 5 nt in their guiding regions and have 13 nt differences in the overlap region (all R to R and Y to Y changes allowing them to guide the generation of the same sequence). The 706–753 population can be further divided into six major sequence groups as indicated. Within each group the different sequence classes differ in the position of the polyU tail (changing the mRNA 5′ border). The different groups, however, are defined by distinct differences in the sequence of the guiding region (nucleotide changes shown in bold and underlined).
Analyses of the gRNAs indicate a number of other interesting features. The shortest and longest gRNAs identified in our search had 24 nt and 61 nt of complementarity to their edited mRNA, respectively. Most of the gRNAs (64%), however, had 38–48 nt of complementarity (Figure 3). In addition, most of the gRNAs had few ‘extra’ nt 5′ or 3′ to the anchor and guiding regions (Figure 4A and B). This was most striking at the 3′ end, where over 50% of the sequence classes contained no non-guiding nucleotides prior to the post-transcriptionally added U-tail. At the 5′ end, 84% of the transcripts had six or fewer nucleotides 5′ to the anchor sequence. In addition, for most of the gRNAs with a large leader sequence, the end of the anchor match was defined by a point mutation, with nucleotides 5′ to the mutation able to base pair with the anchor binding site. Although for some of the gRNAs, a mismatch within a large anchor may be tolerated, for others it signals a possibility for editing anomalies. An example of this is gND7 (152–190), which directs editing of the same region as gND7 (147–199), the initiating gRNA for the 5′ editing domain. gND7 (152–190) has a single T insertion 14 nt from the 5′ end that defines the 5′ anchor border of the gRNA (Figure 5). Analyses of the nucleotides upstream of the T insertion indicate that they can pair with 11 consecutive nucleotides within the HR3 (homology region 3) and possibly also initiate editing of the 5′ domain, directing the generation of a substantially different sequence. The two gRNAs were found in approximately equal numbers.
Figure 3.

Length of gRNA complementarity to fully edited mRNAs. The shortest and longest gRNAs identified had 24 nt and 61 nt of complementarity to their fully edited mRNA. The bulk of the sequence classes (64%) had 38–48 nt of complementarity.
Figure 4.

gRNA characteristics: number of non-matched nucleotides found 5′ to the anchor (A) or 3′ to the guiding region, excluding the U-tail (B). Most of the identified gRNAs had few non-complementary nucleotides.
Figure 5.

Long 5′ non-matched extensions often signal editing anomalies. gND7 (147–199) and gND7 (152–190) were both identified as initiating gRNAs for the 5′ editing domain of ND7. (A) The sequence of the two gRNAs is shown aligned below the edited ND7 mRNA sequence. A single T insertion (bold and underlined) disrupts the anchor of gND7 (152–190). (B) The putative alternative sequence generated if editing initiates with gND7 (152–190). Watson–Crick (|) and G:U base pairs (:) are indicated. The number sign (#) indicates C:A base pairs required for generation of this possible sequence.
In our initial alignments, we identified and aligned the highest scoring gRNAs for each editing region (2 points for canonical Watson–Crick base pairs and 1 point for G-U base pairs). However, population analyses indicate that these ‘best align’ gRNAs (indicated with superscript ‘a’ in Table 1) were often rare transcripts, with the most abundant transcripts having shorter guiding regions.
gRNAs show a strong ATATA initiation bias
Previous characterization of gRNAs suggested that transcript initiation tends to occur 31–32 bp from an imperfect 18 bp inverted repeat, initiating with a 5′ RYAYA motif (7). However, other transcription initiation sequences had been observed (16). This analysis of over 3.5 million transcripts indicates a strong ATATA initiation bias, with over 74% of the transcripts initiating with this sequence. Of the ∼600 major sequence classes identified, the two most common initiation sequences were ATATAT (35%) and ATATAA (21%). Sequence classes initiating with ATATAC and ATATAG were much less common (4.3 and 2.4%, respectively, see Table 3). Unexpectedly, the third most common initiation sequence class was 5′ AAAAAA, with these gRNAs often initiating with a long 5′ adenylate-run (Figure 6A). The number of 5′ A residues involved in anchoring the gRNA varied from 0 [gCR4(504–548)] to 12 [gA6(521-567)]. These gRNAs are interesting because it is difficult to understand how they are selective. For example, the anchor binding sites for both gA6 (521–567) and gCR4 (405–458) are almost identical (Figure 6B and C). For both of these gRNAs, only a single C:G pair is involved in the initial interaction. Interestingly, gA6 (521–567) can continue the editing of two alternative sequences (described fully in the Characteristics of gRNAs for specific mRNAs section, found later in the text).
Table 3.
Most common gRNA initiation sequences
| Initiating Sequence | Number of sequence classes | % | Number of transcripts | % |
|---|---|---|---|---|
| 5′ ATATAT | 214 | 35.2% | 1 320 726 | 37.4% |
| 5′ ATATAA | 128 | 21.1% | 870 728 | 24.7% |
| 5′ AAAAAA | 28 | 4.6% | 45 540 | 1.3% |
| 5′ ATATAC | 26 | 4.3% | 134 862 | 3.8% |
| 5′ ATACAA | 17 | 2.8% | 60 428 | 1.7% |
| 5′ ATATTA | 16 | 2.6% | 31 779 | 0.9% |
| 5′ ATATAG | 15 | 2.4% | 269 306 | 7.6% |
| 5′ ATAAAT | 15 | 2.6% | 23 669 | 0.7% |
| 5′ ATACAT | 13 | 2.2% | 80 453 | 2.2% |
| 5′ ATAAAA | 12 | 2.1% | 38 907 | 1.1% |
| 5′ ATAAAG | 9 | 1.5% | 176 446 | 5.0% |
| 5′ ATACTA | 8 | 1.4% | 58 797 | 1.7% |
The major sequence classes identified were grouped based on the first 6 nt and sorted based on both the number of sequence classes and the total number of transcripts found. Most transcripts (∼74%) initiated with ATATA (includes ATATAT (37.4%)), ATATAA (24.7%, ATATAC (3.8%)) and ATATAG (7.6%).
Figure 6.

Identification of gRNAs that initiate with long A-runs. (A) Examples of identified gRNAs initiating with long A-runs. Sequence complementary to the fully edited mRNA is underlined. (B) Alignment of gCR4(405–548) with its corresponding edited mRNA. (C) Alignment of gA6(521–567) with its corresponding edited mRNA. The gRNA cDNA sequence is shown aligned beneath the fully edited mRNA as described in Figure 1. The partial sequence of the downstream gRNA that directs the creation of the anchor-binding site is also shown. For both of the these gRNAs, the anchor interaction (bold font) involves a single G:C base pair. Two downstream gRNAs were identified that direct editing of the A6 550–570 region. gA6 (549–593) would direct the insertion of 12 U-residues, while gA6(557–593) would direct the insertion of 11 U-residues. gA6(557–593) is much more abundant (∼70 000 versus ∼2500 transcripts identified). (D) Comparison of the protein sequences generated by the conventional 12U-edited (top line) and alternatively 11U-edited A6 transcripts. The alternative protein sequence (double underlined) is 11 AA shorter.
gRNA population numbers
Analyses of the gRNA populations show significant differences in the number of identified gRNA transcripts that guide a specific region. Because the zero mismatch data contained only correctly matched gRNAs, we were able to quantify the total number of identified gRNA transcripts that covered any 1 nt in the fully edited sequence. The data for A6, CR3 and RSP12, which clearly show the large variation in identified gRNAs responsible for the editing of specific regions, are illustrated in Figure 7A–C. The data for all other transcripts can be found in the supplementary files. Although some editing sites are covered by a single gRNA (for example, the initiating gRNA for A6 (gA6-770–822) is represented by a single transcript), other editing sites are covered by hundreds of thousands of gRNA transcripts. The edited RSP12 sequence, nucleotides 203–246, had the highest gRNA transcript coverage, with over 350 000 identified transcripts (Figure 7C and supplementary data). Approximately 340 000 of those transcripts were found in a single sequence class. However, a total of 27 different sequence classes covered this region. In this analysis, the number of gRNAs in any sequence class was determined by sorting gRNAs based on sequence within only the anchor and guiding regions. The 5′ end differences (due to truncation) and differences in the polyU tail length and the interruption of the polyU tail with other nucleotides were ignored. All gRNAs with identical sequence within this region were then summed to determine the total number within the sequence class. In our initial analysis, identical sequences (redundant reads) were collapsed and the number of identical reads recorded. In the sorting of the raw data, it became clear that some gRNA sequences were abundant (highest copy number for any one sequence was 48 097). Because library preparation does include a PCR amplification step, we do note that the some gRNAs may be preferentially amplified. However, in determining population numbers, abundant gRNAs were most often represented by multiple reads with differences in both the 5′ and 3′ regions outside of the anchor/guiding region. In addition, the abundant gRNAs were most often found in large populations with multiple related sequence classes. This suggests that the most abundant gRNAs were associated with high copy number minicircles. More surprising was the number of edited regions covered by low gRNA numbers, especially for those transcripts that are constitutively edited. Interestingly, we note that the initiating gRNA for most of the transcripts were found in low copy numbers. The initiating gRNAs for A6 (1 transcript identified), ND7 (6 transcripts) and ND8 (27 transcripts) were rare (<100 identified transcripts). Although the initiating gRNAs for COIII (111 transcripts), CR4 (511 transcripts), ND3 (545), ND9 (274 transcripts) and RSP12 (128 transcripts) were slightly more abundant, they were still not found in the numbers expected.
Figure 7.

Abundance of gRNA transcripts (y-axis, note log scale) that align to a respective nucleotide in the fully edited mRNA. Both nucleotides and deletion sites in the fully edited mRNA were numbered starting from the 5′ end (+1 = 0). Shaded boxes indicate identified gRNAs that cover specific editing sites. Data include only the identified conventional gRNAs. (A) Identified gRNAs for the A6 edited mRNA. (B) Identified gRNAs for CR3. (C) Identified gRNAs for ribosomal protein subunit 12 (RSP12). All individual data points were designated with open circles. Close overlapping of individual data points generate solid black lines.
Characteristics of gRNA populations for specific mRNAs
ATPase 6
A total of 32 gRNA populations that could guide the editing of A6 were identified. Although the minimum overlap observed was 8 nt, the average overlap was 19 nt, indicative of the extensive overlap observed for many of the gRNA populations (Figure 1). Most of the gRNA populations were reasonably abundant, with two notable exceptions; gA6 (770–822), the initiating gRNA, and gA6 (747–789), which directs the editing just upstream of the initiating guide. The sequence for gA6 (770–822) has been previously identified as gA6-14 (17). A limited search of our mismatch databases did identify an alternative gRNA that could initiate editing for A6 (see Figure 1). Significantly, although the alternative sequence does introduce a number of sequence changes (all downstream of the stop codon), the generated anchor sequence for the next gRNA is maintained. A second region with two identified gRNAs that can generate a sequence anomaly was identified at position 556–567. Four different sequence classes were identified that direct the editing in this region. Three of the sequence classes (gA6 (546–593), gA6(549–593) and gA6(549–592), direct the ‘correct’ insertion of 12 U into this site. However, the most abundant gRNA, gA6 (557–593), would in fact direct the insertion of 11 U instead of the described 12 U in the 556–567 editing site, but correctly edit (insertion of 5 U) the next upstream site (550–554) (Figure 6C). The gRNA that initiates editing in the alternatively edited site is unusual, in that its’ anchor consists of an ‘A’ run with a single G-C pair (gA6 (521–567)). The 11 U sequence decreases the anchor for gA6 (521–567) by a single A-U base pair, suggesting that it could anchor and continue editing for both generated sequences. An analysis of the 11 U open reading frame indicates that the frame shift would generate a new carboxyl terminus that is only 11 AA shorter (Figure 6D).
Cytochrome oxidase III
Complete gRNA coverage for the conventionally edited COIII transcript was obtained with the identification of 40 gRNA populations. Similar to A6, the average overlap of the gRNAs was ∼19 nt (maximum overlap = 36 nt; minimum overlap = 8 nt). Although alternative editing of COIII has been reported, we did not identify a gRNA that could direct editing of the alternative sequence. In COIII, alternative editing involves a gRNA that directs the insertion of two U-residues instead of the conventional three between nucleotides G458 and A462 (18). The sequence changes directed by the alternative gRNA links the open reading frame of the edited 3′ end to an ORF found in the 5′ pre-edited sequence allowing the production of a different protein. In our sequence data, we identified a number of different sequence classes that direct editing of the transition site. gCOIII (456–499) matches the sequence of the gRNA previously identified as directing the conventional COIII editing (insertion of three U-residues). The three most abundant gRNAs in this region, however, all direct editing through nucleotide U461, and would direct the insertion of a single U-residue into the alternatively edited site. No gRNA that could direct the alternative sequence was identified. The previously identified alternative gRNA, which deviates from the conventional sequence only near its 3′ end, was not found in our library.
C-rich regions 3 and 4
The CR3 transcript is small, with extensive editing generating a transcript of ∼310 nt. Interestingly, we were only able to identify gRNAs that direct the editing of the 5′ end of this transcript. gRNAs that matched the published sequence downstream of nucleotide 196 were rare and no transcripts were identified that could direct the editing between nucleotides 275 and 292. We were able to identify a gRNA that could initiate editing of the CR3 transcript, but it would generate a sequence distinctly different from that published (see supplementary data). In contrast, a full complement of gRNAs (18 populations) was identified for the CR4 transcript (average overlap = 16 nt, maximum = 37, minimum = 8). In this study, we used the consensus-edited sequence found for the blood form stage of the parasite (19). In procyclic forms, although the 3′ portion of the transcript is identical to that found in blood forms, the consensus sequence diverges at nucleotide 312, and no consensus was determined upstream of nucleotide 256. It has been previously reported that gRNAs for the developmentally regulated mRNAs are present in both life cycle stages (16). We do note that most (10 of 18) of the populations identified were low abundant populations (<1000 transcripts identified).
Cytochrome B and Murf II
RNA editing of both the CYb and Murf II is limited, occurring only near the 5′ ends of the transcripts (20,21). These transcripts require only two gRNAs for complete editing and both have some interesting and unusual characteristics. In Murf II, one of the gRNAs is known to be maxicircle encoded. The gene is located near the 5′ border of the ND4 gene and appears to be independently transcribed (22). This gRNA guides almost all of the editing required. The search of our database did identify a single gRNA sequence class involved in editing this region [gMurf II (30–79)] that matches the identified maxicircle gene. We did not identify the initiating gRNA for Murf II, which must direct the insertion of a single U-residue and also, we presume, the first few editing site to generate the anchor binding site for gMurfII-2. Because the amount of editing by this gRNA is so limited, it may be that both our size selection and the stringency of our selection precluded our ability to detect this transcript. We did identify both gRNA populations involved in the editing of CYb. gRNAs that can initiate editing of CYb had been previously identified (gCYb-558 and gCYb560A and B) and are unusual because they are not flanked by the 18 bp inverted repeats characteristic of most gRNA genes (7,8,11,23). Interestingly, although the initiating gRNAs identified in our gRNA library are similar to those previously described, only one of the major sequence classes (gCYb (54–91) matches one previously published (gCYb-560A), and it does differ at its 5′ end, in that it initiates with a run of A-residues (Figure 8). Most of the initiating gRNA sequence classes did in fact initiate with a run of A’s. In contrast to the initiating gRNA (over 31 000 transcripts detected), the sequential gRNA (gCYb (32–64) was much less abundant (∼1000 total transcripts detected). It also initiates with a run of A-residues.
Figure 8.

gCYb (54–91) transcript alignment with minicircle MCP23. The minicircle sequence is shown on top with both gCYb(54–91) and the previously identified gCYb-560A aligned underneath.
NADH dehydrogenase subunits 3, 7, 8 and 9
Full complements of gRNAs were not identified for any of the ND subunits. For most of these transcripts, editing is developmentally regulated, with full editing only observed in the bloodsteam stages (24–27). gRNAs that covered all of the fully edited nucleotides for both ND7 and ND9 were identified. Some of the identified gRNAs, however, had distinct mismatches, and it is unclear if these gRNAs would generate the correct sequence (see supplementary data). The ND3 mRNA transcript is edited in two domains with only the large 5′ domain edited to a single consensus sequence (27). The much smaller 3′ domain (nucleotides 375–395) shows several editing patterns, and no gRNAs that span this variable region were identified. We did not, however, search for gRNAs using all of the reported sequence variations. Twenty-five gRNA populations that could direct the editing of ND8 were identified, with good coverage of the edited sequence except for nucleotides 540–554. In addition, a number of the identified gRNAs have mismatches. For example, the rare gND8 (161–198) transcript (<100 transcripts identified) is a perfect match to the published sequence. A near identical gRNA, gND8 (161–187) is abundant (>100 000 transcripts identified) and has a single A:T transversion that introduces a mismatch into the long (20 bp) anchor region. This region is also covered by another abundant gRNA (gND8(158–186)(>150 000 transcripts)). Although this gRNA has several mismatches to the published sequence, the edited sequence it would guide introduces a single amino acid change. Importantly, editing 5′ to the alternative sequence is not affected, maintaining the anchor-binding site for the sequential gRNA (Figure 9).
Figure 9.
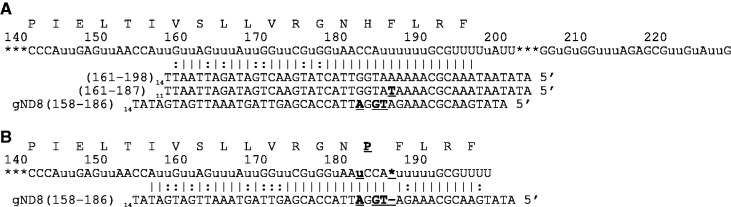
Abundant mismatched gRNA generates edited sequence with a single AA change. (A) Two gRNAs that edited the same region as the rare gND8(161–198) were identified that contain mismatches to the conventional edited sequence of ND8 (mismatches in bold and underlined). The gRNAs are shown aligned beneath the fully edited mRNA as described in Figure 1. (B) gND8(158–186) is the most abundant of the three transcripts, and would generate an edited sequence with a single amino acid change.
Ribosomal Protein S12
A total of 12 gRNA sequence populations are involved in the editing of RSP12 (28). These include one of the most abundant populations [gRSP12(200–246)] identified, with ∼350 000 transcripts in 27 different sequence classes. In contrast to this region, few transcripts were identified that covered nucleotides 122–168. This region is interesting because it does contain a high percentage of C-residues, and the few gRNA transcripts identified all contained C:A mismatches. Because we did not allow for C:A basepairs, it may be that the bulk of the gRNAs that guide this region were not identified.
DISCUSSION
Deep sequencing of the gRNA transcriptome has allowed the identification of a near full complement of gRNAs needed for the extensive editing observed in T. brucei. A total of 642 different major sequence classes were identified, 84% of which are novel (not previously identified). Characterization of this population has identified a number of interesting and unusual features. These include the extreme population differences in the identified gRNAs and the identification of gRNAs that initiate with long A runs. Generation of the gRNA library did include a limited PCR that may exaggerate differences in population number. However, the association of abundant gRNAs with populations containing multiple sequence classes suggests that these represent gRNAs transcribed from amplified abundant minicircles. More surprising to us was the number of identified gRNAs found in low copy number. The high stringency of our initial screen suggests that the editing of these regions may be directed by gRNAs not identified in our initial screen. This postulate is supported by the identification of alternative gRNAs or gRNAs with internal mismatches for some of the low coverage regions. For example, alternative gRNAs were identified for the initiating gRNAs of both A6 and CR3 and a limited search of our mismatch databases did identify a number of abundant gRNAs with internal mismatches (see Figure 9). Unfortunately, the mismatch files are large and difficult to work with. Preliminary inspection of the files indicates that they identify hundreds of thousands of gRNAs with characteristics that suggest they are misaligned. These characteristics include gRNA/mRNA matched regions that contain mostly G:U base pairs, the presence of long 5′ and 3′ extensions outside of the matched regions and the lack of a defined 5′ anchor (the characteristic bias towards Watson-Crick base pairing in the anchor region). We are currently working to refine our search parameters to more heavily weight-specific gRNA characteristics, to sort these files into manageable units. The large numbers of low copy number gRNAs, however, are suggestive of high plasticity within the gRNA encoding minicircles. Previous studies in Leishmania have shown that homologous minicircle sequence class frequencies are extremely variable, even between different isolates from the same strain taken after several years of culture (29,30). In addition, previous studies have indicated that genes encoding highly edited RNAs accumulated mutations at a higher frequency than their unedited homologs in closely related species (31). The rapid evolution of the gRNA populations would explain the rapid changes observed in the protein coding genes. We have in fact identified a number of gRNAs that would generate an mRNA sequence that differs from the consensus sequence determined in the early 1990s. Currently, we are working to determine if we can detect these alternative sequences in the mRNA population.
Deep sequencing of the gRNA population also detected other interesting characteristics. Although the bulk of the gRNAs did initiate with the strong ATATA initiation sequence bias previously reported, a new unexpected class of gRNAs that initiate with long runs of adenylate residues was identified. These gRNAs are interesting because it is difficult to understand how they are selective. The sequential dependence of each gRNA on downstream editing indicates that the overall efficiency is dependent on the efficiency of each gRNA-targeting event. For example, a 90% efficiency rate for each COIII gRNA (40 gRNAs required) would result in <2% fully edited transcripts. We had hypothesized that this evolutionary pressure would select for gRNAs with specific and efficient gRNA targeting characteristics. The identification of a potentially alternatively edited site in A6 (see Figure 6) does suggest that this class of gRNAs may play a significant biological role. The long adenylate anchor of gA6 (521–567) allows it to target both 11U and 12U transcripts, suggesting that both editing events can lead to fully mature and translatable mRNAs. The presence of distinct 5′ truncated gRNAs makes it impossible to determine if the large variability observed in the number of 5′ adenylate residues found in these transcripts is due to 5′ exonuclease activity or to its mechanism of synthesis. Minicircle encoded genes have not been identified for most of the A-run gRNAs. gCYb54-91, however, is a close match to a CYb gRNA encoded on minicircle MCP-23 (23). Interestingly, examination of the primer extension data in the original publication does suggest that this gRNA initiates with multiple adenylates in vivo. The A-run aligns with an A4 stretch on the minicircle, suggesting that it could be produced by a slippage (stuttering) mechanism (32,33). We do note that the existence of 5′–3′ RNA degradation activities in the mitochondria of trypanosomes has not previously been described (34). However, the large numbers of 5′ serial truncated transcripts is suggestive of 5′–3′ exonuclease activity. Although we cannot rule out a contaminating exonuclease from the cytoplasm, a 5′–3′ exonuclease for gRNA recycling may be evolutionarily favorable, as it would remove the anchor targeting sequence first, possibly preventing partially degraded gRNAs from initiating any mRNA editing. Because of the U4 requirement in our initial filter, we would not have identified gRNAs with 3′ truncations.
This is the first study of trypanosomal gRNAs using high-throughput sequencing. In this work, we have defined a near complete set of the gRNAs required for the extensive editing found in T. brucei. The identification of this comprehensive set of gRNAs will allow the characterization of the sequence and structural features important for efficient targeting and should provide insight into the evolution of small RNA targeting strategies.
ACCESSION NUMBERS
SAMN02204165 NCBI's Sequence Read Archive.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R03AI0902 to D.K.]; all undergraduates funded on NSF 04-546 (to J.H., T.T. and J.L.); Interdisciplinary Training for Undergraduates in Biological and Mathematical Sciences. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Ken Stuart and Jason Carnes at Seattle Biomedical Research Institute for supplying the T. brucei strains used in this study. Special thanks to Scooter Nowak for help with computer analyses and thanks to Jeff Landgraf, Kevin Carr and the Michigan State University RTSF genomics core for significant advice and help in the illumina sequencing.
REFERENCES
- 1.Aphasizhev R, Aphasizheva I. Uridine insertion/deletion editing in trypanosomes: a playground for RNA-guided information transfer. Wiley Interdiscip. Rev. RNA. 2011;2:669–685. doi: 10.1002/wrna.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hajduk S, Ochsenreiter T. RNA editing in kinetoplastids. RNA Biol. 2010;7:229–236. doi: 10.4161/rna.7.2.11393. [DOI] [PubMed] [Google Scholar]
- 3.Schneider A. Unique aspects of mitochondrial biogenesis in trypanosomatids. Intern. J. Parasitol. 2001;31:1403–1415. doi: 10.1016/s0020-7519(01)00296-x. [DOI] [PubMed] [Google Scholar]
- 4.Ochsenreiter T, Cipriano M, Hajduk SL. Alternative mRNA editing in trypanosomes is extensive and may contribute to mitochondrial protein diversity. PLoS One. 2008;3:e1566. doi: 10.1371/journal.pone.0001566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lukes J, Hashimi H, Verner Z, Cicova Z. The remarkable mitochondrion of trypanosomes and related flagellates. In: de Souza W, editor. Structures and Organelles in Pathogenic Protists. Microbiology Monographs 17. Berlin Heidelberg: Springer-Verlag; 2010. pp. 227–252. [Google Scholar]
- 6.Steinert M, VanAssel S. Sequence heterogeneity in kinetoplast DNA: Reassociation kinetics. Plasmid. 1980;3:7–17. doi: 10.1016/s0147-619x(80)90030-x. [DOI] [PubMed] [Google Scholar]
- 7.Pollard VW, Rohrer SP, Michelotti EF, Hancock K, Hajduk SL. Organization of minicircle genes for guide RNAs in Trypanosoma brucei. Cell. 1990;63:783–790. doi: 10.1016/0092-8674(90)90144-4. [DOI] [PubMed] [Google Scholar]
- 8.Hong M, Simpson L. Genomic organization of Trypanosoma brucei Kinetoplast DNA minicircles. Protist. 2003;154:265–279. doi: 10.1078/143446103322166554. [DOI] [PubMed] [Google Scholar]
- 9.Simpson L. The genomic organization of guide RNA genes in kinetoplastid protozoa: several conundrums and their solutions. Mol. Biochem. Parasitol. 1997;86:133–141. doi: 10.1016/s0166-6851(97)00037-6. [DOI] [PubMed] [Google Scholar]
- 10.Maslov DA, Simpson L. The polarity of editing within a multiple gRNA-medicated domain is due to formation of anchors for upstream gRNAs by downstream editing. Cell. 1992;70:459–467. doi: 10.1016/0092-8674(92)90170-h. [DOI] [PubMed] [Google Scholar]
- 11.Corell RA, Feagin JE, Riley GR, Strickland T, Guderian JA, Myler PJ, Stuart K. Trypanosoma brucei minicircles encode multiple guide RNAs which can direct editing of extensively overlapping sequences. Nucleic Acids Res. 1993;21:4313–4320. doi: 10.1093/nar/21.18.4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ochsenreiter T, Cipriano M, Hajduk SL. KISS: The kinetoplastid RNA editing sequence search tool. RNA. 2007;13:1–4. doi: 10.1261/rna.232907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madej MJ, Niemann M, Huttenhofer A, Goringer HU. Identification of novel guide RNAs from the mitochondria of Trypanosoma brucei. RNA Biol. 2008;5:84–91. doi: 10.4161/rna.5.2.6043. [DOI] [PubMed] [Google Scholar]
- 14.Stuart K, Panigrahi AK, Schnaufer A. Identification and characterization of trypanosome RNA-editing complex components. Methods Mol. Biol. 2004;265:273–291. doi: 10.1385/1-59259-775-0:273. [DOI] [PubMed] [Google Scholar]
- 15.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 16.Koslowsky DJ, Riley GR, Feagin JE, Stuart K. Guide RNAs for transcripts with developmentally regulated RNA editing are present in both life cycle stages of Trypanosoma brucei. Mol. Cell. Biol. 1992;12:2043–2049. doi: 10.1128/mcb.12.5.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhat GJ, Koslowsky DJ, Feagin JE, Smiley BL, Stuart K. An extensively edited mitochondrial transcript in kinetoplastids encodes a protein homologous to ATPase subunit 6. Cell. 1990;61:885–894. doi: 10.1016/0092-8674(90)90199-o. [DOI] [PubMed] [Google Scholar]
- 18.Ochsenreiter T, Hajduk S. Alternative editing of cytochrome coxidase III mRNA in trypansome mitochondria generates protein diversity. EMBO Rep. 2006;7:1128–1133. doi: 10.1038/sj.embor.7400817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corell RA, Myler P, Stuart K. Trypanosoma brucei mitochondrial CR4 gene encodes an extensively edited mRNA with completely edited sequence only in bloodstream forms. Mol. Biochem. Parasitol. 1994;64:65–74. doi: 10.1016/0166-6851(94)90135-x. [DOI] [PubMed] [Google Scholar]
- 20.Feagin JE, Shaw JM, Simpson L, Stuart K. Creation of AUG initiation codons by addition of uridines within cytochrome b transcripts of kinetoplastids. Proc. Natl Acad. Sci., USA. 1988;85:539–543. doi: 10.1073/pnas.85.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaw JM, Feagin JE, Stuart K, Simpson L. Editing of kinetoplastid mitochondrial mRNAs by uridine addition and deletion generates conserved amino acid sequences and AUG initiation codons. Cell. 1988;53:401–411. doi: 10.1016/0092-8674(88)90160-2. [DOI] [PubMed] [Google Scholar]
- 22.Clement SL, Mingler MK, Koslowsky DJ. An intragenic guide RNA location suggests a complex mechanism for mitochondrial gene expression in Trypanosoma brucei. Eukaryot. Cell. 2004;3:862–869. doi: 10.1128/EC.3.4.862-869.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riley GR, Corell RA, Stuart K. Multiple guide RNAs for identical editing of Trypanosoma brucei apocytochrome b mRNA have an unusual minicircle location and are developmentally regulated. J. Biol. Chem. 1994;269:6101–6108. [PubMed] [Google Scholar]
- 24.Koslowsky DJ, Bhat GJ, Perrollaz AL, Feagin JE, Stuart K. The MURF3 gene of T. brucei contains multiple domains of extensive editing and is homologous to a subunit of NADH dehydrogenase. Cell. 1990;62:901–911. doi: 10.1016/0092-8674(90)90265-g. [DOI] [PubMed] [Google Scholar]
- 25.Souza AE, Myler PJ, Stuart K. Maxicircle CR1 transcripts of Trypanosoma brucei are edited, developmentally regulated and encode a putative iron-sulfur protein homologous to an NADH dehydrogenase subunit. Mol. Cell. Biol. 1992;12:2100–2107. doi: 10.1128/mcb.12.5.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Souza AE, Shu HH, Read LK, Myler PJ, Stuart KD. Extensive editing of CR2 maxicircle transcripts of Trypanosoma brucei predicts a protein with homology to a subunit of NADH dehydrogenase. Mol. Cell. Biol. 1993;13:6832–6840. doi: 10.1128/mcb.13.11.6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Read LK, Wilson KD, Myler PJ, Stuart K. Editing of Trypanosoma brucei maxicircle CR5 mRNA generates variable carboxy terminal predicted protein sequences. Nucleic Acids Res. 1994;22:1489–1495. doi: 10.1093/nar/22.8.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Read LK, Myler PJ, Stuart K. Extensive editing of both processed and preprocessed maxicircle CR6 transcripts in Trypanosoma brucei. J. Biol. Chem. 1992;267:1123–1128. [PubMed] [Google Scholar]
- 29.Simpson L, Thiemann OH, Savill NJ, Alfonzo JD, Maslov DA. Evolution of RNA editing in trypanosome mitochondria. Proc. Natl Acad. Sci. USA. 2000;97:6986–6993. doi: 10.1073/pnas.97.13.6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savill NJ, Higgs PG. A theoretical study of random segregation of minicircles in trypanosomatids. Proc. R. Soc. Lond. B. 1999;266:611–620. doi: 10.1098/rspb.1999.0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Landweber LF, Gilbert W. RNA editing as a source of genetic variation. Nature. 1993;363:179–182. doi: 10.1038/363179a0. [DOI] [PubMed] [Google Scholar]
- 32.Cunningham PR, Weitzmann CJ, Ofengand J. SP6 RNA polymerase stutters when initiating from an AAA… sequence. Nucleic Acids Res. 1991;19:4669–4673. doi: 10.1093/nar/19.17.4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacques J-P, Susskind MM. Pseudo-templated transcription by Escherichia coli RNA polymerase at a mutant promoter. Genes Dev. 1990;4:1801–1810. doi: 10.1101/gad.4.10.1801. [DOI] [PubMed] [Google Scholar]
- 34.Aphasizhev R, Aphasizheva I. Mitochondrial RNA processing in trypanosomes. Res. Microbiol. 2011;162:655–663. doi: 10.1016/j.resmic.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.