

Abstract
Bioassay-guided separation of the South African plant Kniphofia ensifolia for antiplasmodial activity led to the isolation of two new anthraquinones, named kniphofiones A and B (3, 4), together with three known bioactive anthraquinone monomers (1, 2 and 5), and four known bisanthraquinones (6–9). The structures of the two new compounds were elucidated based on analyses of their 1D and 2D NMR spectra and mass spectrometric data. The dimeric compounds 6 and 7 displayed the strongest antiplasmodial activity among all the isolated compounds, with IC50 values of 0.4 ± 0.1 and 0.2 ± 0.1 μM, respectively. The two new compounds displayed modest activities, with IC50 values of 26 ± 4 and 9 ± 1 μM, respectively. Due to the synthetic accessibility of the new compounds and the increased activity shown by the dimeric compounds, a structure-activity relationship study was conducted. As a result, one analogue of kniphofione B (4), the caffeic acid derivative of aloe-emodin, was found to have the highest activity among all the aloe-emodin derivatives, with an IC50 value of 1.3 ± 0.2 μM.
Keywords: Anthraquinone, Antiplasmodial acitivity, Kniphofia ensifolia, Structure-activity relationship
1. Introduction
Malaria remains one of the major infectious diseases that threaten human lives. According to the latest available estimates from the WorldaHealthaOrganization (WHO), there were about 219 million cases of malaria and an estimated 660,000 deaths from malaria in 2010, with about 90% of all of these deaths occurring in sub-Saharan Africa.1 Although current anti-malarial therapies, including mefloquinone and its analogues2 and artemisinin combination therapy combined with insecticide-treated bed nets3 have achieved significant success in controlling the spread of the disease, malaria still remains a burden to humanity due to increasing drug resistance and the lack of multistage therapies to target the complex life cycle of the parasites. The recent emergence of resistance to artemisinin in the Cambodia–Thailand border area is particularly troubling.4 A continuing search for new anti-malarial drugs is thus important to the overall goal of eradicating this scourge of mankind.
As part of a collaborative research project established between Virginia Tech and the Institute for Hepatitis and Virus Research (IHVR) and its the Natural Products Discovery Institute (NPDI),5 we are searching for antiproliferative and antimalarial natural products in the plant extract library of the NPDI. An investigation for antiproliferative compounds from this library yielded several homoisoflavonones and bufadienolides with strong antiproliferative activity,6 and the present work reports the first results from our investigation of extracts with antimalarial activity.
Screening of over 6,900 extracts from the NPDI collection led to the identification of a dichloromethane extract of Kniphofia ensifolia Baker (Asphodelaceae)7 as an extract with promising antiplasmodial activity against the drug-resistant Dd2 strain of Plasmodium falciparum, with an IC50 value of ~ 6 μg/mL. Members of the Asphodelaceae family are widely distributed in Africa, central and western Europe, the Mediterranean basin, Central Asia, and Australia.8 The genus Kniphofia is a rich source of anthraquinones, flavonoids and alkaloids, which are well-known for their broad range of bioactivities, including anticancer and antimalarial activities.9 Although the genus Kniphofia has been well explored, the phytochemistry of K. ensifolia has not previously been investigated, and its extract of was thus selected for bioassay-guided fractionation to isolate its bioactive components.
2. Results and discussions
2.1. Isolation and structure elucidation of bioactive anthraquinones
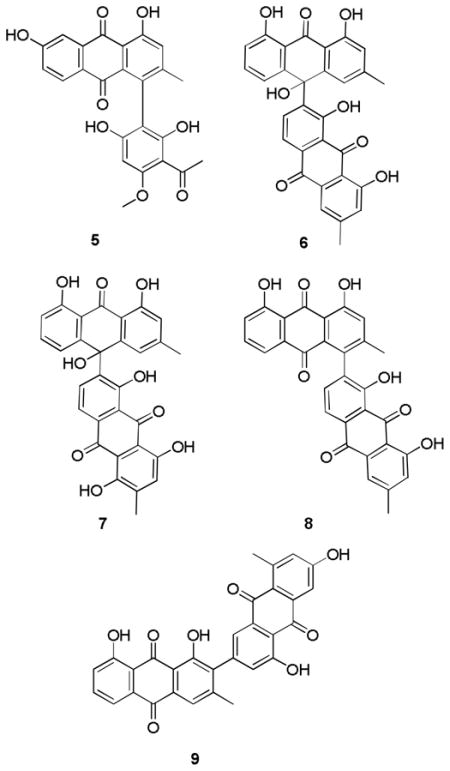
An EtOH extract of the whole plant of K. ensifolia was subjected to liquid-liquid partitioning to give an antiplasmodial CH2Cl2 fraction (IC50 = ~ 6.0 μg/mL). Bioassay-guided separation of the CH2Cl2 fraction, including Sephadex LH-20 size-exclusion chromatography, normal-phase silica gel chromatography, and C18 reverse-phase HPLC, yielded two new anthraquinones, named kniphofiones A and B (3, 4), two known strongly active anthraquinones (6, 7), and five other known anthraquinones (1, 2, 5, 8, 9). The structure elucidation of the new compounds is reported herein.
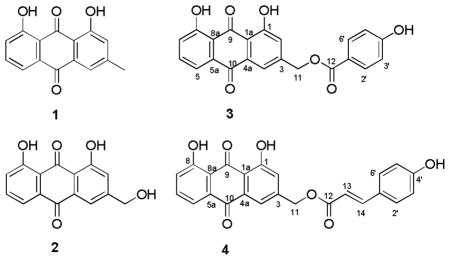
Compounds 1, 2 and 5–9 were identified as chrysophanol (1),10 aloe-emodin (2),11 knipholone (5),12 10-(chrysophanol-7′-yl)-10-hydroxychrysophanol-9-anthrone (6),9b chryslandicin (7),13 asphodeline (8),14 and microcarpin (9)15 respectively, by comparison of their experimental and reported physical and spectroscopic data with literature data.
Kniphofione A (3) was isolated as a yellow-orange powder. Its negative ion HR-ESI-MS revealed a peak for a deprotonated molecular ion at m/z 389.0688 [M-H]−, corresponding to a molecular formula of C22H14O7. Its IR spectrum exhibited absorption bands at 3390 and 1673 cm−1, indicating the presence of hydroxy and conjugated carbonyl groups. The presence of two chelated hydroxy groups was further confirmed by two singlet signals located at δH 12.09 and 12.12 in the 1H NMR spectrum (Table 1). In addition, a set of three coupled aromatic protons in the ABC-type spin system [δH 7.90 (1H, dd, J = 7.9, 1.6 Hz), 7.73 (1H, dd, J = 8.0, 7.9 Hz) and 7.39 (1H, dd, J = 8.0, 1.6 Hz)], two meta-coupled doublets located at δH 7.87 and 7.34 (both, 1H, J = 1.6), as well as methylene protons on an oxygen-bearing carbon at δH 5.44 (2H, singlet), were similar to the corresponding signals of aloe-emodin (2) and of aloe-emodin-type anthraquinones.11, 16 The observation of two carbonyl carbon resonances located at δC 192.7 and 181.6, as well as a methylene carbon signal at δC 64.9 (C-11) in the 13C NMR spectrum, corroborated its structure as an aloe-emodin derivative. The basic skeleton of 1,8-dihydroxy-3-(hydroxymethyl)anthracene-9,10-dione was confirmed by the HMBC correlations between H-2 and C-1a, H-4 and C-10, H-6 and C-8, H-7 and C-8a, and H-11 and C-2 (Figure 1).
Table 1.
NMR Spectroscopic Data for 3 and 4 in CDCl3 (500 MHz)
| 3 | 4 | |||
|---|---|---|---|---|
| posn | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type |
| 1 | - | 162.9, C | - | 162.8, C |
| 1a | - | 115.3, C | - | 115.2, C |
| 2 | 7.87 d (1.6) | 118.6, CH | 7.87 d (1.6) | 118.5, CH |
| 3 | - | 147.0, C | - | 146.9, C |
| 4 | 7.34 d (1.6) | 122.4, CH | 7.36 d (1.6) | 122.4, CH |
| 4a | - | 133.6, C | - | 133.6, C |
| 5 | 7.90 dd (7.9, 1.6) | 120.2, CH | 7.87 dd (7.9, 1.6) | 120.2, CH |
| 5a | - | 134.1, C | - | 133.9, C |
| 6 | 7.73 dd (8.0, 7.9) | 137.4, CH | 7.73 dd (8.0, 7.9) | 137.3, CH |
| 7 | 7.39 dd (8.0, 1.6) | 124.8, CH | 7.35 dd (8.0, 1.6) | 124.8, CH |
| 8 | - | 162.7, C | - | 162.8, C |
| 8a | - | 115.9, C | - | 115.8, C |
| 9 | - | 192.7, C | - | 192.7, C |
| 10 | - | 181.6, C | - | 181.6, C |
| 11 | 5.44 s | 64.9, CH2 | 5.35 s | 64.6, CH2 |
| 12 | - | 165.8, C | - | 166.7, C |
| 13 | - | - | 6.43 d (15.9) | 114.4, CH |
| 14 | - | - | 7.75 d (15.9) | 145.8, CH |
| 1′ | - | 121.8, C | - | 126.9, C |
| 2′ | 8.07 brd (8.8) | 132.0, CH | 7.50 brd (8.5) | 130.0, CH |
| 3′ | 6.92 brd (8.8) | 113.9, CH | 6.88 brd (8.5) | 114.4, CH |
| 4′ | - | 159.6, C | - | 159.5, C |
| 5′ | 6.92 brd (8.8) | 113.9, CH | 6.88 brd (8.5) | 114.4, CH |
| 6′ | 8.07 brd (8.8) | 132.0, CH | 7.50 brd (8.5) | 130.0, CH |
| 1-OH | 12.12 s | - | 12.11 s | - |
| 8-OH | 12.09 s | - | 12.09 s | - |
Figure 1.
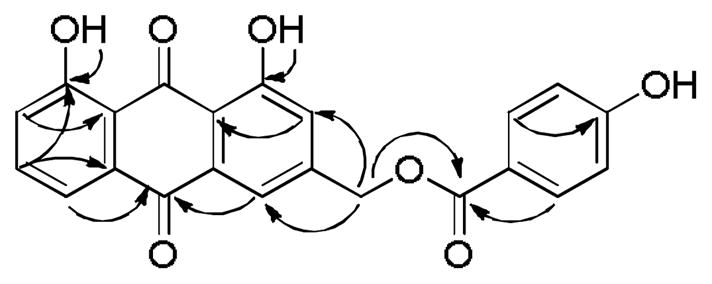
HMBC correlations of compound 3.
Furthermore, the presence of a para-hydroxybenzoate group was suggested by the presence of two doublets at δH 8.07 and 6.92 (2H each, J = 8.8 Hz) in the 1H NMR spectrum, and a set of five carbon signals at δC 165.8 (C-12), 159.6 (C-4′), 132.0 (C-2′/6′), 121.8 (C-1′), and 113.9 (C-3′/5′), in the 13C NMR spectrum.17 The structure of the para-hydroxybenzoate group and its connection to C-11 was further confirmed by the HMBC correlations between H-2′/H-6′ and C-12, H-6′/H-2′ and C-4′, and H2-11 and C-12. Hence, the structure of compound 3 was assigned as (1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-4-hydroxybenzoate.
Kniphofione B (4) was obtained as a light yellow oil. Its molecular formula was deduced to be C24H16O7, based on its protonated molecular ion peak at m/z 417.0986 [M+H]+ and a sodiated molecular ion peak at m/z 439.0806 [M+Na]+ in its positive ion HR-ESI-MS. The 1H-NMR spectrum of compound 4 displayed splitting patterns in the aromatic region similar to that of compound 3, suggesting they have related structures. Comparison of the 13C NMR spectroscopic data of 4 with those of 3 (Table 1) indicated that both compounds shared the same aloe-emodin-type anthraquinone skeleton, but differed in the acyl group attached to C-11. The presence of a trans para-hydroxycinnamate group in 4, as opposed to the para-hydroxybenzoate group in 3, was indicated by the presence of two doublets at δH 7.50 and 6.88 (2H each, J = 8.8 Hz) in the 1H NMR spectrum, a set of five carbon signals at δC 166.7 (C-12), 159.5 (C-4′), 130.0 (C-2′/6′), 126.9 (C-1′), and 114.4 (C-3′/5′) in the 13C NMR spectrum, and trans-coupled doublet proton signals at δH 6.43 and 7.75 (both, 1H, J = 15.9 Hz). The latter signals corresponded by HMQC to the two methine carbon resonances at δC 114.4 (C-13) and 145.8 (C-14).18 The two carbons of the carbon-carbon double bond in the cinnamate group were assigned to C-13 and C-14, based on the HMBC crosspeaks between H-13 and C-1′, H-6′ and C-14, and H-14 and C-12 (Figure 2).
Figure 2.
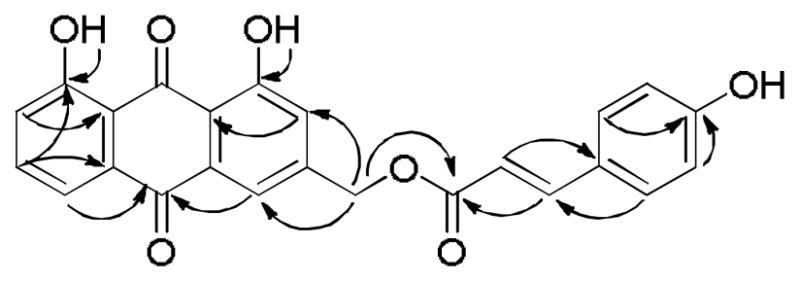
HMBC correlations of compound 4.
The structure of compound 4 was thus determined as (E)-(1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)-methyl-3-(4-hydroxyphenyl) acrylate.
2.2. Biological activities and structure-activity relationships
All the isolated compounds were tested for antiproliferative activity against the A2780 ovarian cancer cell line and for antiplasmodial activity against the Dd2 chloroquine-resistant strain of P. falciparum. As listed in Table 2, the known dimeric compounds (6 and 7) displayed the highest antiplasmodial activity, with IC50 values of 0.4 ± 0.1 and 0.2 ± 0.1 μM, respectively. In addition, both of these dimeric compounds showed modest antiproliferative activity against the A2780 human ovarian cancer cell line, with IC50 values of 6 ± 1 and 4 ± 1 μM, respectively. However, their antiproliferative activity and lack of synthetic accessibility decreased their appeal as lead compounds for potential new antimalarial agents.
Table 2.
Bioactivities of the isolated anthraquinones (1–9).
| Compound | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | Artemisinin | |
| Dd2 IC50 (μM) | 58 | 55 | 26 ± 4 | 9 ± 1 | 1.1± 0.2 | 0.4 ± 0.1 | 0.2 ± 0.1 | 10 ± 2 | 10 ± 2 | 0.007 |
| A2780 IC50 (μM) | > 60 | 26 ± 2 | > 60 | > 60 | 8 ± 2 | 6 ± 1 | 4 ± 1 | >60 | >60 | ND |
For these reasons, the two modestly active new compounds (3 and 4, IC50 = 26 ± 4 and 9 ± 1 μM, respectively) were more attractive for a study of structure-activity relationships due to their low cytotoxicities (IC50 > 60 μM to the A2780 ovarian cancer cell line) and synthetic accessibility. Both compounds are C-11 acylated derivatives of aloe-emodin (2), and so a number of C-11 acylated aloe-emodin derivatives were prepared by standard methods to determine the feasibility of developing derivatives with improved antiplasmodial activity.
Antiplasmodial activity data on the isolated natural products showed that compound 4, the para-hydroxy cinnamate derivative of aloe-emodin (2), is about six times more potent than 2, and this compound was thus selected as the lead compound for the present SAR study. Various acyl derivatives of aloe-emodin were then prepared by reaction of the commercially available natural product aloe-emodin (2) with a variety of organic acids, with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI) as the coupling reagent. The reaction products were purified by column chromatography and their structures were confirmed by 1H and 13C NMR spectroscopy, as well as by HR-ESI-MS. The resulting compounds and their antiplasmodial activities are listed in Table 3. All the compounds were also tested for antiproliferative activity against A2780 cells, but none of them had IC50 values less than 60 μM.
Table 3.
Antiplasmodial activity of natural and synthetic aloe-emodin derivatives
| Cmpd | Structure | IC50 (μM) |
|---|---|---|
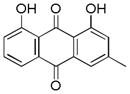
| 1 |
|
~ 58 |
| 2 |
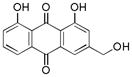
|
~ 55 |
| 3 |
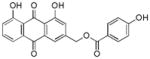
|
26 ± 4 |
| 4 |
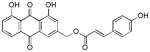
|
9 ± 1 |
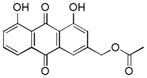
| 10 |
|
~ 42 |
| 11 |
|
~ 60 |
| 12 |
|
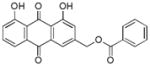
31 ± 2 |
| 13 |
|
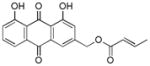
~ 45 |
| 14 |
|
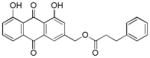
~ 36 |
| 15 |
|
18 ± 2 |
| 16 |
|
22 ± 2 |
| 17 |
|
5 ± 1 |
| 18 |
|
12 ± 2 |
| 19 |
|
30 ± 2 |
| 20 |
|
14 ± 2 |
| 21 |
|
16 ± 2 |
| 22 |
|
1.3 ± 0.2 |
| 23 |
|
2.7 ± 0.4 |
| 24 |
|
2.5 ± 0.9 |
| 25 |
|
6 ± 2 |
| 26 |
|
8 ± 2 |
| 27 |
|
9 ± 2 |
| 28 |
|
1.9 ± 0.3 |
Inspection of the results of this study led to the following conclusions:
-
Acetylation of the primary alcohol of aloe-emodin slightly increased antiplasmodial activity, while acetylation of the hydrogen-bonded hydroxy group had the reverse effect.
The C-11 monoacetate (10, IC50 = ~ 42 μM) displayed slightly better antiplasmodial activity than aloe-emodin (IC50 = ~ 55 μM), while the mixture of the C-1, C-11 and C-8, C-11 diacetates (11, IC50 = ~ 60 μM) was slightly less potent than aloe-emodin. This result suggested that the presence of the hydrogen-bonded hydroxy groups might be important for antiplasmodial activity, and so acylation of the primary alcohol (C-11) of aloe-emodin was selected as the modification of choice for investigation.
-
The phenyl group and the non-aromatic carbon-carbon double bond are both important functional groups for increasing the antiplasmodial activity.
To explore whether acylation of the primary alcohol might increase potency, the C-11 crotonate, benzoate, phenylpropionate and cinnamate derivatives were synthesized. Cinnamate 15 (IC50 = 18.4 ± 2.4 μM) displayed the highest activity among the four; benzoate 12 (IC50 = 31.5 ± 1.5 μM), phenylpropionate 14 (IC50 = ~ 36 μM), and crotonate 13, (IC50 = ~ 45 μM) were all less potent. Although cinnamate 15 displayed the highest activity among all four acylated derivatives, it was only approximately half as potent as the 4-hydroxy-cinnamate 4 (kniphofione B), indicating that the hydroxy group at the C-4′ of the phenyl group is a contributor to the higher activity.
-
The position and number of oxygenations on the phenyl group influences antiplasmodial activity.
The influence of the type of oxygenation at C-4′ of the phenyl ring on antiplasmodial activity was investigated by the synthesis of 4-methoxybenzoate 16 (IC50 = 22 ± 2 μM), 4-methoxycinnamate 17 (IC50 = 5 ± 1 μM), 4-acetoxycinnamate 18 (IC50 = 12 ± 2 μM), and 4-acetoxybenzoate 19 (IC50 = 30 ± 2 μM). 4-Methoxycinnamate derivative 17 was more potent than kniphofione B (4) and 18, with hydroxycinnamate and acetoxycinnamate groups, respectively. Similar results were observed for the corresponding benzoate derivatives. The methoxy group is thus important for activity and was selected as the oxygenated moiety for further study.
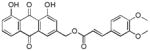
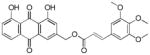
Compounds 22 and 23 were then synthesized to investigate the influence of the number of methoxy groups on the phenyl ring of the cinnamate on potency. The 3,4-dimethoxycinnamate 22 (IC50 = 1.3 ± 0.2 μM) was more potent than both the 4-methoxycinnamate 17 (IC50 = 5 ± 1 μM) and the 3,4,5-trimethoxy-cinnamate 23 (IC50 = 2.7 ± 0.4 μM), suggesting that two methoxy groups on the cinnamate provides the best potency.
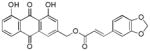
Finally, to understand what effect the positions of the methoxy groups on the cinnamate phenyl ring might have, 2,3-dimethoxy-cinnamate (26), 2,4- dimethoxy-cinnamate (25), 2,5-dimethoxy-cinnamate (27), and 3,5-dimethoxy-cinnamate (24) were synthesized and evaluated. 3,4-Dimethoxy-cinnamate (22) was the most potent of these compounds, and a similar response was observed from 3,4-methylenedioxy-cinnamate 28 (IC50 = 1.9 ± 0.3 μM), suggesting that alkylation on the 3′ and 4′ positions of the phenyl group favorably influences antiplasmodial activity.
In summary, this SAR study showed that esterification of the primary hydroxyl group of aloe-emodin (2) with various carboxylic acids increased its antiplasmodial activity, with the most potent analogue being the 3,4-dimethylcaffeic acid derivative (22), with an IC50 value of 1.3 ± 0.2 μM (Table 3). This analogue displays approximately 7 and 20 times the potency of the two new natural products 3 and 4, and over 40 times than that of aloe-emodin (2).
Although aloe-emodin (2) has previously been identified as an anticancer agent,19 and might thus be thought to have little potential for development as an antiplasmodial agent, our bioassay data indicated that acylation of the primary alcohol of aloe-emodin resulted in the increase of antiplasmodial activity (Dd2 assay), accompanied by a decrease of antiproliferative activity (A2780 assay). As an example, acylation of the C-11 hydroxyl group of aloe-emodin to give the p-coumaroyl derivative 4 increases its antiplasmodial activity sixfold while decreasing its antiproliferative activity to the A2780 cell line at least twofold. This observation indicates that further modifications of the C-11 hydroxyl group of aloe-emodin have the potential to yield antiplasmodial agents with little or no antiproliferative activity.
3. Experimental Section
3.1. General Experimental Procedures
UV and IR spectroscopic data were measured on a Shimadzu UV-1201 spectrophotometer and a MIDAC M-series FTIR spectrophotometer, respectively. NMR spectra were recorded in CDCl3 on Bruker Avance 500 or 600 spectrometers. The chemical shifts are given in δ (ppm), and coupling constants (J) are reported in Hz. Mass spectra were obtained on an Agilent 6220 LC-TOF-MS in the positive and negative ion mode.
3.2. Antiproliferative Bioassays (A2780 assay)
Antiproliferative activities were obtained at Virginia Tech against the drug-sensitive A2780 human ovarian cancer cell line as previously described.20
3.3. Antiplasmodial Bioassays (Dd2 assay)
The effect of each fraction and pure compound on parasite growth of the P. falciparum Dd2 strain was measured in a 72 h growth assay in the presence of drug as described previously with minor modifications.21 Briefly, ring stage parasite cultures (200 μL per well, with 1% hematocrit and 1% parasitemia) were then grown for 72 h in the presence of increasing concentrations of the drug in a 5.05% CO2, 4.93% O2, and 90.2% N2 gas mixture at 37 °C. After 72 h in culture, parasite viability was determined by DNA quantitation using SYBR Green I (50 μL of SYBR Green I in lysis buffer at 0.4 μL of SYBR Green I/mL of lysis buffer). The half-maximum inhibitory concentration (IC50) calculation was performed with GraFit software using nonlinear regression curve fitting. IC50 values are the average of three independent determinations with each determination in duplicate and are expressed ± SEM.
3.4. Plant Material
Plant collection of K. ensifolia Baker (Asphodelaceae) was made in April 1999 on top of Mariepskop in the Pilgrims Rest district, State of Mpumalanga, South Africa, by Prof. P. C. Zietsman under the auspices of the New York Botanical Garden, accession number Z03796a. A voucher specimen is deposited in the New York Botanical Garden and also at BLFU (Bloemfontein Free University, South Africa)
3.5. Extraction and Isolation
The dried and powdered whole plant (100 g) of Kniphofia ensifolia was exhaustively extracted with EtOH (2 × 1 L) in two 24-hour percolation steps; successive partition of the concentrated extract with hexanes and CH2Cl2 gave an active CH2Cl2 fraction. A 0.75 g sample of the original EtOH extract, designated 60031-9D, was shipped to Virginia Tech for bioassay-guided isolation. A 0.60 g portion of this sample (IC50 = ~ 6.0 μg/mL against P. falciparum strain Dd2) was suspended in aqueous MeOH (MeOH-H2O 9:1, 100 mL), and extracted with hexanes (3 × 100 mL). The aqueous layer was diluted to 60% MeOH (v/v) with H2O and extracted with CH2Cl2 (3 × 150 mL). The hexanes fraction was evaporated in vacuo to leave 107.7 mg of material with an IC50 value of 5 ~ 10 μg/mL. The residue from the CH2Cl2 fraction (20.7 mg) was the most active fraction with an IC50 value of 2.5 ~ 5 μg/mL. The remaining aqueous MeOH fraction had an IC50 value of greater than 10 μg/mL. The hexanes and CH2Cl2 fractions were combined due to the similarity of their TLC patterns.
The combined hexanes and CH2Cl2 fractions were divided into five fractions by Sephadex LH-20 size exclusion open column chromatography. The most active fraction (F4, 28.6 mg) had an IC50 value of 2.5 μg/mL. Fraction F4 was then applied to a silica gel column eluted with hexanes: EtOAc = 7:3 to give six fractions. Fraction 4-1 was evaporated to give compound 1 (0.3 mg, IC50 = ~ 58 μM) and fractions 4–5 were evaporated to give compound 5 (2.6 mg, IC50 = 1.1 ± 0.2 μM). The most active fraction 4-2 (1.8 mg, IC50 = 0.27 μg/mL), was subjected to HPLC on a C18 column using a MeCN and H2O gradient as the solvent system to yield the four known active compounds 6 (0.3 mg, IC50 = 0.43 ± 0.14 μM), 7 (0.9 mg, IC50 = 0.20 ± 0.08 μM), 8 (0.3 mg, IC50 = 10.2 ± 1.6 μM) and 9 (0.3 mg, IC50 = 9.6 ± 1.8 μM), with retention times of 21.3, 26.0, 28.5 and 31.2 min, respectively. HPLC of fraction 4-3 (2.2 mg, IC50 = 4.3 μg/mL) on a C18 column gave compounds 2 (0.5 mg, IC50 = ~ 55 μM) 3 (0.7 mg, IC50 = 25.6 ± 3.6 μM) and 4 (1.0 mg, IC50 = 8.9 ± 1.2 μM), with retention times of 8.5, 18.0 and 21.2 min, respectively.
3.6. (1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-4-hydroxybenzoate (3, kniphofione A)
Yellow-orange powder. UV (MeOH) λmax (ε) 257 (1.6), 289 (0.5), 432 (0.5); IR νmax cm−1: 3390, 2915, 2343, 1673, 1621, 1450, 1277, 1110 cm−1; 1H NMR (600 MHz, CDCl3), and 13C NMR (150 MHz, CDCl3), see Table 1; HR-ESI-MS m/z 389.0688 [M-H]− (calcd for C22H13O7, 389.0667).
3.7. (E)-(1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-3-(4-hydroxyphenyl) acrylate (4, kniphofione B)
Yellow-orange powder. UV (MeOH) λmax (ε) 259 (1.2), 289 (1.4), 310 (0.9), 433 (0.6); IR νmax cm−1: 3390, 2916, 2362, 1706, 1627, 1603, 1450, 1268, 1161 cm−1; 1H NMR (600 MHz, CDCl3), and 13C NMR (150 MHz, CDCl3), see Table 1; HR-ESI-MS m/z 439.0806 [M+Na]+ (calcd for C24H16NaO7, 439.0788) and 417.0986 [M+H]+ (calcd for C24H17O7, 417.0969).
3.8. General procedure for synthesis of ester derivatives of aloe-emodin22
Substituted benzoic acids or cinnamic acids (0.1 mmol) in dry CH2Cl2 (0.2 mL) were treated with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI, 20.6 mg, 0.1 mmol) and DMAP (1.0 mg, 0.006 mmol). The mixtures were stirred at room temperature for 5 minutes. Aloe-emodin (2, 5.4 mg, 0.02 mmol) was added, and stirring was continued at room temperature until the starting compound was consumed. The resulting solution was diluted with EtOAc (10 mL) and concentrated on a rotary evaporator. The final benzoate and cinnamate derivatives of aloe-emodin were purified by using preparative TLC (silica gel, 500 m; hexane/EtOAc, 4:1).
3.9. (1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl acetate (10)
Yellow-orange powder; HR-ESI-MS m/z 313.0711 [M+H]+ (calcd for C17H13O6, 313.0707); 1H NMR (500 MHz, CDCl3) δH 12.01 (s, 1H), 11.98 (s, 1H), 7.78 (dd, J = 7.5, 1.1 Hz, 1H), 7.72 (d, J = 1.6 Hz, 1H), 7.63 (dd, J = 8.2, 7.6 Hz, 1H), 7.25 (dd, J = 8.4, 1.1 Hz, 1H), 7.20 (d, J = 1.6 Hz, 1H), 5.12 (s, 2H), 2.12 (s, 3H); 13C NMR (126 MHz, CDCl3) δC 192.7, 181.5, 170.5, 162.8, 162.6, 146.4, 137.3, 133.9, 133.51, 124.8, 122.4, 120.2, 118.5, 115.8, 115.3, 64.7, 20.8.
3.10. Mixture of (1-acetoxy-8-hydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl acetate and (8-acetoxy-1-hydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl acetate (11)
Yellow-orange powder; HR-ESI-MS m/z 355.0815 [M+H]+ (calcd for C19H15O7, 355.0812); 1H NMR (500 MHz, CDCl3) δH 12.57 (s, 1H), 12.55 (s, 1H), 8.28 (dd, J = 7.8, 1.3 Hz, 1H), 8.22 (d, J = 1.0 Hz, 1H), 7.82 (dd, J = 8.0, 7.9 Hz, 1H), 7.81 (dd, J = 7.5, 1.2 Hz, 1H), 7.75(d, J = 1.6 Hz, 1H), 7.66 (dd, J = 8.3, 7.6 Hz, 1H), 7.43 (dd, J = 8.0, 1.3 Hz, 1H), 7.40 (d, J = 1.8 Hz, 1H), 7.26 (d, J =1.0 Hz, 1H), 5.24 (s, 2H), 5.17 (s, 2H), 2.48 (s, 3H), 2.48 (s, 3H), 2.19 (s, 3H), 2.18 (s, 3H).
3.11. (1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl benzoate (12)
Yellow-orange powder; HR-ESI-MS m/z 375.0866 [M+H]+ (calcd for C22H15O6, 375.0863); 1H NMR (500 MHz, CDCl3) δH 12.11 (s, 1H), 12.06 (s, 1H), 8.12 (dd, J = 8.4, 1.3 Hz, 2H), 7.89 (d, J = 1.6 Hz, 1H), 7.85 (dd, J = 7.5, 1.1 Hz, 1H), 7.71 (dd, J = 8.2, 7.6 Hz, 1H), 7.61 (tt, J = 7.5, 1.3 Hz, 1H), 7.49 (t, J = 7.8, 2H), 7.32 (dd, J = 8.4, 1.0 Hz, 1H),, 5.46 (s, 2H); 13C NMR (125 MHz, CDCl3) δC 192.7, 181.5, 166.0, 162.8, 162.6, 146.6, 137.3, 133.9, 133.8, 133.5, 130.1, 129.8, 128.6, 124.8, 122.4, 120.2, 118.5, 115.8, 115.3, 65.1.
3.12. (E)-(1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-but-2-enoate (13)
Yellow-orange powder; HR-ESI-MS m/z 339.0865 [M+H]+ (calcd for C19H15O6, 339.0863); 1H NMR (500 MHz, CDCl3) δH 12.09 (s, 1H), 12.07 (s, 1H), 7.85 (dd, J = 7.6, 1.1 Hz, 1H), 7.80 (d, J = 1.6 Hz, 1H), 7.70 (dd, J = 8.2, 7.8 Hz, 1H), 7.32 (dd, J = 8.4, 1.1 Hz, 1H), 7.29 (d, J = 1.6 Hz, 1H), 7.10 (dd, J = 15.5, 7.0 Hz, 1H), 5.96 (dd, J = 15.6, 1.6 Hz, 1H), 1.94 (dd, J = 6.9, 1.6 Hz, 3H); 13C NMR (125 MHz, CDCl3) δC 192.7, 181.6, 166.4, 162.8, 162.6, 146.8, 146.4, 137.3, 133.8, 133.6, 124.8, 122.3, 121.9, 120.2, 118.4, 115.8, 115.2, 64.4, 18.2.
3.13. (1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-3-phenylpropanoate (14)
Yellow-orange powder; HR-ESI-MS m/z 403.1146 [M+H]+ (calcd for C24H19O6, 403.1176); 1H NMR (500 MHz, CDCl3) δH 12.07 (s, 1H), 12.06 (s, 1H), 7.85 (dd, J = 7.6, 1.1 Hz, 1H), 7.76 (d, J = 1.6 Hz, 1H), 7.70 (dd, J = 8.3, 7.8 Hz, 1H), 7.32 (dd, J = 8.4, 1.1 Hz, 1H), 7.29 (d, J = 1.6 Hz, 1H), 7.28 (m, 2H), 7.21 (m, 3H), 5.18 (s, 2H), 3.01 (d, J = 7.8 Hz, 2H), 2.77 (d, J = 7.8 Hz, 2H); 13C NMR (125 MHz, CDCl3) δC 192.7, 181.5, 172.4, 162.8, 162.6, 146.4, 140.1, 137.3, 133.9, 133.5, 128.6, 128.3, 126.4, 124.8, 122.5, 120.2, 118.6, 115.8, 115.3, 64.7, 35.7, 30.9.
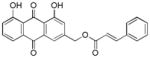
3.14. (1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl cinnamate (15)
Yellow-orange powder; HR-ESI-MS m/z 401.1031 [M+H]+ (calcd for C24H17O6, 401.1020); 1H NMR (500 MHz, CDCl3) δH 12.03 (s, 1H), 12.00 (s, 1H), 7.79 (dd, J = 7.5, 1.1 Hz, 1H), 7.79 (d, J = 1.6 Hz, 1H), 7.73 (d, J = 16.0 Hz, 1H), 7.64 (dd, J = 8.3, 7.6 Hz, 1H), 7.50 (m, 2H), 7.35 (m, 3H), 7.27 (d, J = 1.6 Hz, 1H), 7.25 (dd, J = 8.4, 1.1 Hz, 1H), 6.48 (d, J = 16.0 Hz, 1H), 5.27 (s, 2H); 13C NMR (125 MHz, CDCl3) δC 192.7, 181.6, 166.4, 162.8, 162.6, 146.7, 146.2, 137.3, 134.1, 133.9, 133.6, 130.6, 129.0, 128.3, 124.8, 122.4, 120.2, 118.6, 117.0, 115.4, 115.3, 64.7.
3.15. (1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-4-methoxybenzoate (16)
Yellow-orange powder; HR-ESI-MS m/z 405.0971 [M+H]+ (calcd for C23H17O7, 405.0969); 1H NMR (500 MHz, CDCl3) δH 12.10 (s, 1H), 12.07 (s, 1H), 8.08 (d, J = 8.9 Hz, 2H), 7.88 (d, J = 1.5 Hz, 1H), 7.85 (dd, J = 7.5, 1.0 Hz, 1H), 7.70 (dd, J = 8.1, 7.8 Hz, 1H), 7.37 (d, J = 1.5 Hz, 1H), 7.32 (dd, J = 8.4, 1.0 Hz, 1H), 6.96 (d, J = 8.9 Hz, 2H), 5.42 (s, 2H), 3.88 (s, 3H); 13C NMR (125 MHz, CDCl3) δC 192.7, 181.6, 165.7, 164. 9, 162.8, 162.6, 146.9, 137.3, 134.1, 133.6, 131.9, 124.8, 122.3, 121.7, 120.2, 118.5, 115.8, 115.3, 113.8, 64.8, 55.5.
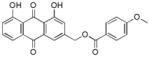
3.16. (E)-(1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-3-(4-methoxyphenyl) acrylate (17)
Yellow-orange powder; HR-ESI-MS m/z 431.1133 [M+H]+ (calcd for C25H19O7, 431.1125); 1H NMR (500 MHz, CDCl3) δH 12.02 (s, 1H), 12.00 (s, 1H), 7.79 (dd, J = 7.4, 1.1 Hz, 1H), 7.78 (d, J = 1.6 Hz, 1H), 7.68 (d, J = 16.0 Hz, 1H), 7.63 (dd, J = 8.3, 7.7 Hz, 1H), 7.45 (d, J = 8.8 Hz, 2H), 7.27 (d, J = 1.6 Hz, 1H), 7.25 (dd, J = 8.4, 1.1 Hz, 1H), 6.86 (d, J = 8.8 Hz, 2H), 6.34 (d, J = 16.0 Hz, 1H), 5.26 (s, 2H), 3.78 (s, 3H); 13C NMR (125 MHz, CDCl3) δC 192.7, 181.6, 166.7, 162.8, 161.7, 159.8, 146.9, 145.8, 137.3, 133.9, 133.6, 130.0, 126.9, 124.8, 122.4, 120.2, 118.5, 118.2, 115.8, 115.3, 114.4, 64.6, 55.5.
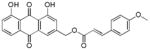
3.17. (E)-(1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-3-(4-acetoxy phenyl)acrylate (18)
Yellow-orange powder; HR-ESI-MS m/z 459.1079 [M+H]+ (calcd for C26H19O8, 459.1074); 1H NMR (600 MHz, CDCl3) δH 12.09 (s, 1H), 12.06 (s, 1H), 7.86 (dd, J = 7.4, 1.1 Hz, 1H), 7.85 (d, J = 1.6 Hz, 1H), 7.76 (d, J = 16.0 Hz, 1H), 7.70 (dd, J = 8.3, 7.7 Hz, 1H), 7.58 (d, J = 8.7 Hz, 2H), 7.35 (d, J = 1.6 Hz, 1H), 7.32 (dd, J = 8.4, 1.1 Hz, 1H), 7.15 (d, J = 8.6 Hz, 2H), 6.50 (d, J = 16.0 Hz, 1H), 5.33 (s, 2H), 2.32 (s, 3H); 13C NMR (150 MHz, CDCl3) δC 192.8, 181.6, 169.2, 166.3, 162.9, 162.7, 152.5, 146.7, 145.1, 137.4, 134.0, 133.7, 132.0, 129.5, 124.9, 122.6, 122.3, 120.3, 118.7, 117.3, 115.9, 115.4, 64.9, 21.2.
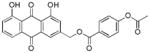
3.18. (1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl 4-acetoxybenzoate (19)
Yellow-orange powder; HR-ESI-MS m/z 433.0935 [M+H]+ (calcd for C24H17O8, 433.0918); 1H NMR (600 MHz, CDCl3) δH 12.09 (s, 1H), 12.05 (s, 1H), 8.15 (d, J = 8.9 Hz, 2H), 7.87 (d, J = 1.6 Hz, 1H), 7.85 (dd, J = 7.5, 1.0 Hz, 1H), 7.70 (dd, J = 8.1, 7.8 Hz, 1H), 7.36 (d, J = 1.6 Hz, 1H), 7.32 (dd, J = 8.4, 1.0 Hz, 1H), 7.22 (d, J = 8.9 Hz, 2H), 5.44 (s, 2H), 2.33 (s, 3H); 13C NMR (150 MHz, CDCl3) δC 192.7, 181.6, 167.7, 164.3, 162.8, 162.6, 158.4, 143.5, 137.4, 134.1, 133.5, 131.5, 127.0, 124.9, 122.5, 121.9, 120.3, 118.6, 115.7, 115.3, 65.3, 21.3.
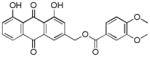
3.19. (1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl 3,4-dimethoxybenzoate (20)
Yellow-orange powder; HR-ESI-MS m/z 435.1084 [M+H]+ (calcd for C24H19O8, 435.1074); 1H NMR (500 MHz, CDCl3) δH 12.10 (s, 1H), 12.06 (s, 1H), 7.89 (d, J = 1.6 Hz, 1H), 7.85 (dd, J = 7.5, 1.1 Hz, 1H), 7.78 (dd, J = 8.4, 2.0 Hz, 1H), 7.77 (dd, J = 8.2, 7.8 Hz, 1H), 7.60 (d, J = 2.0 Hz, 1H), 7.36 (d, J = 1.6 Hz, 1H), 7.32 (dd, J = 8.4, 1.1 Hz, 1H), 6.93 (d, J = 8.5 Hz, 1H), 5.43 (s, 2H), 3.96 (s, 6H) 13C NMR (125 MHz, CDCl3) δC 192.7, 181.6, 165.8, 162.8, 162.6, 153.4, 148.8, 146.9, 137.3, 133.91, 133.5, 124.8, 124.0, 122.4, 121.8, 120.2, 118.5, 115.8, 115.3, 112.1, 110.4, 65.0, 56.1.
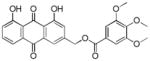
3.20. (1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-3,4,5-trimethoxybenzo ate (21)
Yellow-orange powder; HR-ESI-MS m/z 465.1187 [M+H]+ (calcd for C25H21O9, 465.1180); 1H NMR (500 MHz, CDCl3) δH 12.09 (s, 1H), 12.05 (s, 1H), 7.89 (d, J = 1.6 Hz, 1H), 7.85 (dd, J = 7.5, 1.1 Hz, 1H), 7.70 (dd, J = 8.2, 7.7 Hz, 1H), 7.35 (s, 2H), 7.33 (d, J = 1.6 Hz, 1H), 7.32 (dd, J = 8.4, 1.1 Hz, 1H), 5.44 (s, 2H), 3.93 (s, 9H); 13C NMR (125 MHz, CDCl3) δC 192.7, 181.5, 165.7, 162.8, 162.6, 159.8, 153.1, 146.6, 137.4, 134.0, 133.5, 124.8, 124.3, 122.5, 120.2, 118.5, 115.8, 115.4, 107.1, 65.2, 61.0, 56.3.
3.21. (E)-(1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-3-(3,4-dimethoxy phenyl)acrylate (22)
Yellow-orange powder; HR-ESI-MS m/z 461.1245 [M+H]+ (calcd for C26H21O8, 461.1231); 1H NMR (500 MHz, CDCl3) δH 12.02 (s, 1H), 11.99 (s, 1H), 7.78 (d, J = 1.6 Hz, 1H), 7.78 (dd, J = 7.6, 1.1 Hz, 1H), 7.66 (d, J = 15.9 Hz, 1H), 7.63 (dd, J = 8.3, 7.6 Hz, 1H), 7.27 (d, J = 1.6 Hz, 1H), 7.25 (dd, J = 8.4, 1.1 Hz, 1H), 7.07 (dd, J = 8.3, 1.9 Hz, 1H), 7.02 (d, J = 1.9 Hz, 1H), 6.82 (d, J = 8.3 Hz, 1H), 6.35 (d, J = 15.9 Hz, 1H), 5.26 (s, 2H), 3.87 (s, 3H), 3.86 (s, 3H). 13C NMR (125 MHz, CDCl3) δC 192.7, 181.7, 166.7, 162.8, 162.7, 151.4, 149.3, 146.9, 146.1, 137.3, 133.9, 133.6, 127.1, 124.8, 123.0, 122.4, 120.2, 118.5, 115.8, 115.3, 114.6, 111.0, 109.6, 64.6, 56.0, 55.9.
3.22. (E)-(1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-3-(3,4,5-trimethoxy phenyl)acrylate (23)
Yellow-orange powder; HR-ESI-MS m/z 491.1352 [M+H]+ (calcd for C27H23O9, 491.1337); 1H NMR (500 MHz, CDCl3) δH 12.04 (s, 1H), 11.99 (s, 1H), 7.80 (d, J = 1.6 Hz, 1H), 7.79 (dd, J = 7.6, 1.1 Hz, 1H), 7.64 (dd, J = 8.3, 7.6 Hz, 1H), 7.63 (d, J = 15.9 Hz, 1H), 7.28 (d, J = 1.6 Hz, 1H), 7.26 (dd, J = 8.4, 1.1 Hz, 1H), 6.73 (s, 2H), 6.39 (d, J = 15.9 Hz, 1H), 5.27 (s, 2H), 3.84 (s, 6H), 3.83 (s, 3H); 13C NMR (125 MHz, CDCl3) δC 192.7, 181.6, 166.3, 162.8, 162.6, 153.5, 146.7, 146.1, 140.4, 137.3, 133.9, 133.5, 129.6, 124.8, 122.4, 120.2, 118.5, 116.2, 115.8, 115.3, 105.4, 64.7, 61.0, 56.2.
3.23. (E)-(1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-3-(3,5-dimethoxy phenyl)acrylate (24)
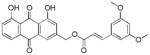
Yellow-orange powder; HR-ESI-MS m/z 461.1224 [M+H]+ (calcd for C26H21O8, 461.1231); 1H NMR (600 MHz, CDCl3) δH 12.09 (s, 1H), 12.06 (s, 1H), 7.86 (dd, J = 7.6, 1.1 Hz, 1H), 7.85 (d, J = 1.6 Hz, 1H), 7.70 (dd, J = 8.3, 7.6 Hz, 1H), 7.70 (d, J = 15.9 Hz, 1H), 7.33 (d, J = 1.6 Hz, 1H), 7.32(dd, J = 8.4, 1.1 Hz, 1H), 6.70 (d, J = 2.2 Hz, 2H), 6.52 (t, J = 2.3 Hz, 1H), 6.51(d, J = 15.9 Hz, 1H), 5.33 (s, 2H), 3.83 (s, 6H); 13C NMR (150 MHz, CDCl3) δC 192.8, 181.7, 166.4, 162.7, 161.2, 146.8, 146.3, 137.5, 136.1, 134.1, 133.7, 124.9, 122.6, 120.3, 118.7, 117.7, 116.0, 115.5, 106.2, 103.2, 77.2, 64.9, 55.5.
3.24. (E)-(1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-3-(2,4-dimethoxy phenyl)acrylate (25)
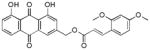
Yellow-orange powder; HR-ESI-MS m/z 461.1212 [M+H]+ (calcd for C26H21O8, 461.1231); 1H NMR (600 MHz, CDCl3) δH 12.08 (s, 1H), 12.07 (s, 1H), 8.01 (d, J = 16.1 Hz, 1H), 7.85 (dd, J = 7.7, 1.1 Hz, 1H), 7.85 (d, J = 1.6 Hz, 1H), 7.70 (dd, J = 8.2, 7.7 Hz, 1H), 7.47 (d, J = 8.6 Hz, 1H), 7.34 (d, J = 2.3 Hz, 1H), 7.32 (dd, J = 8.4, 1.1 Hz, 1H), 6.54 (d, J = 16.1 Hz, 1H), 6.53, 6.53, 6.52 (dd, J = 8.6, 2.3 Hz, 1H), 6.47 (d, J = 2.3 Hz, 1H), 5.32 (s, 2H), 3.89 (s, 3H), 3.85 (s, 3H); 13C NMR (150 MHz, CDCl3) δC 192.7, 181.6, 167.3, 163.0, 162.6, 160.1, 147.2, 146.8, 141.6, 137.2, 133.8, 133.6, 130.9, 124.7, 122.4, 120.1, 118.6, 118.3, 116.4, 115.9, 114.8, 105.3, 98.5, 64.4, 55.5.
3.25. (E)-(1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-3-(2,3-dimethoxy phenyl)acrylate (26)
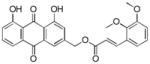
Yellow-orange powder; HR-ESI-MS m/z 461.1242 [M+H]+ (calcd for C26H21O8, 461.1231); 1H NMR (600MHz, CDCl3) δH 12.08 (s, 1H), 12.06 (s, 1H), 8.12 (d, J = 16.2 Hz, 1H), 7.86 (d, J = 1.6 Hz, 1H), 7.85 (dd, J = 7.4, 1.1 Hz, 1H), 7.70 (dd, J = 8.3, 7.6 Hz, 1H), 7.34 (d, J = 1.6 Hz, 1H), 7.32 (dd, J = 8.4, 1.1 Hz, 1H), 7.20 (dd, J = 7.9, 1.2 Hz, 1H), 7.08 (t, J = 8.0 Hz, 1H), 6.97 (dd, J = 8.1, 1.3 Hz, 1H), 6.59 (d, J = 16.2 Hz, 1H), 5.34 (s, 2H), 3.89 (s, 6H); 13C NMR (150 MHz, CDCl3) δC 192.7, 181.5, 166.6, 162.8, 162.6, 153.2, 148.7, 146.8, 141.0, 137.3, 133.9, 133.6, 128.3, 124.7, 124.2, 122.4, 120.2, 119.4, 118.5, 118.3, 115.8, 115.3, 114.3, 64.7, 61.4.
3.26. (E)-(1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methyl-3-(2,5-dimethoxy phenyl)acrylate (27)
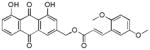
Yellow-orange powder; HR-ESI-MS m/z 461.1211 [M+H]+ (calcd for C26H21O8, 461.1231); 1H NMR (600 MHz, CDCl3) δ 12.07 (s, 1H), 12.06 (s, 1H), 8.07 (d, J = 16.1 Hz, 1H), 7.86 (d, J = 1.6 Hz, 1H), 7.85 (dd, J = 7.4, 1.1 Hz, 1H), 7.70 (dd, J = 8.3, 7.6 Hz, 1H), 7.34 (d, J = 1.6 Hz, 1H), 7.32 (dd, J = 8.4, 1.1 Hz, 1H), 7.07 (d, J = 3.0 Hz, 1H), 6.93 (dd, J = 9.0, 3.0 Hz, 1H), 6.87 (d, J = 9.0 Hz, 1H), 6.62 (d, J = 16.1 Hz, 1H), 5.33 (s, 2H), 3.86 (s, 3H), 3.81 (s, 3H); 13C NMR (150 MHz, CDCl3) δC 192.8, 181.7, 166.9, 163.0, 162.7, 153.7, 153.2, 147.1, 141.6, 137.4, 134.1, 133.7, 124.9, 123.8, 122.6, 120.3, 118.7, 117.8, 116.0, 115.4, 113.5, 112.7, 77.2, 64.8, 56.2.
3.27. (E)-(1,8-dihydroxy-9,10-dioxo-9,10-dihydroanthracen-3-yl)methy-3-(3,4-methylene dioxyphenyl)acrylate (28)
Yellow-orange powder; HR-ESI-MS m/z 445.0912 [M+H]+ (calcd for C25H17O8, 445.0918); H NMR (500 MHz, CDCl3) H NMR (500 MHz, CDCl3) δH 12.09 (s, 1H), 12.07 (s, 1H), 7.86 (d, J = 1.6 Hz, 1H), 7.85 (dd, J = 7.4, 1.1 Hz, 1H), 7.70 (dd, J = 8.3, 7.6 Hz, 1H), 7.69 (d, J = 15.9 Hz, 1H), 7.33 (d, J = 1.6 Hz, 1H), 7.32 (dd, J = 8.4, 1.1 Hz, 1H), 7.07 (d, J = 1.7 Hz, 1H), 7.04 (dd, J = 8.0, 1.7 Hz, 1H), 6.83 (d, J = 8.0 Hz, 1H), 6.37 (d, J = 15.9 Hz, 1H), 6.02 (s, 2H), 5.32 (s, 2H); 13C NMR (125 MHz, CDCl3) δC 192.8, 181.7, 166.7, 163.0, 162.8, 150.1, 148.6, 147.0, 141.6, 137.5, 134.0, 133.7, 128.7, 125.0, 124.9, 122.6, 120.3, 118.7, 116.0, 115.4, 115.1, 108.8, 106.8, 101.8, 64.8.
Acknowledgments
This project was supported by the National Center for Complementary and Alternative Medicine under award 3U01TW000313-19S1 as a supplement to Cooperative Agreement U01 TW000313-19 from the Fogarty International Center, the National Cancer Institute, the National Science Foundation, the National Heart, Lung and Blood Institute, the National Institute of Mental Health, the Office of Dietary Supplements, and the Office of the Director of NIH, with the International Cooperative Biodiversity Groups. This work was also supported by The Jeffress Memorial Trust of Virginia (J-1058). These supports are gratefully acknowledged. This work was also supported by the National Science Foundation under Grant no. CHE-0619382 for purchase of the Bruker Avance 500 NMR spectrometer and Grant no. CHE-0722638 for the purchase of the Agilent 6220 mass spectrometer. We thank Mr. Bill Bebout for obtaining the mass spectra. We gratefully acknowledge Dr. Dennis Stevenson and Daniel Atha of the New York Botanical Garden and Dr. P. C. Zietsman of the National Museum (Bloemfontein, South Africa) for the provision of plant material. J.D.B. is a recipient of a scholarship from the National Science Foundation S-STEM project (DUE-0850198)
Footnotes
Notes
The authors declare that there are no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.WHO. World Malaria Report 2012. World Health Organization; Geneva, Switzerland: 2012. p. 59. [Google Scholar]
- 2.Boudreau EF, Pang LW, Chaikummao S, Witayarut C. Southeast Asian J Trop Med Public Health. 1991;22:183. [PubMed] [Google Scholar]
- 3.Bhattarai A, Ali AS, Kachur SP, Mårtensson A, Abbas AK, Khatib R, Al-Mafazy AW, Ramsan M, Rotllant G, Gerstenmaier JF, Molteni F, Abdulla S, Montgomery SM, Kaneko A, Björkman A. PLoS Med. 2007;4:e309. doi: 10.1371/journal.pmed.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dondorp AM, Fairhurst RM, Slutsker L, MacArthur JR, Breman JG, Guerin PJ, Wellems TE, Ringwald P, Newman RD, Plowe CV. N Engl J Med. 2011;365:1073. doi: 10.1056/NEJMp1108322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jarvis L. Chem Eng News. 2012;90(8):30. [Google Scholar]
- 6.Dai Y, Harinantenaina L, Brodie PJ, Goetz M, Shen Y, TenDyke K, Kingston DGI. J Nat Prod. 2013 doi: 10.1021/np300900a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark VR, Barker NP, Mucina L. S Afr J Bot. 2009;75:196. [Google Scholar]
- 8.Chase MW, De Bruijn AY, Cox AV, Reeves G, Rudall PJ, Johnson MAT, Eguiarte LE. Ann Bot (Oxford, U K) 2000;86:935. [Google Scholar]
- 9.a) Habtemariam S. Fitoterapia. 2010;81:1013. doi: 10.1016/j.fitote.2010.06.021. [DOI] [PubMed] [Google Scholar]; b) Wube AA, Bucar F, Asres K, Gibbons S, Rattray L, Croft SL. Phytother Res. 2005;19:472. doi: 10.1002/ptr.1635. [DOI] [PubMed] [Google Scholar]; c) Blitzke T, Porzel A, Masaoud M, Schmidt J. Phytochemistry. 2000;55:979. doi: 10.1016/s0031-9422(00)00269-7. [DOI] [PubMed] [Google Scholar]
- 10.García-Sosa K, Villarreal-Alvarez N, Lübben P, Peña-Rodríguez LM. J Mex Chem Soc. 2006;50:76. [Google Scholar]
- 11.Danielsen K, Aksnes DW, Francis GW. Magn Reson Chem. 1992;30:359. [Google Scholar]
- 12.Dagne E, Steglich W. Phytochemistry. 1984;23:1729. doi: 10.1016/0031-9422(95)00704-0. [DOI] [PubMed] [Google Scholar]
- 13.Dagne E, Berhanu E, Steglich W. Bull Chem Soc Ethiop. 1987:32. [Google Scholar]
- 14.Van Wyk BE, Yenesew A, Dagne E. Biochem Syst Ecol. 1995;23:805. [Google Scholar]
- 15.Fujitake N, Suzuki T, Fukumoto M, Oji Y. J Nat Prod. 1998;61:189. doi: 10.1021/np9703050. [DOI] [PubMed] [Google Scholar]
- 16.Conner JM, Gray AI, Reynolds T, Waterman PG. Phytochemistry. 1989;28:3551. [Google Scholar]
- 17.Chang CL, Zhang LJ, Chen RY, Kuo LMY, Huang JP, Huang HC, Lee KH, Wu YC, Kuo YH. J Nat Prod. 2010;73:1482. doi: 10.1021/np100181c. [DOI] [PubMed] [Google Scholar]
- 18.Hosny M, Rosazza JPN. J Nat Prod. 1999;62:853. doi: 10.1021/np980566p. [DOI] [PubMed] [Google Scholar]
- 19.a) Pecere T, Gazzola MV, Mucignat C, Parolin C, Vecchia FD, Cavaggioni A, Basso G, Diaspro A, Salvato B, Carli M, Palù G. Cancer Res. 2000;60:2800. [PubMed] [Google Scholar]; b) Srinivas G, Babykutty S, Sathiadevan PP, Srinivas P. Med Res Rev. 2007;27:591. doi: 10.1002/med.20095. [DOI] [PubMed] [Google Scholar]
- 20.Pan E, Harinantenaina L, Brodie PJ, Callmander M, Rakotonandrasana S, Rakotobe E, Rasamison VE, TenDyke K, Shen Y, Suh EM, Kingston DGI. Bioorg Med Chem. 2011;19:4224. doi: 10.1016/j.bmc.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Bennett TN, Paguio M, Gligorijevic B, Seudieu C, Kosar AD, Davidson E, Roepe PD. Antimicrob Agents Chemother. 2004;48:1807. doi: 10.1128/AAC.48.5.1807-1810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Antimicrob Agents Chemother. 2004;48:1803. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Harinantenaina L, Bowman JD, Brodie PJ, Slebodnick C, Callmander MW, Rakotobe E, Randrianaivo R, Rasamison VE, Gorka A, Roepe PD, Cassera MB, Kingston DGI. J Nat Prod. 2013;76:388. doi: 10.1021/np300750q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rivero-Cruz B, Rivero-Cruz I, Rodríguez-Sotres R, Mata R. Phytochemistry. 2007;68:1147. doi: 10.1016/j.phytochem.2007.02.026. [DOI] [PubMed] [Google Scholar]