


Abstract
Alternative splicing regulates developmentally and tissue-specific gene expression programs, disruption of which have been implicated in numerous diseases. Muscleblind-like 1 (MBNL1) regulates splicing transitions, which are disrupted on loss of MBNL1 function in myotonic dystrophy type 1 (DM1). One such event is MBNL1-mediated activation of insulin receptor exon 11 inclusion, which requires an intronic enhancer element downstream of exon 11. The mechanism of MBNL1-mediated activation of exon inclusion is unknown. We developed an in vitro splicing assay, which robustly recapitulates MBNL1-mediated splicing activation of insulin receptor exon 11 and found that MBNL1 activates removal of the intron upstream of exon 11 upon binding its functional response element in the downstream intron. MBNL1 enhances early spliceosome assembly as evidenced by enhanced complex A formation and binding of U2 small nuclear ribonucleoprotein auxiliary factor 65 kDa subunit (U2AF65) on the upstream intron. We demonstrated that neither the 5′ splice site nor exon 11 sequences are required for MBNL1-activated U2AF65 binding. Interestingly, the 5′ splice site is required for MBNL1-mediated activation of upstream intron removal, although MBNL1 has no effect on U1 snRNA recruitment. These results suggest that MBNL1 directly activates binding of U2AF65 to enhance upstream intron removal to ultimately activate alternative exon inclusion.
INTRODUCTION
The majority of vertebrate genes are interrupted by introns, which are recognized and removed by the basal splicing machinery, the spliceosome, through recognition of multiple cis-acting sequence elements within the pre-mRNA. These RNA elements are recognized by RNA and protein components of the spliceosome. The mammalian spliceosome consists of five U-rich snRNA (small nuclear RNA)-containing small nuclear ribonucleoprotein (snRNP) complexes: U1, U2, U4, U5 and U6. These complexes assemble in a step-wise manner to define splice sites and ultimately catalyze intron removal and exon joining (1,2). The early (E) spliceosome complex consists of the U1 snRNP bound at the 5′ splice site and U2 auxiliary factor (U2AF) bound at the polypyrimidine tract and 3′ splice site. Next, the first ATP-dependent complex, complex A, forms by recruitment of the U2 snRNP to the branch site. The U4/U6.U5 tri-snRNP is then recruited, and rearrangements of RNA and protein contacts result in the removal of U1 and U4 from the complex, resulting in the formation of a catalytically activated complex. Following formation of the lariat intermediate, the 5′ exon is joined with the 3′ exon to form spliced mRNA and the lariat intron is degraded.
Alternative splicing is a pervasive mechanism of gene expression regulation occurring in >95% of human multi-exon genes (3–5). Alternative splicing events can be coordinately regulated by RNA-binding proteins that bind to specific motifs often located proximal to the regulated splice sites to result in developmentally and tissue-specific programs of gene expression (6). A 5′ and 3′ splice site may be recognized across an exon as described by the exon definition model in which assembly of spliceosomal complexes at one splice site enhances assembly of spliceosomal complexes at the splice site across the exon (7–10). A single splicing regulator can act as an activator or repressor depending on the target pre-mRNA. This context-dependent activity is thought to arise, at least in part, from the location of the regulatory cis-element relative to the regulated exon (11,12). The mechanism of splicing repression has been characterized for several alternative splicing events. neuro-oncological ventral antigen (Nova) impedes complex E formation by binding within the 5′ end of the intron disrupting binding of U1 snRNA to the 5′ splice site (12). hnRNP L and hnRNP A1 coordinately cause steric hindrance of U6 snRNA binding to the 5′ splice site by promoting aberrant U1 snRNA binding with exonic sequences upstream of the 5′ splice site of CD45 exon 4 (13). Polypyrimidine tract binding protein (PTB) represses c-src N1 exon inclusion by binding a portion of U1 snRNA disrupting contacts with downstream spliceosomal components, resulting in disruption of intron definition (14,15). Furthermore, PTB binds an exonic silencer and disrupts cross-exon communication by interfering with the ability of the U1 snRNP bound at the 5′ splice site to recruit U2AF65 to the upstream polypyrimidine tract (16). Muscleblind-like 1 (MBNL1) represses cardiac troponin T (cTNT) exon 5 by binding to a structured intronic silencer, which competes with U2AF65 binding to the single-stranded polypyrimidine tract (17).
Mechanisms of activated exon inclusion generally involve interactions between a splicing regulator and a protein component of early spliceosome complexes. For example, CUGBP, Elav-like family member 2 (CELF2) binds an intronic element downstream of cTNT exon 5 and activates inclusion by promoting U2 snRNP assembly through direct protein–protein interactions with two U2 snRNP components (18). T cell-restricted intracellular antigen 1 (TIA1) binds intronic enhancer elements downstream of the 5′ splice site of Fas exon 6 and interacts with the U1-C protein, stabilizing binding of the U1 snRNP to the 5′ splice site (19). This promotes U2AF65 recruitment to the polypyrimidine tract of the intron upstream of exon 6 through exon definition (16). Nova enhances spliceosome complex A formation on the downstream intron through binding intronic enhancers downstream of the alternative exon (12). Sam68 binds the upstream intron and recruits U2AF65 to the polypyrimidine tract of the CD44 V5 exon through a protein–protein interaction (20).
MBNL is a family of three CCCH-type zinc finger-containing RNA-binding proteins that regulates alternative splicing both positively and negatively during development (11,21–25). RNA sequencing, high-throughput cross-linking immunoprecipitation and microarray analysis of Mbnl1 knockout mice demonstrated that Mbnl1 regulates >900 splicing events in skeletal muscle, heart and brain (11,26). Loss of MBNL1 function through its sequestration on expanded CUG repeat RNA leads to pathogenic features in myotonic dystrophy type 1 (DM1) (27–30). One splicing event that is relevant to DM1 clinical features is MBNL1-mediated activation of exon 11 inclusion of the insulin receptor (IR). Adult DM1 skeletal muscle tissue shows a reversion to the embryonic splicing pattern in which IR exon 11 is predominantly skipped due to loss of MBNL1 function. Skipping of exon 11 produces a protein isoform lacking a 12 amino acid portion of the intracellular domain resulting in diminished signaling capacity and insulin insensitivity in DM1 (31,32). We previously showed that MBNL1 activates IR exon 11 inclusion by directly binding an intronic splicing enhancer element within the intron downstream of exon 11 (33,34). However, the mechanism by which MBNL1 communicates with the basal splicing machinery to activate exon inclusion is unknown.
In this study, we have characterized the mechanism by which MBNL1 activates inclusion of IR exon 11. We previously showed that a 273-nt region of the IR pre-mRNA containing exon 11 is necessary and sufficient for MBNL1-mediated regulation. MBNL1 binds a 30-nt region containing three YGCY(U/G)Y motifs that lies 93-nt downstream of IR exon 11, and mutation of this sequence disrupts MBNL1 binding and splicing activation (33). To investigate the mechanism of IR exon 11 activation, we developed an in vitro splicing assay using the minimal MBNL1-resposive region of the IR pre-mRNA and recombinant MBNL1. We show that MBNL1 binds to the intron downstream of exon 11 and promotes removal of the upstream intron. We hypothesized that MBNL1 promotes definition of exon 11 by activating assembly of spliceosome components at the splice sites flanking exon 11. Consistent with this hypothesis, we found that MBNL1 enhances spliceosome complex A on the intron upstream of exon 11 and enhances binding of U2AF65 to the polypyrimidine tract of the upstream intron. Strikingly, this response was intact regardless of whether the intron upstream of exon 11 was complete or contained only the 3′-most 52 nt, indicating that the 5′ splice site of the upstream intron is not required. The ability of MBNL1 to promote U2AF65 binding requires neither the 5′ splice site of exon 11 nor the binding of factors within exon 11, suggesting that the mechanism involves a cross-exon effect. Although MBNL1 had no effect on binding of U1 snRNA to the 5′ splice site, we demonstrated that blocking the 5′ splice site disrupts the ability of MBNL1 to activate splicing of the upstream intron. This result suggests that the 5′ splice is involved in MBNL1-mediated splicing activation downstream of U1 snRNP recruitment. This is the first characterization of the mechanism used by MBNL1 to activate alternative splicing and indicates that MBNL1 promotes inclusion of IR exon 11 by stimulating an early step of spliceosome assembly on the upstream intron.
MATERIALS AND METHODS
RNA constructs and oligonucleotide sequences
E11TNI67 contains a genomic segment of IR pre-mRNA (52 nt of the upstream intron, the entire 36 nt exon and 185 nt of the downstream intron) between chicken skeletal troponin I (sTNI) exons 6 and 7 downstream of the T7 promoter. For constructs defective in MBNL1 binding, we incorporated a mutation in the MBNL1 response element, which abolishes binding to the 30-nt RNA fragment in a gel-shift assay (33). To generate single-intron substrates, DNA templates for transcription were generated by polymerase chain reaction (PCR) from the E11TNI67 plasmid using the following primers: upstream intron substrate forward (ATGACACAGACCTGAAGC), reverse (AAGAGCAGAGCATGAGTT); downstream intron substrate forward (TGTCTAATGAAGTTCCCTCT), reverse including a 5′ splice site (GCCCAACTTACCTGTGAACTTGCCCCTCAGGT). The psoralen cross-linking substrate was generated using forward (GGACCCTAGGTATGACTC) and reverse (CTGCTCTCCAGCACAGCT) primers. Forward primers also contained the T7 promoter sequences at the 5′-end. For the substrate in which the 5′ splice site of exon 11 was mutated to disrupt U1 snRNA binding, we mutated the 5′-most nucleotides of the intron from GT→CG. Primers used for reverse transcriptase (RT)-PCR were forward (GCTTCATGCCAAGATAGA) and reverse (AGAGGTGGCCTCTTGAAC). Morpholino oligonucleotides (MOs) (GeneTools, LLC) were complementary to specific RNA sequences: 5′ splice site-spanning MO (5′ GGGTCGCACAGGTGAGTCATACCTA), exon 11-spanning MO (5′ GTCCTCGGCACCAGTGCCTGAAGAG) and non-targeting control MO (5′ ATCCATACTGAGTGGACACGCTGGG). The 5′ splice site MO is complementary to the last 3 nucleotides of exon 11 and the first 22 nucleotides of the downstream intron. The exon 11-spanning MO is complementary to the central 25 nt of exon 11. The polypyrimidine tract of the upstream intron was mutated from 5′ gttccctctgtcctcaaaggcgttggttttgtttccacag 3′ to 5′ gAtcAAAcGgGcAtGaaaggcgttggAAtGgGAtAcacag 3′ (mutated residues are capitalized).
In vitro transcription
DNA templates used for in vitro transcription of RNA were generated either by FspI-mediated digestion of E11TNI67 or by preparative-scale PCR using E11TNI67 as a template. The T7 promoter sequence was present in the plasmid immediately upstream of DNA sequence to be transcribed or was placed at the 5′-end of the forward PCR primer. RNA was uniformly radiolabeled with 32P-UTP. Following transcription, RNA was treated with DNaseI (Ambion) and gel isolated.
In vitro splicing assay
Uniformly-radiolabeled RNA was incubated under the following conditions for in vitro splicing: 8 nM RNA substrate, 0.8 mM dithiothreitol, 1.7 mM magnesium acetate, 1.7 mM ATP, 17 mM phospho-creatine, 1 U/μl RNasin Plus (Promega) and 2.5% polyvinyl alcohol (PVA). The percentage of the reaction comprising HeLa nuclear extract (NE; 10.5 μg/μl, CilBiotech) for each substrate was 27.5% for E11TNI67 and the downstream intron substrate and 17.5% for the upstream intron substrate and the isolated exon substrate. Samples with addition of recombinant MBNL1 contained 25 ng/μl of human transcript variant 6 protein purified from HEK293 cells (OriGene, TP323216). Splicing reactions were conducted in a total volume of 15 μl and were incubated for indicated time points at 30°C. Splicing reactions were stopped by digestion with proteinase K (Ambion) followed by RNA purification and electrophoresis on 8 M urea 6% polyacrylamide gels. Gels were exposed to a film to obtain autoradiograms or to a phosphorimager screen for quantification (Typhoon Trio phosphorimager, GE Healthcare). When splicing assays were conducted in the presence of a MO, it was added to a final concentration of 1 µM.
Lariat de-branching assay
To identify bands suspected to be lariat intermediates, we conducted in vitro splicing assays, as described earlier, purified RNA and separated RNA species by urea PAGE. Next, we gel-isolated RNA bands and incubated the RNA in the presence of HeLa cytoplasmic S100 extracts under the same conditions as described for the in vitro splicing assay. The S100 extracts were prepared as described (35). Reactions were incubated at 30°C for 1 h.
Spliceosome complex formation assay
In vitro splicing was carried out as described above using the upstream intron substrate. To stop assembly, reactions were placed on ice and heparin was added to a final concentration of 2 μg/μl. Reactions were incubated in the presence of heparin at 30°C for 5 min and immediately loaded onto 0.75-mm non-denaturing 4% acrylamide–0.4% agarose composite gels. Gels were run at 225 V at room temperature in 1× Tris-glycine running buffer for 3 h.
Ultraviolet-mediated protein–RNA cross-linking
Splicing reactions were carried out as described earlier except that PVA was replaced by Dignam buffer DG, and the total reaction volume was scaled up to 30 μl. The upstream intron RNA substrate and the isolated exon substrate were labeled with 32P-UTP and 32P-GTP to increase the radioactive signal. Reactions were incubated at 30°C for 30 min and then placed as drops onto parafilm over an aluminum block in an ice bath placed 4 cm under an ultraviolet (UV) light. Samples were irradiated at 254 nm for 10 min. RNase A (Sigma Aldrich) was added to a final concentration of 1 μg/μl, and digestions were incubated at 37°C for 30 min. Ten percent of this sample was removed for sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of samples before immunoprecipitation.
Immunoprecipitation and western blotting
For one 30 μl cross-linking reaction, 20 μl of protein G dynabeads (Invitrogen) was combined with 3 μg of anti-U2AF65 monoclonal antibody (clone MC3, Santa Cruz Biotechnology) in 10 volumes of 1× binding buffer (50 mM HEPES, pH 7.8, 100 mM NaCl, 5 mM MgOAc, 0.05% NP-40) and incubated for 1 h at room temperature while rotating gently. Samples were added to the resin in a total volume of 150-μl binding buffer and incubated at room temperature for 2 h followed by 5 washes with 10 volumes of binding buffer. Bound proteins were obtained by boiling the resin in protein loading buffer. Following 10% SDS-PAGE, proteins were transferred to an Immobilon-FL polyvinylidene difluoride membrane (PVDF, Millipore) and exposed to film or a phosphorimager screen or analyzed by western blotting. For western blotting, U2AF65 was detected using a rabbit polyclonal antibody (clone H-300, Santa Cruz Biotechnology) and an AlexaFluor 680-conjugated goat anti-rabbit secondary antibody (Invitrogen) for quantitative fluorescence detection. MBNL1 was detected with a rabbit polyclonal antibody (Bethyl). hnRNP A1 was detected with a mouse monoclonal antibody (Sigma Aldrich). The fluorescent signal was read and quantified by an Odyssey infrared imager (Li-Cor).
Psoralen-mediated RNA–RNA cross-linking
The RNA substrate used for psoralen cross-linking contained the T7 promoter, 5 nt of exon 11 and 130 nt of the downstream intron including the MBNL1 response element and was uniformly labeled with 32P-UTP and 32P-GTP. Before conducting a splicing assembly assay, NEs were depleted of U1 snRNA. Twenty microliters of NE was digested with 1 μg of DNA oligonucleotide [complimentary to nucleotides 1-12 of U1 snRNA or R5S, a non-specific control oligonucleotide (36)] and 1 μl of RNase H (5 U/μl, New England Biolabs) in the presence of ATP for 1 h at 30°C.
After RNase H digestion, a splicing assay was performed as described earlier including 25% HeLa NEs and 25 ng/μl AMT-psoralen (Sigma). Reactions were incubated at 30°C for 30 min and then placed as drops onto parafilm over an aluminum block in an ice bath placed 4 cm under a UV light covered with a thin glass plate. Samples were irradiated at 365 nm for 15 min. RNA was purified as described earlier and analyzed by urea PAGE followed by autoradiography.
RESULTS
MBNL1 activates inclusion of IR exon 11 in vitro
To characterize the mechanism by which MBNL1 activates splicing, we developed an in vitro splicing assay that recapitulates MBNL1-mediated activation of exon inclusion. We previously demonstrated that a human IR genomic segment containing the 36 nt exon with 52 and 185 nt of the upstream and downstream introns, respectively, is sufficient to mediate MBNL1-activated inclusion of exon 11 in vivo (33). This IR genomic segment was inserted into a chicken sTNI in vitro splicing reporter, E11TNI67, such that it is flanked by two heterologous exons and flanking intronic regions (Figure 1A). This substrate contains the MBNL1 response element located 93-nt downstream of exon 11 (33). In vitro splicing of the uniformly radiolabeled E11TNI67 pre-mRNA was quantified following display of products and intermediates on a denaturing urea gel (Figure 1B, quantified in Figure 1C). In addition, we conducted RT-PCR of RNA extracted from in vitro splicing reactions using primers complementary to the two flanking exons (Figure 1D, quantified in Figure 1E). The identities of the RT-PCR products are indicated and were verified by sequencing gel-isolated bands (data not shown). Using both assays, addition of recombinant human MBNL1 consistently increased exon 11 inclusion by 40 percentage points (Figure 1B–E). To determine whether activation of exon inclusion required binding of MBNL1 to the MBNL response element, we tested an identical pre-mRNA containing mutations in all three MBNL1 binding motifs within the 30-nt MBNL1 response element (Figure 1A). Specifically, each of the three MBNL1 binding motifs within this region was mutated to AUAAUA, a sequence which has been shown to abolish binding of MBNL1 to RNA (25). We previously showed that these three mutations completely abolished binding of MBNL1 in vitro and MBNL1 responsiveness in vivo (33). This mutation in E11TNI67 prevented activation of exon 11 inclusion in the presence of recombinant MBNL1 (Figure 1B–E). These results indicate that MBNL1-mediated activation of IR exon 11 is robust in vitro and is mediated by MBNL1 binding to its cognate target sequence in the downstream intron.
Figure 1.

MBNL1 activates IR exon 11 inclusion in vitro. (A) E11TNI67 contains a 273-nt region of the human IR pre-mRNA inserted into a sTNI splicing reporter as shown. (B) Autoradiograms of E11TNI67 RNA purified from splicing reactions and run on a denaturing gel. Two exposures of the gels are shown to visualize the exon 11 inclusion product (left) and activated appearance of the spliced upstream intron intermediate (right) by recombinant MBNL1 (rMBNL1). Mutation of the MBNL1 response element (E11TNI67 mut) disrupts activation of exon 11 inclusion. The identities of the prominent bands are based on size and sequence of cDNAs generated from isolated bands. (C) Bands in (B) were quantified using a phosphorimager, and the percentage exon inclusion was calculated as [100 × E11 inclusion signal/(E11 inclusion signal + E11 skipping signal)]. (*P < 0.05 as calculated by a two-tailed Student’s t-test). (D) RT-PCR performed on RNA purified from in vitro splicing reactions using primers in the flanking exons (shown as arrows). DNA species were analyzed by non-denaturing PAGE followed by ethidium bromide staining. The identities of the indicated bands were confirmed by sequencing. (E) Quantification of percentage exon 11 inclusion from RT-PCR products in (D) by densitometry (*P < 0.05).
MBNL1 promotes exon 11 inclusion by activating removal of the upstream intron
MBNL1 could promote IR exon 11 inclusion by activating removal of the downstream intron to which it is bound (consistent with intron definition), and/or by activating removal of the intron upstream of exon 11 by cross-exon communication (consistent with exon definition). To determine whether MBNL1 promotes removal of the upstream and/or downstream intron, we first analyzed the splicing intermediates of the E11TNI67 pre-mRNA. MBNL1 enhanced the level of the intermediate in which the upstream, but not the downstream, intron had been removed [the 430-nt band in Figure 1B (right) and the 427-nt band in Figure 1D) indicating that MBNL1 promotes removal of the upstream intron. To directly test this, we generated two-exon pre-mRNA substrates, containing either the upstream or the downstream intron (Figure 2A). MBNL1 activated splicing of the upstream, but not the downstream, intron (Figure 2B and C). Furthermore, the MBNL1 response element was required for MBNL1-enhanced splicing of the upstream intron (Figure 2B). Importantly, mutation of the MBNL1 response element in the downstream intron substrate did not decrease the efficiency of intron removal (Figure 2C). These results indicate that MBNL1 does not promote splicing of the intron to which it is bound. Taken together, these data reveal that MBNL1 activates exon 11 inclusion by first promoting removal of the intron upstream of exon 11. This suggests that MBNL1 promotes communication from its response element in the downstream intron, across the upstream exon, resulting in definition of the exon.
Figure 2.

MBNL1 activates removal of the upstream, but not the downstream, intron. (A) Single-intron substrates containing the upstream or downstream intron were generated by PCR amplification of a portion of E11TNI67. The expected spliced products are shown on the right. (B) Autoradiogram of the RNA products and intermediates of splicing of the upstream intron (WT) and upstream intron mutant (mut), as confirmed by RT-PCR and sequencing of gel-isolated bands (data not shown) as well as a lariat de-branching assay in the presence of HeLa S100 extracts (Supplementary Figure S1). Quantification of splicing using a phosphorimager [100 × (signals from two splicing intermediates + signal from spliced product)/total signals of all RNA isoforms present] (*P < 0.05) (n = 6) (bottom). (C) Addition of rMBNL1 had no effect on splicing of the downstream intron.
MBNL1 activates spliceosome complex a formation on the upstream intron
We next sought to determine whether MBNL1 activates an early step of spliceosome assembly on the upstream intron. The pre-mRNA containing the MBNL1-responsive upstream intron was incubated under splicing conditions, and complexes were separated using native gel electrophoresis. Over time, we saw the appearance of the ATP-dependent complex A that migrated slowly in relation to the non-specific complex H (Figure 3A). Addition of MBNL1 to the splicing reactions enhanced formation of complex A. Importantly, the ability of MBNL1 to promote complex A formation was disrupted in the identical RNA containing the mutation of the MBNL1 response element in the downstream intron (Figure 3A and B). We observed that the activation of complex A formation by rMBNL1 was robust at 1 min and also exhibited dose-dependence (Supplementary Figure S2). We conclude that MBNL1 promotes removal of exon 11 by activating assembly of spliceosomal complex A. These results indicate that MBNL1 promotes cross-exon communication to enhance assembly of early spliceosome complexes to promote splicing of the intron upstream of exon 11.
Figure 3.
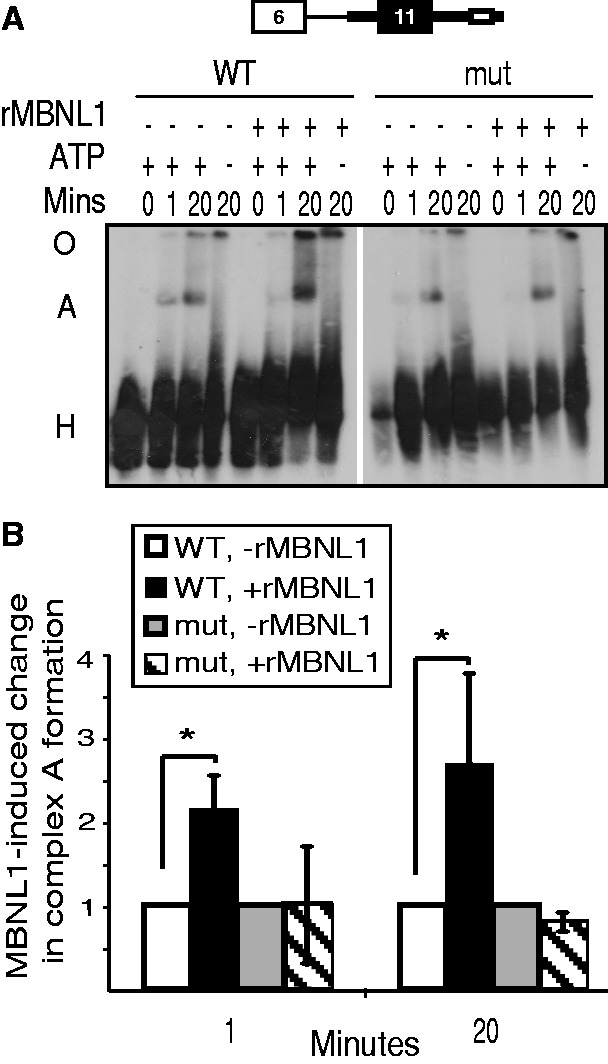
MBNL1 activates complex A formation on the upstream intron. (A) Autoradiogram of spliceosome complexes assembled on the upstream intron substrate visualized following native gel electrophoresis. rMBNL1 activates complex A (A) assembly on the upstream intron substrate, but not the substrate containing a mutated MBNL1 binding site (mut). The non-specific H complex is indicated (H). (B) Phosphorimager quantification of the percentage of RNA in complex A [( = radioactive signal in complex A/(radioactive signal in complex A + complex H)] was used to calculate the MBNL1-induced fold change in complex A formation (*P < 0.05). The RNA–protein complexes retained in the well/origin (O) were not included in the quantification.
MBNL1 promotes binding of U2AF65 upstream of exon 11
Our observation that MBNL1 activates assembly of complex A suggests that MBNL1 promotes assembly of early spliceosomal proteins on the pre-mRNA. We used a UV cross-linking assay on two uniformly radiolabeled IR exon 11 substrates to identify RNA–protein interactions promoted by MBNL1. One substrate was the upstream intron pre-mRNA and the other contained only exon 11, 52 nt of the upstream intron and 185 nt of the downstream intron (Figure 4A). For both substrates, we also tested the identical RNA containing the mutated MBNL1 response element. Following cross-linking, RNase-treated samples were separated by SDS-PAGE. By autoradiography, we observed a 42-kDa band interacting with both wild-type substrates corresponding in size to recombinant MBNL1, and this band was absent from the substrates containing the mutant MBNL1 binding sites (Figure 4A). We also saw a faint band that appeared to be enhanced by MBNL1, and that was the expected size of U2AF65 (Figure 4A). To specifically determine whether MBNL1 enhanced binding of U2AF65, we performed immunoprecipitation with a U2AF65 antibody. Immunoprecipitated proteins were separated by SDS-PAGE, transferred to a PVDF membrane and the level of RNA binding was quantified using a phosphorimager and normalized to the amount of U2AF65 protein detected on the same membrane by western blotting using a fluorescently labeled secondary antibody (Figure 4B). This quantitative analysis demonstrated that MBNL1 enhanced binding of U2AF65 to the upstream intron even on the substrate containing the isolated exon and partial flanking introns. MBNL1 did not affect hnRNP A1 cross-linking to the same two substrates, indicating the specificity of the U2AF65 response to MBNL1 (Supplementary Figure S3). In addition, mutation of the polypyrimidine tract or complete removal of the intron 10 segment abolished cross-linking of U2AF65 to both pre-mRNA substrates in the presence or absence of rMBNL1 (Supplementary Figure S4) demonstrating that the U2AF65 cross-link represents binding to the polypyrimidine tract of intron 10. Furthermore, enhanced binding of U2AF65 by MBNL1 is dependent on the intact MBNL1 response element in the downstream intron. These results indicate that MBNL1 promotes assembly of spliceosome complexes on an isolated exon, with flanking intronic segments consistent with the exon definition model.
Figure 4.
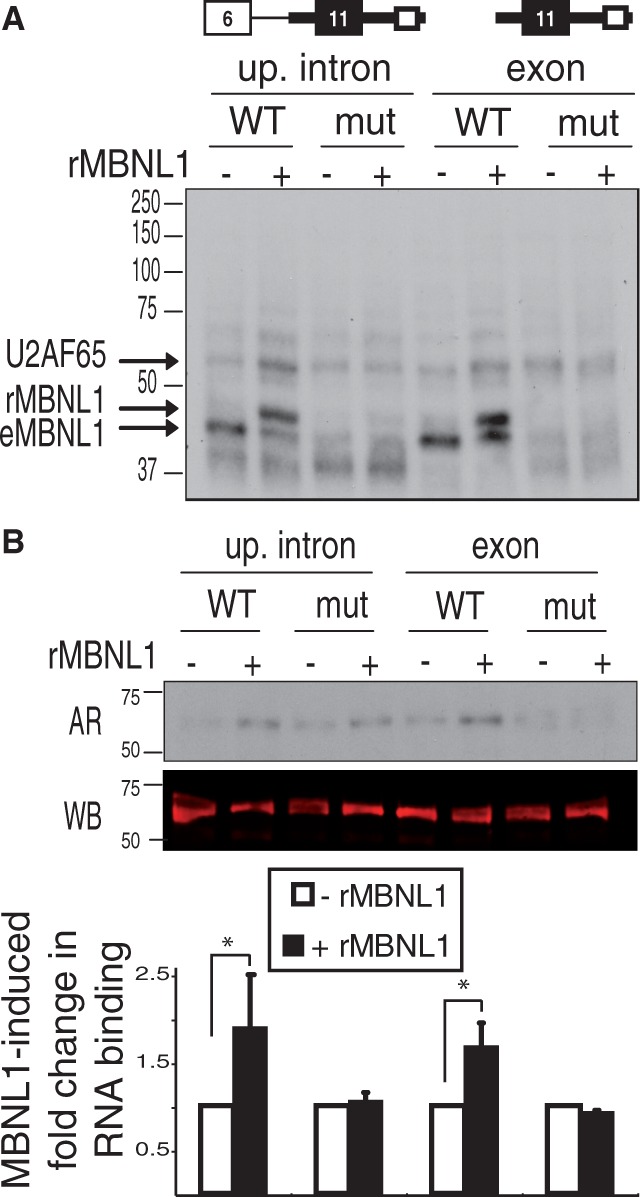
MBNL1 activates UV cross-linking of U2AF65 to two IR exon 11 substrates dependent on the MBNL1 response element. (A) UV cross-linking of splicing reactions using the upstream intron substrate and the isolated exon with surrounding intronic sequences with (mut) or without (WT) mutated MBNL1 binding sites. Cross-linked proteins were visualized by SDS-PAGE followed by autoradiography. The expected sizes of U2AF65, recombinant MBNL1 (rMBNL1) and endogenous MBNL1 (eMBNL1) are indicated. Molecular weights in kilodaltons are shown on the left. (B) U2AF65 was immunoprecipitated following UV cross-linking. An autoradiogram (AR) of immunoprecipitated samples subjected to SDS-PAGE (top). A western blot (WB) for U2AF65 (middle). Quantification of the MBNL1-induced fold change in U2AF65 bound to RNA calculated by first normalizing the phosphorimager quantification of the RNA signal to the fluorescent signal from the western blot (*P < 0.05) (bottom).
To test whether MBNL1 interacts with U2AF65, we immunoprecipitated U2AF65 from HeLa NEs treated with RNase A followed by western blotting for MBNL1. We were unable to detect co-immunoprecipitation of MBNL1 with U2AF65 (Supplementary Figure S5). We obtained the same result by the reciprocal co-immunoprecipitation test (data not shown). These data suggest that MBNL1 activates binding of U2AF65 to the upstream intron through a mechanism other than a direct and stable protein–protein interaction.
Splicing activation by MBNL1 does not require binding of factors within exon 11
We next tested whether activated splicing of the upstream intron or enhanced binding of U2AF65 to the upstream 3′ splice site by MBNL1 required assembly of a complex spanning exon 11. To accomplish this, we used an MO to block the central 25 nt of exon 11 in an in vitro splicing assay using the RNA substrate containing the upstream intron. We first confirmed that the MO bound to the RNA by demonstrating protection of 25-nt RNA fragment from RNase A digestion (Supplementary Figure S6A). Inclusion of the MO blocked progression of the splicing reaction past the first catalytic step, resulting only in accumulation of the lariat intermediate formed after cleavage at the 5′ splice site, whereas a control MO had no effect (Figure 5A, quantified in Figure 5B). The splicing inhibitory effect of antisense blockage of internal portions of a 3′ exon has been observed previously (37). Using the exon 11 blocking MO, we observe that only the first catalytic step of splicing occurs. As shown above (Figure 2B), MBNL1 activates appearance of the lariat intermediate resulting from the first catalytic step. The exon 11 MO did not disrupt the ability of MBNL1 to activate this intermediate (Figure 5A and B), confirming our earlier results that MBNL1 activates an early step of spliceosome assembly. In addition, these data demonstrate that MBNL1 activates upstream intron removal across the exon without the requirement for exon-binding factors.
Figure 5.
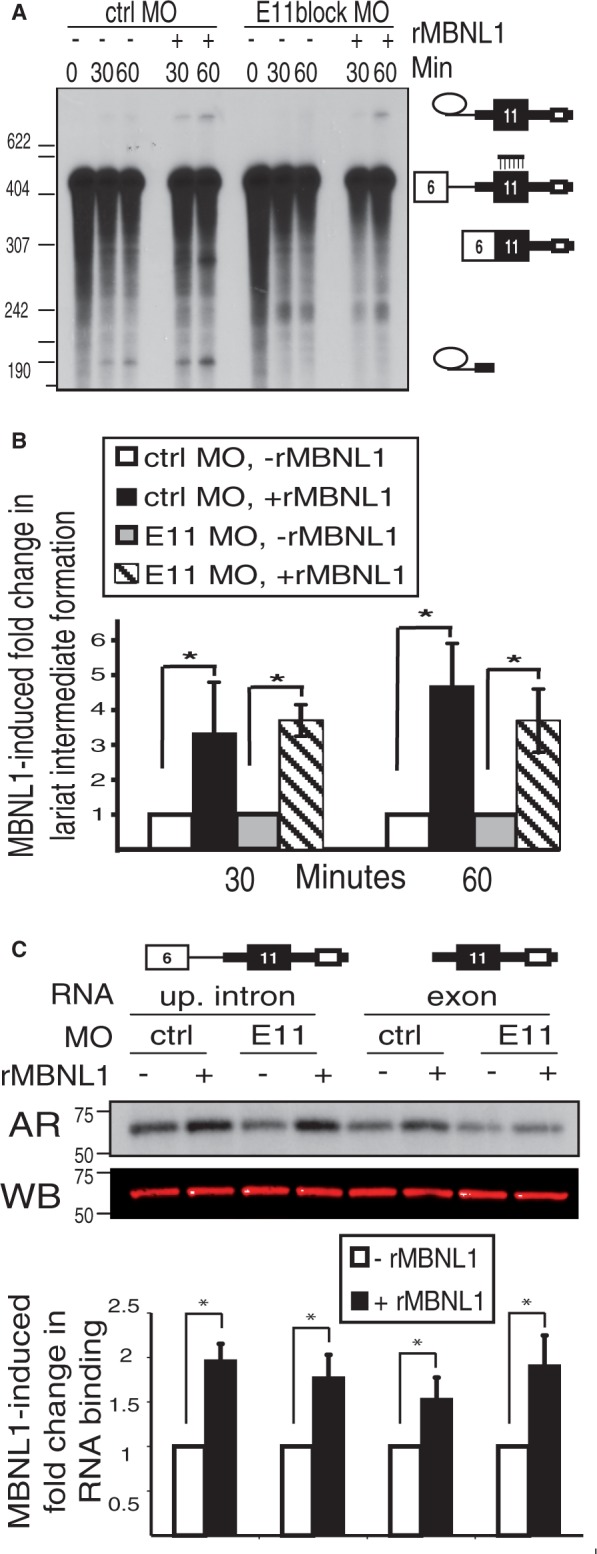
Access to exon 11 sequences is not required for MBNL1-mediated activation of upstream intron splicing or U2AF65 binding. (A) The upstream intron RNA substrate was spliced in the presence of an exon 11-spanning (E11block) or control (ctrl) MO and visualized by autoradiography. The location of the exon 11 MO is indicated on the right. The ∼240-nt band appears in some experiments using both MOs in the presence and absence of ATP, suggesting they are non-specific degradation products (data not shown). (B) Quantification of the MBNL1-induced fold change in lariat intermediate formation using the phosphorimager signal for the percentage lariat intermediate (compared with total RNA isoforms in the lane). (*P < 0.05) (C) UV cross-linking followed by immunoprecipitation for U2AF65 (as described in Figure 4B) in the presence of the exon 11 MO (E11). An autoradiogram (AR) and western blot for U2AF65 (WB) are shown. The bottom panel shows quantification of the MBNL1-induced fold change in RNA binding (normalized to protein levels as in 4B, *P < 0.05).
Furthermore, we tested whether enhanced binding of U2AF65 by MBNL1 is affected by the exon 11 MO. We found that the exon 11 MO did not disrupt MBNL1-mediated activation of U2AF65 binding to the upstream intron using the substrate containing the complete intron or the substrate containing only the isolated exon (Figure 5C). Taken together, these data indicate that MBNL1 activates U2AF65 binding and splicing of the upstream intron independent of assembly of complexes within exon 11.
MBNL1-mediated splicing of the upstream intron requires the 5′ splice site of exon 11
The MBNL1 response element lies in the intron downstream of IR exon 11, and we have demonstrated that MBNL1 activates splicing of the intron upstream of IR exon 11 (Figure 2). Binding of the U1 snRNP at the 5′ splice site is known to enhance assembly of spliceosome components at the 3′ splice site upstream of the exon via exon definition (9). Therefore, we wanted to determine whether MBNL1-mediated activation of exon 11 splicing was dependent on the 5′ splice site of exon 11. We used a 5′ splice site-blocking MO to test whether the 5′ splice site of exon 11 is required for MBNL1 to activate splicing of the intron upstream of exon 11. The same concentration of a scrambled MO was used as a control. We confirmed that the 5′ splice site MO, but not the control MO, inhibited use of the exon 11 5′ splice site by demonstrating that it completely blocks splicing of the downstream intron in a two-exon substrate (Supplementary Figure S6B). When included in a splicing reaction of the upstream intron RNA, the 5′ splice site MO led to a lower basal level of splicing. This result was not surprising, as the presence of a 5′ splice site enhances removal of the upstream intron (9). While the control MO had no inhibitory effect on activation of splicing by MBNL1, the 5′ splice site MO prevented MBNL1-activated splicing of the upstream intron (Figure 6A, quantified in Figure 6B).
Figure 6.
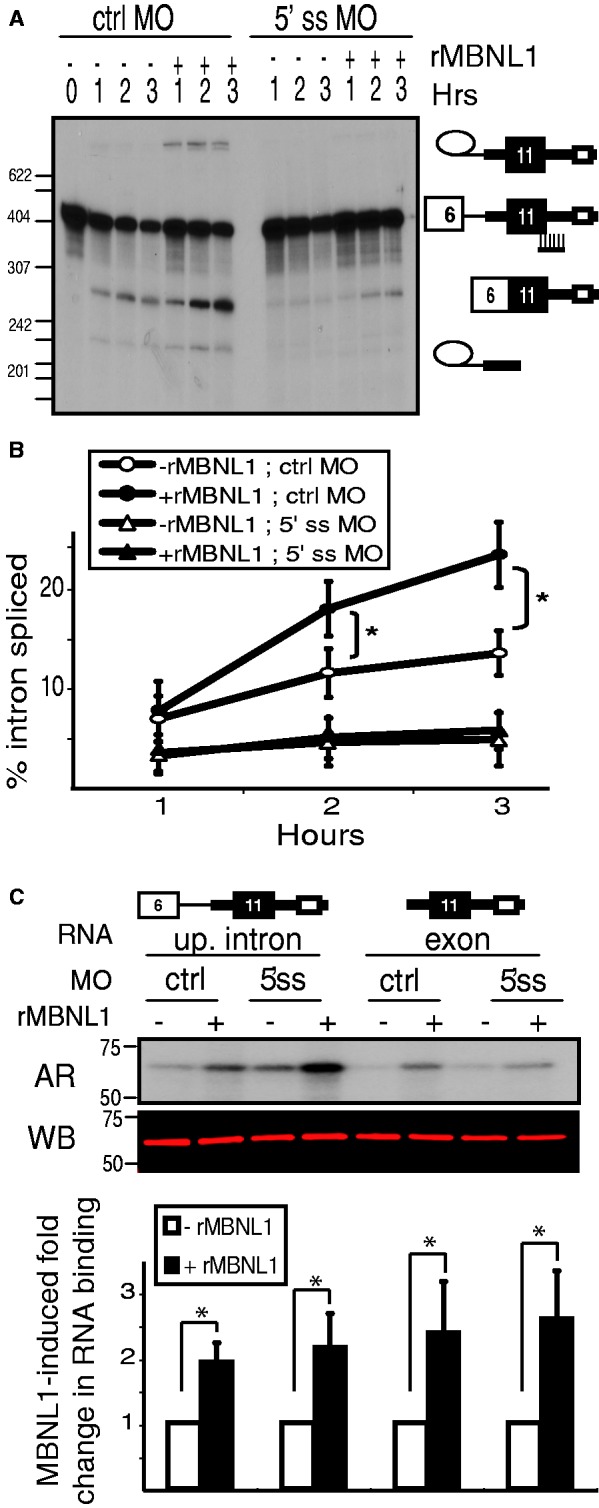
The ability of MBNL1 to activate splicing of the upstream intron is disrupted by blocking the 5′ splice site of exon 11. (A) The upstream intron RNA substrate was spliced in the presence of a 5′ splice site-blocking MO (indicated on the right), displayed by urea PAGE and visualized by autoradiography. (B) We calculated the percentage of the upstream intron spliced [100 × (phosphorimager signals from two splicing intermediates + signal from spliced product)/total signal of all isoforms present]. (*P < 0.05) (C) UV cross-linking analysis on the upstream intron substrate and the isolated exon substrate was conducted as described in Figure 4, except in the presence of a control (ctrl) MO or a 5′ splice site-blocking (5′ss) MO. U2AF65 was immunoprecipitated from UV cross-linking reactions as described in Figure 4B. The autoradiogram, western blot and quantification were conducted as described in Figure 4B. (*P < 0.05).
We next tested whether the enhanced binding of U2AF65 mediated by MBNL1 required the 5′ splice site of exon 11. We tested the effect of the 5′ splice MO on U2AF65 recruitment using both the upstream intron substrate and the isolated exon substrate. We performed UV cross-linking of each of these substrates in splicing reactions in the presence of the 5′ splice site-blocking MO. Following immunoprecipitation with a U2AF65 antibody, we observed that MBNL1 mediated activation of U2AF65 binding even in the presence of the 5′ splice site MO (Figure 6C). We confirmed this result using an RNA substrate with a mutated 5′ splice site (without using MOs) (Supplementary Figure S7). These results indicate that MBNL1 can activate binding of U2AF65 to the upstream intron using a mechanism that is independent of the 5′ splice site downstream of exon 11. Combined with the result that blocking exon 11 does not disrupt the MBNL1 response, these data indicate that MBNL1 is able to directly promote assembly of U2AF65 on the upstream intron.
To test whether the requirement for the 5′ splice site for MBNL1-activated upstream intron splicing is due to enhanced U1 snRNP binding by MBNL1, we conducted psoralen-mediated RNA–RNA cross-linking to detect binding of U1 snRNA to the pre-mRNA. We used a uniformly radiolabeled RNA substrate containing the minimal region of interest (the 5′ splice site of exon 11 and the downstream intron extending to 5 nt downstream of the MBNL1 response element) (Figure 7). This RNA was incubated under splicing conditions in the presence of the photochemical cross-linker AMT-psoralen. Reactions were UV-cross-linked followed by RNA purification and separation by urea PAGE. NEs used in this assay were first treated with RNase H and a DNA oligonucleotide to digest U1 snRNA or a non-targeting control oligonucleotide. On digestion of U1 snRNA, we saw disappearance of a prominent cross-linked band identifying it as the pre-mRNA-U1 snRNA cross-link. However, this band did change in intensity in response to addition of MBNL1 (Figure 7). We confirmed that binding of MBNL1 and U2AF65 to IR pre-mRNA is not disrupted in the presence of psoralen in a UV cross-linking assay (data not shown). These results indicate that MBNL1 does not directly enhance U1 snRNA binding to the 5′ splice site of IR exon 11. Taken together, these findings indicate that activation of IR exon 11 splicing by MBNL1 is mediated by enhanced U2AF65 binding to the upstream intron. Although the 5′ splice site of exon 11 is required for MBNL1-mediated activation of upstream intron splicing, our results indicate that MBNL1 does not affect the recruitment of U1 snRNP to the 5′ splice site.
Figure 7.
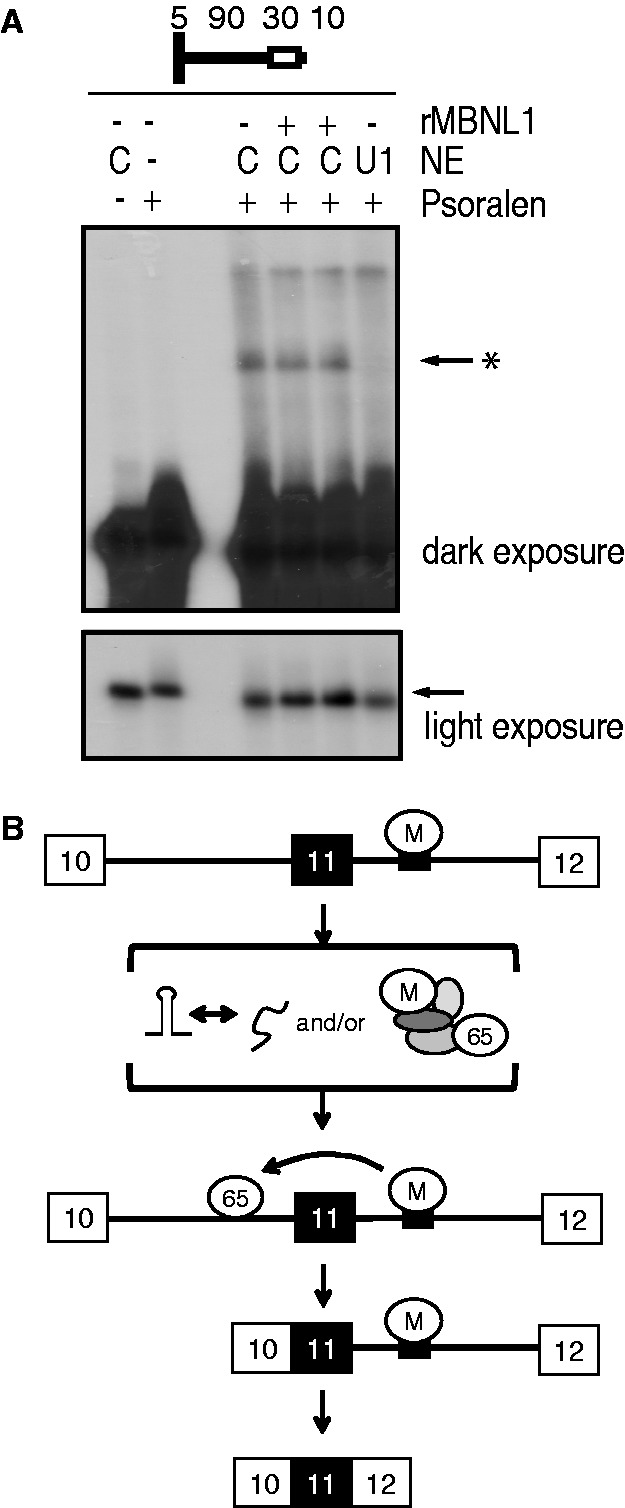
MBNL1 does not activate binding of U1 snRNA to the 5′ splice site of exon 11. (A) A psoralen cross-linking assay was conducted using a shortened RNA substrate comprising the 5′ splice site (including the last 5 nt of E11) and a portion of the downstream intron including the MBNL1 response element (top). Before assembly of the splicing assay, NEs were treated with an oligo to digest U1 snRNA (U1) or the R5S control oligo (C). The light exposure shows the level of un-cross-linked precursor RNA (arrow). The dark exposure shows U1 snRNA-containing cross-link (arrow*). (B) A model for MBNL1-mediated activation of IR exon 11 inclusion. MBNL1 binds its response element in the downstream intron (small black box) and activates U2AF65 binding on the upstream intron. This activation might involve modulation of RNA secondary structure and/or formation of a protein complex, which bridges U2AF65 and MBNL1.
DISCUSSION
MBNL1 splicing regulatory activity is context-dependent such that its influence on alternative exon inclusion depends on the pre-mRNA target. The mechanism by which MBNL1 represses splicing of cTNT exon 5 has been characterized previously (17). The work presented here represents the first characterization of the mechanism by which MBNL1 communicates with the basal splicing machinery to activate exon inclusion. We found that MBNL1 activates early spliceosome assembly across exon 11 directly on the upstream intron (Figure 2B). Specifically, MBNL1 activates U2AF65 binding and spliceosome complex A formation on the intron upstream of exon 11 (Figures 3 and 4). We confirmed that MBNL1 activates early spliceosome assembly before the first catalytic step of splicing using an exon 11-blocking MO, which prevented progression to the second catalytic step (Figure 5A). We propose that MBNL1 activates recognition of exon 11 by directly promoting assembly of complexes on intronic regions upstream of exon 11, thus ‘defining’ the exon. Exon definition can result from a mechanism in which the U1 snRNP first assembles at the 5′ splice site, which subsequently recruits spliceosomal complexes (U2AF65) across the exon to the upstream 3′ splice site through a network of protein–protein interactions spanning the exon (9). However, several pieces of evidence indicate that MBNL1-mediated activation of U2AF65 binding is through a direct mechanism, rather than by initially activating U1 snRNP assembly at the 5′ splice site of exon 11. First, MBNL1 does not enhance binding of U1 snRNA to the 5′ splice site of exon 11 (Figure 7) and consequently does not activate splicing of the downstream intron to which it is bound (Figure 2C). Second, we prevented binding of factors within exon 11 using an exon-spanning MO. We did not observe disruption of MBNL1-activated upstream intron splicing or of MBNL1-activated U2AF65 binding (Figure 5). Third, we prevented binding of factors (such as U1 snRNP) at the 5′ splice site of exon 11 using a 5′ splice site-spanning MO. We did not observe disruption of MBNL1-activated U2AF65 binding (Figure 6C). Together, these results indicate that the ability of MBNL1 to activate binding of U2AF65 to the upstream intron does not depend on binding events at the 5′ splice site or within exon 11. We also observed that blocking the 5′ splice site of exon 11 disrupts the ability of MBNL1 to activate upstream intron splicing (Figure 6A and B). This could indicate that the mechanism activated by MBNL1 requires the 5′ splice site in steps subsequent to U1 snRNP recruitment.
The direct contacts induced by MBNL1 to result in this effect are yet unknown. Important future studies will determine whether MBNL1 promotes formation of a protein complex that recruits and/or stabilizes U2AF65 on the upstream intron, although we did not observe co-immunoprecipitation of MBNL1 and U2AF65 (Supplementary Figure S5). Alternatively, MBNL1 could promote a secondary structure, which favors spliceosome assembly. In the case of cTNT exon 5 repression, MBNL1 binds a stem-loop structure at the polypyrimidine tract upstream of exon 5, which prevents U2AF65 binding to the single-stranded polypyrimidine tract (17). MBNL1 is known to bind stem-loop RNA structures (38). It is possible that MBNL1 binds to and enhances the stability of a stem-loop structure within the IR pre-mRNA promoting pre-mRNA rearrangements that enhance binding of U2AF65 to the polypyrimidine tract upstream of exon 11 (Figure 7B). Modulation of RNA secondary structure has been indicated in several other splicing regulatory mechanisms (39). It will be important to determine whether mechanistic features of MBNL1-mediated splicing activation are conserved across various RNA targets. High-throughput RNA sequencing and cross-linking immunoprecipitation-sequencing analyses revealed an ‘RNA map’ for hundreds of MBNL1-regulated splicing events (11). These studies suggested that the architecture of the RNA target impacts the splicing regulatory potential of MBNL1. Specifically, the location of the MBNL1 response element relative to the alternative exon determines whether MBNL1 will act as a splicing activator or as a repressor. Important future investigations will identify whether MBNL1 activates U2AF65 recruitment to the upstream intron through modulation of RNA secondary structure and/or a U2AF65-binding auxiliary protein complex and whether these mechanistic features are shared with other MBNL1-activated exons.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
A National Science Foundation Graduate Research Fellowship [DGE0642350 to G.V.E.]; National Institutes of Health (NIH) [R01HL045565 and R01AR045653 to T.A.C.]. Funding for open access charge: NIH [R01HL045565].
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Dr. Eric Wagner for providing HeLa cell cytoplasmic fraction with which we prepared S100 extracts. They thank Dr. Ioannis Grammatikakis and Dr. Ravi Singh for helpful insights and suggestions during the preparation of this manuscript. They thank Dr. Ioannis Grammatikakis and Donnie Bundman for generating several plasmid constructs used in this study.
REFERENCES
- 1.Will CL, Luhrmann R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2010;3 doi: 10.1101/cshperspect.a003707. pii: a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoskins AA, Moore MJ. The spliceosome: a flexible, reversible macromolecular machine. Trends Biochem. Sci. 2012;37:179–188. doi: 10.1016/j.tibs.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science. 2012;338:1593–1599. doi: 10.1126/science.1228186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 5.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalsotra A, Cooper TA. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011;12:715–729. doi: 10.1038/nrg3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robberson BL, Cote GJ, Berget SM. Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol. Cell. Biol. 1990;10:84–94. doi: 10.1128/mcb.10.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berget SM. Exon recognition in vertebrate splicing. J. Biol. Chem. 1995;270:2411–2414. doi: 10.1074/jbc.270.6.2411. [DOI] [PubMed] [Google Scholar]
- 9.Hoffman BE, Grabowski PJ. U1 snRNP targets an essential splicing factor, U2AF65, to the 3′ splice site by a network of interactions spanning the exon. Genes Dev. 1992;6:2554–2568. doi: 10.1101/gad.6.12b.2554. [DOI] [PubMed] [Google Scholar]
- 10.De Conti L, Baralle M, Buratti E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip. Rev. RNA. 2012;4:49–60. doi: 10.1002/wrna.1140. [DOI] [PubMed] [Google Scholar]
- 11.Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–724. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, Gaasterland T, Blencowe BJ, Darnell RB. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- 13.Chiou NT, Shankarling G, Lynch KW. hnRNP L and hnRNP A1 induce extended U1 snRNA interactions with an exon to repress spliceosome assembly. Mol. Cell. 2013;49:972–982. doi: 10.1016/j.molcel.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma S, Maris C, Allain FH, Black DL. U1 snRNA directly interacts with polypyrimidine tract-binding protein during splicing repression. Mol. Cell. 2011;41:579–588. doi: 10.1016/j.molcel.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma S, Falick AM, Black DL. Polypyrimidine tract binding protein blocks the 5′ splice site-dependent assembly of U2AF and the prespliceosomal E complex. Mol. Cell. 2005;19:485–496. doi: 10.1016/j.molcel.2005.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Izquierdo JM, Majos N, Bonnal S, Martinez C, Castelo R, Guigo R, Bilbao D, Valcarcel J. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol. Cell. 2005;19:475–484. doi: 10.1016/j.molcel.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 17.Warf MB, Diegel JV, von Hippel PH, Berglund JA. The protein factors MBNL1 and U2AF65 bind alternative RNA structures to regulate splicing. Proc. Natl Acad. Sci. USA. 2009;106:9203–9208. doi: 10.1073/pnas.0900342106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goo YH, Cooper TA. CUGBP2 directly interacts with U2 17S snRNP components and promotes U2 snRNA binding to cardiac troponin T pre-mRNA. Nucleic Acids Res. 2009;37:4275–4286. doi: 10.1093/nar/gkp346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forch P, Puig O, Martinez C, Seraphin B, Valcarcel J. The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5′ splice sites. EMBO J. 2002;21:6882–6892. doi: 10.1093/emboj/cdf668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tisserant A, Konig H. Signal-regulated Pre-mRNA occupancy by the general splicing factor U2AF. PLoS One. 2008;3:e1418. doi: 10.1371/journal.pone.0001418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- 22.Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, Cooper TA. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl Acad. Sci. USA. 2008;105:20333–20338. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Artero R, Prokop A, Paricio N, Begemann G, Pueyo I, Mlodzik M, Perez-Alonso M, Baylies MK. The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2. Dev. Biol. 1998;195:131–143. doi: 10.1006/dbio.1997.8833. [DOI] [PubMed] [Google Scholar]
- 24.Begemann G, Paricio N, Artero R, Kiss I, Perez-Alonso M, Mlodzik M. muscleblind, a gene required for photoreceptor differentiation in Drosophila, encodes novel nuclear Cys3His-type zinc-finger-containing proteins. Development. 1997;124:4321–4331. doi: 10.1242/dev.124.21.4321. [DOI] [PubMed] [Google Scholar]
- 25.Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA. Muscleblind proteins regulate alternative splicing. EMBO J. 2004;23:3103–3112. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, Hall MP, Shiue L, Swanson MS, Thornton CA, et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 2010;17:187–193. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 28.Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 29.Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS, Thornton CA. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 2001;10:2165–2170. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- 30.Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moxley RT, III, Griggs RC, Goldblatt D, VanGelder V, Herr BE, Thiel R. Decreased insulin sensitivity of forearm muscle in myotonic dystrophy. J. Clin. Invest. 1978;62:857–867. doi: 10.1172/JCI109198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 33.Grammatikakis I, Goo YH, Echeverria GV, Cooper TA. Identification of MBNL1 and MBNL3 domains required for splicing activation and repression. Nucleic Acids Res. 2010;39:2769–2780. doi: 10.1093/nar/gkq1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sen S, Talukdar I, Liu Y, Tam J, Reddy S, Webster NJ. Muscleblind-like 1 (Mbnl1) promotes insulin receptor exon 11 inclusion via binding to a downstream evolutionarily conserved intronic enhancer. J. Biol. Chem. 2010;285:25426–25437. doi: 10.1074/jbc.M109.095224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krainer AR, Maniatis T, Ruskin B, Green MR. Normal and mutant human beta-globin pre-mRNAs are faithfully and efficiently spliced in vitro. Cell. 1984;36:993–1005. doi: 10.1016/0092-8674(84)90049-7. [DOI] [PubMed] [Google Scholar]
- 36.Seiwert SD, Steitz JA. Uncoupling two functions of the U1 small nuclear ribonucleoprotein particle during in vitro splicing. Mol. Cell. Biol. 1993;13:3135–3145. doi: 10.1128/mcb.13.6.3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munroe SH. Antisense RNA inhibits splicing of pre-mRNA in vitro. EMBO J. 1988;7:2523–2532. doi: 10.1002/j.1460-2075.1988.tb03100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Warf MB, Berglund JA. MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. RNA. 2007;13:2238–2251. doi: 10.1261/rna.610607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McManus CJ, Graveley BR. RNA structure and the mechanisms of alternative splicing. Curr. Opin. Genet. Dev. 2011;21:373–379. doi: 10.1016/j.gde.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]