

Abstract
This study was performed to determine the hepatotoxicity of di(2-ethylhexyl)phthalate (DEHP) in relation to selenium status. In 3-week-old Sprague-Dawley rats, selenium deficiency was induced by a ≤0.05 selenium mg/kg. A selenium supplementation group was given 1 mg selenium/kg diet for 5 weeks. Di(2-ethylhexyl)phthalate-treated groups received 1000 mg/kg dose by gavage during the last 10 days of the experiment. Histopathology, peroxisome proliferation, catalase (CAT) immunoreactivity and activity and apoptosis were assessed. Activities of antioxidant selenoenzymes [glutathione peroxidase 1 (GPx1), glutathione peroxidase 4 (GPx4), thioredoxin reductase (TrxR1)], superoxide dismutase (SOD), and glutathione S-transferase (GST); aminotransferase, total glutathione (tGSH), and lipid peroxidation (LP) levels were measured. Di(2-ethylhexyl)phthalate caused cellular disorganization while necrosis and inflammatory cell infiltration were observed in Se-deficient DEHP group (DEHP/SeD). Catalase activity and immunoreactivity were increased in all DEHP-treated groups. Glutathione peroxidase 1 and GPx4 activities decreased significantly in DEHP and DEHP/SeD groups, while GST activities decreased in all DEHP-exposed groups. Thioredoxin reductase activity increased in DEHP and DEHP/SeS, while total SOD activities increased in all DEHP-treated groups. Lipid peroxidation levels increased significantly in SeD (26%), DEHP (38%) and DEHP/SeD (71%) groups. Selenium supplementation partially ameliorated DEHP-induced hepatotoxicity; while in DEHP/SeD group, drastic changes in hepatic histopathology and oxidative stress parameters were observed.
Keywords: antioxidant enzymes, di(ethylhexyl)phthalate, liver, oxidative stress, selenium deficiency, selenium supplementation
Reactive oxygen species (ROS) are free radicals, excessive productions of which lead to oxidative stress. The unbalance between oxidants and antioxidants within the cell in the favour of oxidants may subsequently cause deleterious effects including loss of cell function or cell death (Nose 2000). Many environmental chemicals are able to generate ROS which may contribute to the pathophysiology of many disease conditions, including inflammatory diseases, neurodegenerative disorders and cancer. A shift in the pro-oxidant–antioxidant balance has been proposed as a factor involved in carcinogenesis (Oberley et al. 2000). Phthalate esters are considered as endocrine disruptors with peroxisome proliferator (PP) activity. They have increasingly attracted substantial attention because of their high production volume and widespread use, particularly in a variety of polyvinyl chloride (PVC)-based consumer products, food containers and medical devices (Akingbemi et al. 2004). Phthalates easily migrate from plastic matrix into the product content as well as into the air, water and soil causing human exposures by various means (Clark et al. 1991). Diet, particularly fatty food is the main source of phthalate exposure for the general public (Clark et al. 1991; Wormuth et al. 2006). Other sources such as personal care products also make an important contribution to overall exposure levels (Fromme et al. 2007). Moreover, low-molecular-weight phthalate esters, including di(2-ethylhexyl)phthalate (DEHP), are used to make coatings for oral medications, and they are considered to be high exposure sources (Hernández-Díaz et al. 2009).
Biological effects of DEHP, the most abundantly produced and widely used phthalate ester (Latini 2005), are of major concern. Di(2-ethylhexyl)phthalate is known as a reproductive toxicant in rodents. Like other PPs, phthalates cause peroxisome proliferation, cell proliferation and induction of lipid metabolizing enzymes in the liver and hepatocytes cultured in vitro (Moody & Reddy 1978; Rusyn et al. 2006). Di(2-ethylhexyl)phthalate was also shown to be hepatocarcinogenic in both sexes in rodents causing hepatocellular carcinomas and adenomas, and the incidence of liver tumours is known to be dependent both on dose of DEHP and duration of exposure (Kluwe et al. 1985). Although PP-activated receptor α (PPARα) activation and its sequelae have been suggested to constitute the predominant mode of action for hepatocarcinogenesis by phthalates, it does not seem to be a sole causative factor. As with other PPs, it is assumed that ROS might be associated with tumorigenic mechanism of phthalates. This assumption is based on the fact that various cellular proteins that are induced by PP chemicals in hepatocyte organelles (peroxisomes, mitochondria and microsomes) are prone to formation of H2O2 and other oxidants (Gazouli et al. 2002; O'Brien et al. 2005). The oxidative stress elicited by phthalates is proposed to be associated with impaired mitochondrial function, and there is evidence that phthalates increase ROS in the rodent liver before peroxisomal oxidases are induced (Rusyn et al. 2001). In addition, Kupffer cells have been suggested to be a potential source of oxidants in rat and mouse liver after treatment with DEHP or other PPs (Rose et al. 2000; Rusyn et al. 2001). It appears that oxidant-related molecular events could interact with other pathways activated by PPs in vivo in rodent liver, and therefore, understanding their precise mechanism of action is critically important (Rusyn et al. 2006). However, it is still a moot point whether ROS production induction is a one of the major pathways or whether elimination of ROS is not efficiently achieved in the series of events induced by phthalates.
Concern over human exposure comes from the toxicological effects of phthalates in rodents (Klaunig et al. 2003), although the mechanisms underlying these phthalate-induced effects and the health risk for humans remain controversial (Rusyn et al. 2001). Some studies suggested that PPARα-dependent mechanisms leading to hepatocarcinogenesis are low relevance to humans (Melnick 2001; Willhite 2001). However, such discrepancy necessitates more research to elucidate the difference in the mechanism of action of phthalate in rodent and human livers.
Selenium (Se) is one of the key essential trace elements against oxidative stress (Zwolak & Zaporowska 2012). It is the important component of cellular antioxidant defence and is involved in the modulation of intracellular redox equilibrium with its some 25 forms of cellular selenoproteins, particularly with glutathione peroxidases (GPx), and thioredoxin reductases (TrxR) (Oberley et al. 2000; Steinbrenner & Sies 2009). Selenium is actively involved in many fundamental biological processes ranging from immune functions to apoptosis and protection and repair of DNA (Ganther 1999; Jablonska et al. 2009). The requirement for Se and its beneficial role in human health have been known for several decades. An imbalance between high oxidative stress and decreased antioxidative defence has been suggested to cause a variety of liver diseases (Zhu et al. 2012). Therefore, antioxidants, including Se, can be beneficial to minimize the detrimental effects of oxidative stress-producing toxicant in liver.
Based on this background and taking into account the oxidative potential of phthalate esters in rodents, this study was designed to investigate the hepatic toxicity of subacute DEHP exposure in rat liver. Hepatic histopathology and electron microscopy (EM) were performed, and catalase (CAT) immunohistochemistry along with CAT activity and hepatic oxidant/antioxidant parameters was evaluated. By conducting the experiments in normal, Se-deficient and Se-supplemented rats, the effects of Se status on the hepatic effects of DEHP were also investigated.
Methods
Chemicals and reagents
All chemicals, including DEHP (purity 99%), were purchased from Sigma-Aldrich (St. Louis, MO, USA). Bouin's fixative solution, glutaraldehyde solution, Masson's trichrome (M-S), Mayer's haematoxylin solution, phosphate-buffered saline (PBS) and Periodic acid-Schiff (PAS) kit, colorimetric assay kits for TrxR1 and total glutathione (tGSH) were also from Sigma-Aldrich. Sep-Pak C18 cartridge was obtained from Waters (Milford, MA, USA). Entellan was obtained from Merck (Darmstadt, Germany). Biotinylated secondary antibody (Histostain Plus Broad Spectrum) and streptavidin–peroxidase complex (Histostain Plus Broad Spectrum) were from Invitrogen (Carlsbad, CA, USA). 3,3′-Diaminobenzidine (DAB) was purchased from Zymed (San Francisco, CA, USA). Cell Death Detection Kit (AP), TUNEL dilution buffer and fast red tablets were obtained from Roche Applied Science (Roche, Mannheim, Germany). Anti-rat monoclonal catalase (CAT) was from Abcam (San Francisco, CA, USA). Araldite/Epon812 was from EMS (Hatfield, PA, USA). All animal feed (A03/R03 base) were supplied by Scientific Animal Food and Engineering (SAFE) Laboratories (Augy, France).
Animals and treatment
Male Sprague-Dawley (SD) rats, 3 weeks old, supplied from Hacettepe University Experimental Animals Laboratory, were used in the experiments. Animals were divided randomly in six groups of six of each, and each group was housed in plastic cages with stainless steel grid tops. The cages were placed in a room with controlled temperature (23 °C), humidity (50%) and a 12-h light–dark cycle. Body weights (bw) were monitored weekly, including before the first dose of DEHP treatment. Feeding period was 5 weeks, and animals were allowed to access ad libitum feed and drinking water.
Ethical approval
The animals were treated humanely and with regard for alleviation of suffering, and the study was approved by Hacettepe University Ethical Committee.
Experimental groups
(i) Control group (C) was fed regular diet (0.15 mg/kg Se), (ii) Se-supplemented group (SeS) was fed Se-supplemented diet (1 mg/kg Se), (iii) Se-deficient group (SeD) was fed Se-deficient diet (≤0.05 mg/kg Se), (iv) DEHP-treated group (DEHP) was fed regular diet (0.15 mg/kg Se) and received 1000 mg/kg DEHP during the last 10 days by intragastric gavage (i.g.), (v) Se-supplemented DEHP group (DEHP/SeS) was fed Se-supplemented diet (1 mg/kg Se) and received 1000 mg/kg DEHP during the last 10 days by i.g., (vi) Se-deficient DEHP group (DEHP/SeD) was fed Se-deficient diet (≤0.05 mg/kg Se) and received 1000 mg/kg DEHP during the last 10 days by i.g.
Di(2-ethylhexyl)phthalate was dissolved in corn oil, and the animals in C, SeS and SeD groups received equivalent amount of the vehicle by i.g. during the last 10 days. Twenty-four hours after the last dose of DEHP treatment or vehicle administration, animals were weighed and sacrificed by decapitation under thiopental anaesthesia. Venous blood samples were obtained, and liver tissues were removed. Liver tissues to be used for oxidant/antioxidant parameters were frozen immediately in liquid nitrogen, divided into pieces and stored at −80 °C until the preparation of tissue homogenates. Liver tissues for histopathological evaluation, EM and CAT immunohistochemistry were processed as indicated below.
Histopathological evaluation and electron microscopy
One lobe of liver was divided into three pieces. First piece was used for histopathological evaluation, second for EM, while third piece was used for CAT immunohistochemistry. Briefly, first piece of the fresh tissue sample was rapidly fixed in Bouin's fixative solution, then dehydrated through graded alcohols and embedded in paraffin blocks. Sections (5 μm) were cut and stained with haematoxylin and eosin (H&E), M-S and PAS according to the standard protocols. The second piece of the fresh tissue sample was fixed in 2.5% glutaraldehyde solution in phosphate buffer, pH 7.4 for 4 h and postfixed for 1 h in 1% osmium tetroxide solution in 0.1M phosphate buffer. After washing in phosphate buffer, sample was dehydrated in a graded series of alcohols, treated with propylene oxide and embedded in Araldite/Epon812. After heat polymerization, sections were cut using a microtome. Semi-thin sections were stained with methylene blue–azure II and examined using a light microscope (Leica DM6000B, Wetzlar, Germany) with a Leica DC490 digital camera. Ultrathin sections (Leica ultracut R) were double-stained with uranyl acetate and lead citrate (Leica EM AC20). These sections were examined in JEOL-JEM 1400 EM (Tokyo, Japan) and photographed by CCD camera (Gatan Inc., Pleasanton, CA, USA). The number of peroxisomes was counted in eight random representative ultrastructural photomicrographs per sample in each group, and the average for each group was calculated.
Catalase immunohistochemistry
The third piece of liver tissue was immediately frozen in the liquid nitrogen. Six-to eight-μm-thick cryostat sections were cut and fixed in acetone for 10 min, then air-dried for at least 30 min. Endogenous peroxidase was blocked by incubation in 10% H2O2 in PBS for 10 min at 4 °C. Unspecific binding was blocked using rat serum at a dilution of 1:10 for 30 min at room temperature. Then, sections were incubated for 60 min with anti-rabbit CAT (1:50 dilution) primary antibody. After washing three times for 5 min each with PBS, sections were incubated with biotinylated secondary antibody for 30 min, followed by streptavidin–peroxidase complex for 10 min at room temperature. After washing in PBS, peroxidase activity was revealed by incubation for 8 min with DAB and counterstained with Mayer's haematoxylin. Following washing with tap water, sections were dehydrated through graded alcohols and cleared in xylene prior to mounting with Entellan. Negative controls were performed by omitting incubation with the primary antibody. All slides were examined and photographed using a light microscope (Leica DM6000B) with a Leica DC490 digital camera. The intensity of CAT immunoreactivity was semi-quantitatively evaluated according to the following intensity categories: 0, no staining; 1+, weak but detectable staining; 2+, moderate or distinct staining; 3+, intense staining. For each tissue, a histological score (H-SCORE) value was determined using the formula H-SCORE = ΣPi (i + l), where i represents the intensity scores, and Pi is the corresponding percentage of cells. Five randomly selected areas were evaluated under a light microscope (40× magnification) in each slide, and the percentage of the cells for each intensity within these areas was determined by two investigators blinded to the type and source of the tissues. The mean value of two investigators' score was used for graphical and statistical calculations.
Determination of biochemical parameters
Venous blood samples were collected into heparinized tubes, and plasma was separated after centrifugation at 800 g for 15 min. Plasma alanine transaminase (ALT) and aspartate transaminase (AST) activities were measured by an automatic analyser (Perkin Elmer, Waltham, MA, USA).
Preparation of liver homogenates
Liver homogenates were prepared in a volume of ice-cold buffer containing Tris (10 mM), diethylenetriaminepentaacetic acid (1 mM) and phenylmethanesulphonyl fluoride (1 mM; adjusted to pH 7.4) using a Teflon pestle homogenizer to obtain 10% (w/v) whole homogenate. After centrifugation at 1500 g, 4 °C, for 10 min, thiobarbituric acid reactive substance (TBARS) concentration was measured in the supernatant. The rest of the supernatants were recentrifugated at 9500 g, 4 °C for 20 min, and the antioxidant enzyme activities and thiol groups were determined in the supernatant. For the measurement of tGSH levels, the whole homogenate was diluted (5:1) with metaphosphoric acid (6%), centrifuged at 1500 g, 4 °C, for 10 min, and the supernatant was used. All spectrophotometric measurements were performed using a spectrophotometer (Uvikon 860, Kontron Instruments, France).
Determination of antioxidant enzyme activities
The activity of GPx1 was measured in a coupled reaction with glutathione reductase (GR) as described earlier (Flohé & Günzler 1984) using an automatic analyser (Hitachi 904; B Braun Science Tec, France). The assay of GPx4 activity was based on the same reaction as GPx1 determination, but freshly synthesized phosphatidylcholine hydroperoxide (PCOOH) was used as a substrate and the concomitant oxidation of NADPH was monitored spectrophotometrically at 340 nm as described by Maiorino et al. (1990). Phosphatidylcholine hydroperoxide was synthesized using the method of Maiorino et al. (1990) and Weitzel et al. (1990) with some modification. Thioredoxin reductase activity was determined colorimetrically using the Thioredoxin Reductase Assay kit as described previously (Arnér et al. 1999). Catalase activity was determined according to Aebi (1974). The total superoxide dismutase (SOD) and Mn-SOD activities were determined by monitoring the auto-oxidation of pyrogallol at 420 nm according to Marklund and Marklund (1974) with some modifications. One unit of total SOD activity was defined as the amount of enzyme required to inhibit the rate of pyrogallol auto-oxidation by 50%. Potassium cyanide (3 mM) was used to inhibit both Cu,Zn-SOD and extracellular SOD, resulting in detection of only MnSOD activity. Cu,Zn-SOD activity was then determined by subtracting Mn-SOD activity from total SOD activity. Glutathione S-transferase activity was determined according to Habig et al. (1974).
Determination of total glutathione levels
Total glutathione contents of the samples were assessed by Glutathione Assay Kit using a kinetic assay in which catalytic amounts of GSH caused a continuous reduction of 5,5′-dithiobis-(2-nitrobenzoic) acid (DTNB) to TNB at 412 nm (Akerboom & Sies 1981). Quantification was achieved by parallel measurements of a standard curve of known tGSH concentrations, and results were expressed in nmol/mg protein.
Determination of lipid peroxidation
Lipid peroxidation (LP) in liver tissues was quantified measuring the concentration of TBARS by a spectrofluorometric assay as described by Richard et al. (1992) using a Hitachi F4500 spectrofluorometer (B Braun Science Tec), and the level of TBARS was expressed as μmol/g tissue.
Protein determination
Protein concentrations were determined by the standard method of Lowry et al. (1951) using an automatic analyser (Hitachi 904; B Braun Science Tec).
TUNEL assay
Apoptosis in liver tissues was detected by enzymatic labelling of DNA strand breaks using terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labelling (TUNEL) assay with Cell Death Detection kit according to the manufacturers' instructions. Briefly, paraffin sections (5 μm) from liver tissues were deparaffinized. Thereafter, rehydratated slides were washed twice in PBS for 5 min and incubated with the permeabilization solution (0.1% Triton X-100 in 0.1% sodium citrate) for 8 min at 4 °C. After washing twice with PBS for 5 min, the labelling reaction was performed using 50 μl TUNEL reagent for each sample, except negative control, in which reagent without enzyme was added and incubated for 1 h at 37 °C. After washing with PBS, slides were incubated with converter reagent for 30 min at 37 °C. Following the washing step, colour development for localization of cells containing labelled DNA strand breaks was performed by incubating the slides with Fast Red substrate solution for 10 min. A positive control for detection of DNA fragmentation was included in each experiment, by adding deoxyribonuclease I solution grade I (1500 U/ml in 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 1 mg/ml bovine serum albumin) and incubating for 30 min at 37 °C, previously to the TdT reaction. A negative control was also performed in which no terminal deoxynucleotidyl transferase (TdT) enzyme was present in the reaction mixture, and sections were incubated in label solution only for 1 h at 37 °C. All slides were examined and photographed using Leica DM6000B microscope with a Leica DC490 digital camera. Red-labelled TUNEL-positive apoptotic cells were counted in fifteen random fields in each slide from liver tissues at a microscopic magnification of ×400. The data were expressed as the mean number of TUNEL-positive cells/field.
Statistics
The results were expressed as mean ± standard error of mean (SEM). The differences among the groups were evaluated with Kruskal–Wallis one-way analysis of variance, followed by Mann–Whitney U test using a Statistical Package for Social Sciences Program (spss) version 17.0. P-values <0.05 were considered as statistically significant.
Results
Absolute and relative liver weights
All animals appeared to remain healthy throughout the experiments. Significant differences in the food intake were not observed between the groups. There was no significant alteration in body weights in any of the groups before the DEHP treatment started. After 10 days of DEHP exposure, the weight gain in all DEHP-exposed groups was found significantly lower (P < 0.05) than the control group and Se supplementation was found to be partially protective, as shown elsewhere (Erkekoglu et al. 2011a). At the termination of the experiments, absolute liver weight significantly increased only in DEHP/SeD group (52%, P > 0.05), whereas relative liver weights were increased in all DEHP-exposed groups compared with control (35%, P > 0.05) (Table 1).
Table 1.
Effects of di(2-ethylhexyl)phthalate (DEHP) and selenium status on mean and relative liver weights in Study Groups
| Mean liver weight (g) | Relative liver weight (g/100 g bw) | |
|---|---|---|
| Control | 8.74 ± 0.25a | 4.71 ± 0.38a |
| SeS | 10.06 ± 0.35a | 5.00 ± 0.07a |
| SeD | 9.57 ± 0.20a | 4.82 ± 0.14a |
| DEHP | 10.82 ± 0.09a | 6.37 ± 0.09b |
| DEHP/SeS | 11.05 ± 0.51a | 6.39 ± 0.0 9b |
| DEHP/SeD | 13.27 ± 0.42b | 6.35 ± 0.08b |
All results were given as mean ± SEM of n = 6 animals.
Columns that do not share same letters (superscripts) are significantly different from each other (P < 0.05).
Histopathological evaluation and electron microscopy
Light microscopic examination of liver tissues revealed normal morphology with hepatic cells arranged in cords and sinusoids in control, SeS and SeD groups (Figure 1a–c). Increased eosinophilia in the cytoplasm of the hepatocytes was observed in DEHP group (Figure 1d). In DEHP-exposed Se-supplemented animals, normal liver architecture was maintained (Figure 1e), whereas in DEHP-exposed Se-deficient group, a marked focal necrosis and inflammatory cell infiltration was noted (Figure 1f).
Figure 1.
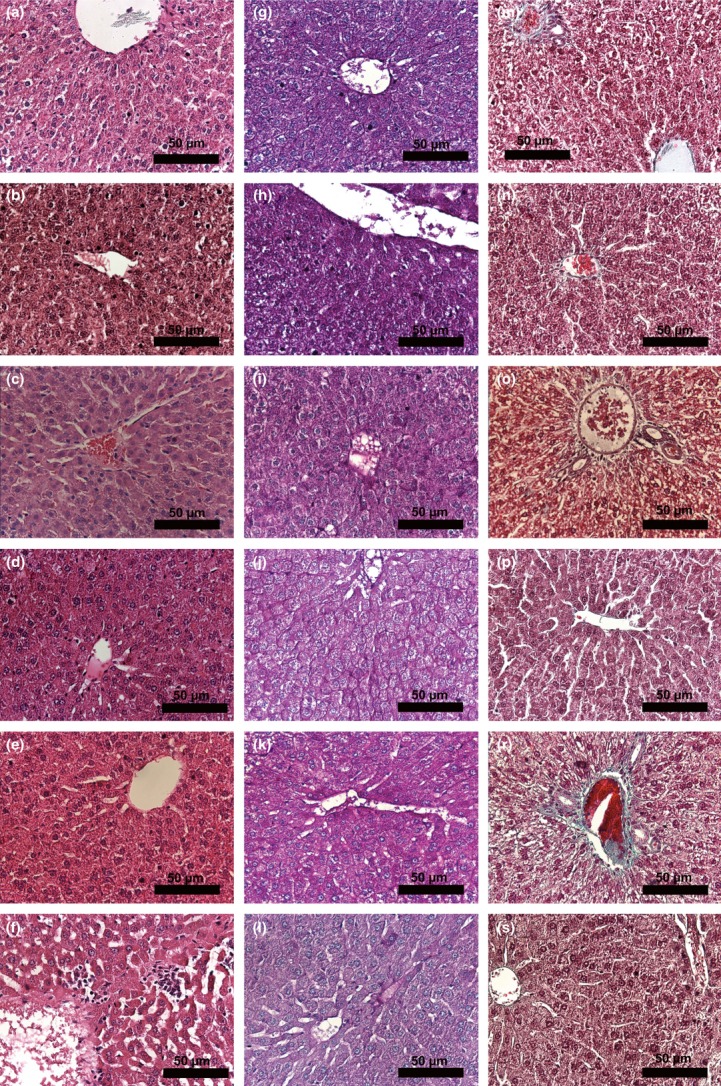
Histopathology of the study groups. Histopathology of liver tissues in study groups was determined by light microscopy using haematoxylin and eosin (a–f), Periodic acid-Schiff (PAS) (g–l) and Masson's trichrome (M-S) stains (×400). a, g, m: Control group; b, h, n: SeS group; c,i, o: SeD group d, j, p: di(2-ethylhexyl)phthalate (DEHP) group; e, k, r: DEHP/SeS group; f, l, s: DEHP/SeD group. Micrographs show cytoplasmic eosinophilia in DEHP (d); focal necrosis and inflammatory cell infiltration in DEHP/SeD (f); decrease in glycogen content in DEHP (i), SeD (j) and DEHP/SeD (l) groups.
The glycogen storage of the hepatic cells as examined in PAS-stained sections of liver is shown in Figure 1g–l. Glycogen granules were found to increase in SeS group (Figure 1h) but decreased slightly in SeD group (Figure 1i). While granules were evident in DEHP/SeS group (Figure 1k), marked decreases were observed in DEHP (Figure 1j) and DEHP/SeD (Figure 1l) groups.
The collagen fibres in the connective tissue examined by Masson's trichrome staining showed that there was no fibrosis in any of the groups (Figure 1m–s).
Electron microscopy examination of hepatocytes in control and SeS groups showed normal morphology (Figure 2a,b). SeS group was similar to control group regarding the structure of the nucleus, arrangement of endoplasmic reticulum (ER) and the other organelles, but a slight increase in the number of peroxisomes was observed (Figure 2b), whereas in SeD group, the number of peroxisomes were found to be decreased (Figure 2c). Dilatation and destruction of ER, loss of Golgi apparatus arrangement, glycogen depletion, size enlargement in mitochondria and fragmentation in their crista were observed in DEHP group (Figure 2d). The cytoplasm was filled with damaged mitochondria in addition to the increased number of peroxisomes. In DEHP/SeS group, some of the mitochondria showed usual morphology with prominent cristae, but mitochondrial damage was also determined and the number of peroxisomes was found to increase (Figure 2e). In DEHP/SeD group, the mitochondria were pleomorphic and showed homogenous texture without prominent cristae formation. The membranous organelles such as ER and golgi were also damaged, and the number of peroxisomes increased (Figure 2f). Lipid droplets were observed in DEHP and DEHP/SeD groups (data not shown).
Figure 2.
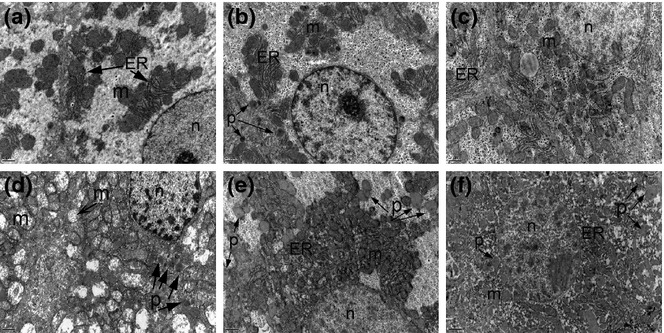
Electron micrographs of liver tissues in the study groups. Uranyl acetate–lead citrate, a, b, c, d, e, f ×12000. a: Control; b: SeS; c: SeD; d: di(2-ethylhexyl)phthalate (DEHP); e: DEHP/SeS; f: DEHP/SeD groups. Nucleus (n), endoplasmic reticulum (ER), mitochondria (m), peroxisome (p) were marked. Normal ultrastructural features of hepatocytes with the normal arrangement of ER and mitochondria are observed in C and SeS groups (a and b). The glycogen granules are prominent in the cytoplasm of SeS group (c). The cytoplasm is filled with damaged mitochondria in DEHP group (d). The crista of mitochondria cannot be observed in DEPH/SeD group (e).
The peroxisome counts in the study groups are summarized in Figure 3. The average number of peroxisomes per micrograph increased significantly (P < 0.05) in DEHP (100%), DEHP/SeS (100%) and DEHP/SeD (50%) groups compared with the control value, while a significant (50%) decrease was observed in SeD group.
Figure 3.
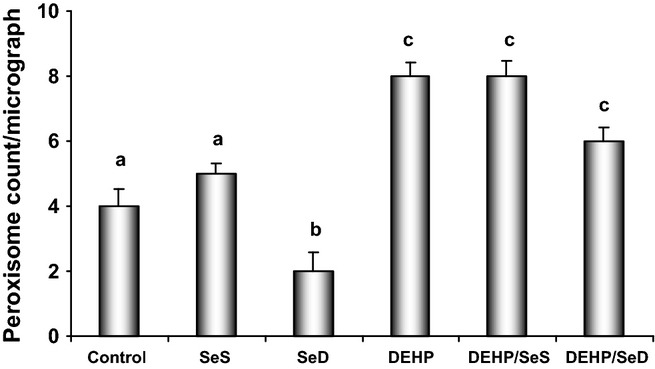
The Peroxisome count in hepatocytes in the study groups. Bars represent the average number of peroxisomes counted in eight random representative ultrastructural photomicrographs in each group. All results were given as mean ± SEM of n = 6 animals. Bars that do not share same letters (superscripts) are significantly different from each other (P < 0.05). Experimental groups for 5 week were on the following: (c) regular diet (0.15 mg/kg Se); (SeS) Se-supplemented diet (1 mg/kg Se); (SeD) Se-deficient diet (≤0.05 mg/kg Se); di(2-ethylhexyl)phthalate (DEHP) regular diet (0.15 mg/kg Se) and received 1000 mg/kg, i.g. DEHP for the last 10 days; (DEHP/SeS) Se-supplemented diet (1 mg/kg Se) and received 1000 mg/kg, i.g. DEHP for the last 10 days; (DEHP/SeD) Se-deficient diet (≤0.05 mg/kg Se) and received 1000 mg/kg, i.g. DEHP for the last 10 days. Significantly higher number of peroxisomes was detected in DEHP, DEHP/SeS and DEHP/SeD groups compared with control group.
Catalase immunocytochemistry
As shown in Figure 4, CAT immunoreactivity in the cytoplasm of hepatocytes was determined as dark brown dots. The dots tend to unite and showed widespread distribution in the cytoplasm of hepatocytes in all groups. The immunoreactivity was moderate in control and SeS groups (Figure 4a,b), weak in SeD group (Figure 4c) whereas in DEHP-exposed groups, strong immunoreactivity of CAT was determined (Figure 4d–f). This was in consistence with the increase in CAT activity observed in the same groups (Table 4). The H-SCORE for CAT immunoreactivity determined as described above was not different than the control group in SeS group, whereas found to decrease significantly (P < 0.05) in the SeD (41%) and increase in DEHP (29%), DEHP/SeS (30%) and DEHP/SeD (17%) groups (Figure 5).
Figure 4.

The catalase immunoreactivity of hepatocytes in the study groups. Original magnifications a–f: ×400. a: Control; b: SeS; c: SeD; d: di(2-ethylhexyl)phthalate (DEHP); e: DEHP/SeS; f: DEHP/SeD groups. Catalase (CAT) immunoreactivity in the cytoplasm of hepatocytes was determined as dark brown dots. Strong CAT immunoreactivity is seen in DEHP, DEHP/SeS and DEHP/SeD groups.
Table 4.
Effects of di(2-ethylhexyl)phthalate (DEHP) and selenium status on hepatic antioxidant enzyme activities and thiobarbituric acid reactive substance (TBARS) levels
| Total SOD (U/mg protein) | Mn-SOD (U/mg protein) | Cu,Zn-SOD (U/mg protein) | CAT (U/mg protein) | TBARS (μmol/g protein) | |
|---|---|---|---|---|---|
| Control | 28.50 ± 1.12a | 2.52 ± 0.11a | 25.98 ± 1.05a | 883.16 ± 77.97a | 0.34 ± 0.02a |
| SeS | 28.09 ± 1.95a | 2.47 ± 0.30a | 25.62 ± 1.67a | 1146.30 ± 184.01ac | 0.33 ± 0.02a |
| SeD | 39.43 ± 2.17b | 6.46 ± 0.63b | 32.97 ± 0.98b | 678.79 ± 69.12b | 0.43 ± 0.03b |
| DEHP | 45.68 ± 2.89bc | 5.32 ± 0.49b | 40.36 ± 2.69c | 1522.82 ± 36.54 cd | 0.47 ± 0.06b |
| DEHP/SeS | 45.30 ± 3.82bc | 3.51 ± 0.23c | 41.79 ± 3.79c | 1661.46 ± 74.68d | 0.33 ± 0.02a |
| DEHP/SeD | 51.27 ± 2.88c | 4.41 ± 0.84bc | 46.86 ± 2.13c | 999.88 ± 26.52a | 0.58 ± 0.04c |
Thiobarbituric acid reactive substances (TBARS) were measured as an indicator of lipid peroxidation.
All results were given as mean ± SEM of n = 6 animals.
Columns that do not share the same letters are significantly different from each other.
Figure 5.
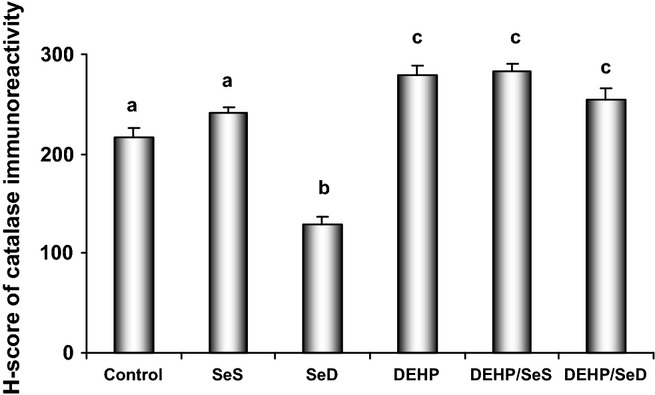
The histological score (H-SCORE) analysis of catalase in the hepatocytes in the study groups. For each tissue, a H-SCORE value was determined using the formula H-SCORE = ΣPi (i + l), where i represents the intensity scores and Pi is the corresponding percentage of cells. All results were given as mean ± SEM of n = 6 animals. Bars that do not share same letters (superscripts) are significantly different from each other (P < 0.05). Experimental groups for 5 week were on the following: (c) regular diet (0.15 mg/kg Se); (SeS) Se-supplemented diet (1 mg/kg Se); (SeD) Se-deficient diet (≤0.05 mg/kg Se); di(2-ethylhexyl)phthalate (DEHP) regular diet (0.15 mg/kg Se) and received 1000 mg/kg, i.g. DEHP for the last 10 days; (DEHP/SeS) Se-supplemented diet (1 mg/kg Se) and received 1000 mg/kg, i.g. DEHP for the last 10 days; (DEHP/SeD) Se-deficient diet (≤0.05 mg/kg Se) and received 1000 mg/kg, i.g. DEHP for the last 10 days.
Biochemical parameters
The levels of plasma ALT and AST are given in Table 2. While there was no difference in SeS and SeD groups than that of control, ALT levels markedly increased in all DEHP-exposed groups (102% in DEHP, 97% in DEHP/SeS and 92% in DEHP/SeD, P < 0.05) compared with control. Aspartate transaminase levels were also markedly higher in DEHP and DEHP/SeD groups (56% and 64% respectively) vs. control group. Se supplementation in DEHP-treated group provided a marked decrease (25%, P < 0.05) in AST levels compared with DEHP group.
Table 2.
Effects of di(2-ethylhexyl)phthalate (DEHP) and selenium status on plasma biochemical parameters in study groups
| ALT (U/l) | AST (U/l) | |
|---|---|---|
| Control | 25.00 ± 2.39a | 123.00 ± 7.38a |
| SeS | 21.14 ± 3.39a | 129.86 ± 13.61a |
| SeD | 29.83 ± 3.65a | 135.67 ± 0.88a |
| DEHP | 50.50 ± 1.71b | 191.83 ± 9.24b |
| DEHP/SeS | 49.17 ± 8.20b | 143.17 ± 23.87a |
| DEHP/SeD | 48.00 ± 2.85b | 201.17 ± 10.08b |
ALT, alanine transaminase; AST, aspartate transaminase.
All results were given as mean ± SEM of n = 6 animals.
Columns that do not share same letters (superscripts) are significantly different from each other (P < 0.05).
Antioxidant enzymes and glutathione
Activity of hepatic antioxidant selenoenzymes is shown in Table 3. Glutathione peroxidase 1 activity did not change in SeS and DEHP/SeS groups but, significantly decreased in Se-deficient animals with or without DEHP treatment (93%, P < 0.05). A significant decrease was also observed in DEHP group (17%, P < 0.05).
Table 3.
Effects of di(2-ethylhexyl)phthalate (DEHP) and selenium status on hepatic selenoenzyme and glutathione S-transferase (GST) activities and total glutathione (tGSH) levels
| GPx1 (U/g protein) | GPx4 (mU/mg protein) | TrxR1 (U/mg protein) | GST (μmol/mg protein/min) | tGSH (nmol/mg protein) | |
|---|---|---|---|---|---|
| Control | 944 ± 0.29a | 5.73 ± 0.13a | 0.16 ± 0.02a | 0.31 ± 0.02a | 13.40 ± 0.52a |
| SeS | 955 ± 29a | 6.11 ± 0.30a | 0.21 ± 0.02b | 0.30 ± 0.02a | 14.56 ± 0.86a |
| SeD | 67 ± 3b | 5.33 ± 0.15a | 0.07 ± 0.01c | 0.30 ± 0.02a | 11.74 ± 0.73b |
| DEHP | 787 ± 19c | 4.46 ± 0.12b | 0.28 ± 0.01bd | 0.21 ± 0.01b | 11.46 ± 0.58b |
| DEHP/SeS | 859 ± 19a | 5.63 ± 0.25a | 0.32 ± 0.02d | 0.23 ± 0.02b | 12.20 ± 0.56ab |
| DEHP/SeD | 64 ± 4b | 4.25 ± 0.32b | 0.11 ± 0.02c | 0.12 ± 0.01c | 10.96 ± 0.58b |
All results were given as mean ± SEM of n = 6 animals.
Columns that do not share the same letters (superscripts) are significantly different from each other (P < 0.05).
Glutathione peroxidase 4 activity did not change neither by Se supplementation nor by Se deficiency, but decreased in DEHP and DEHP/SeD groups (13%, and 15% respectively; P < 0.05), whereas the same activity as control was maintained in DEHP/SeS group (Table 3).
Thioredoxin reductase activity was found to increase significantly in SeS, DEHP and DEHP/SeS groups (31%, 75% and 100% respectively; P < 0.05) compared with control group. In SeD and DEHP/SeD groups, there were marked decreases (56%, 32% respectively; P < 0.05) compared with control (Table 3).
Glutathione S-transferase activities showed marked decreases in all DEHP-exposed groups (DEHP, 32%; DEHP/SeS, 26%; and DEHP/SeD, 61%; P < 0.05) vs. control and the decrease in DEHP/SeD group was significantly different than DEHP group (43%, P < 0.05) (Table 3).
Compared with control, tGSH levels were not different in SeS and DEHP/SeS groups, but significantly decreased in SeD, DEHP and DEHP/SeD groups (12%, 15% and 18% respectively; P < 0.05) (Table 3).
The data of the other antioxidant enzyme activities are presented in Table 4. Total SOD activity did not change in SeS group, but increased in SeD, DEHP, DEHP/SeS and DEHP/SeD groups (38%, 60%, 59% and 80% respectively; P < 0.05).
Catalase activity did not change significantly in SeS groups, whereas in SeD group, a significant decrease (23%) was observed. On the other hand, in DEHP and DEHP/SeS groups, CAT activity increased markedly compared with. control (72% and 88% respectively; P < 0.05) (Table 4).
Lipid peroxidation
Thiobarbituric acid reactive substance levels, measured as an indicator of LP, were not different from control in SeS and DEHP/SeS groups (Table 4). However, DEHP treatment, and Se deficiency with and without DEHP treatment increased TBARS levels significantly (SeD, 26%; DEHP, 38%; DEHP/SeD, 71%; P < 0.05). The marked increase in DEHP/SeD group showed the augmenting effect of Se deficiency on the oxidant stress-inducing effect of DEHP, whereas the unchanged TBARS level in DEHP/SeS group compared with control indicated a protective effect of Se supplementation.
Apoptosis
As shown in the micrographs of Figure 6, increase in apoptotic cells was observed only in SeD group vs. control, while a few apoptic cells were detected in control and SeS groups. When the data were quantitated and expressed as the mean number of TUNEL-positive cells/field, significant increase was determined only in SeD animals (44%, P < 0.05) while there was no difference in apoptic cell counts any of the groups compared with control (Figure 7).
Figure 6.
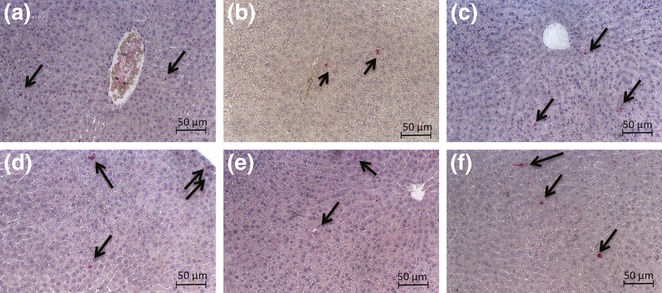
Micrographs for the apoptosis of hepatocytes using terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labelling (TUNEL) assay. Original magnifications a, b, c, d, e, f: ×200. a: Control; b: SeS; c: SeD; d: di(2-ethylhexyl)phthalate (DEHP); e: DEHP/SeS; f: DEHP/SeD groups. Apoptosis of hepatocytes was detected by TUNEL assay using Cell Death Detection kit according to the manufacturers' instructions. Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labelling-positive (apoptotic) cells are indicated by arrow.
Figure 7.
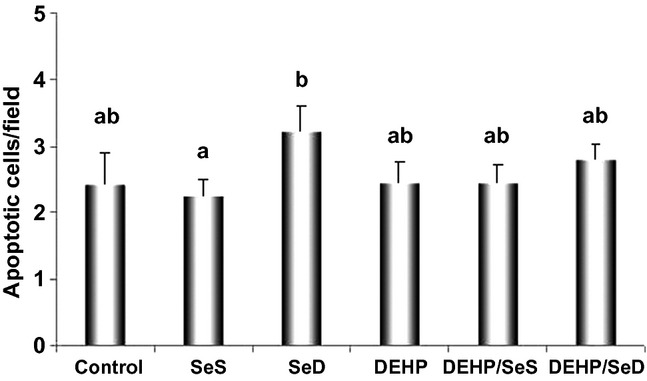
Quantitative analysis of terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labelling (TUNEL)-positive cells in rat livers. Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labelling-positive apoptotic cells were counted in 15 random fields, and the data were expressed as the mean number of TUNEL-positive cells/field. All results were given as mean ± SEM of n = 6 animals. Bars that do not share same letters (superscripts) are significantly different from each other (P < 0.05). Experimental groups for 5 week were on the following: (c) regular diet (0.15 mg/kg Se); (SeS) Se-supplemented diet (1 mg/kg Se); (SeD) Se-deficient diet (≤0.05 mg/kg Se); (DEHP) regular diet (0.15 mg/kg Se) and received 1000 mg/kg, i.g. DEHP for the last 10 days; (DEHP/SeS) Se-supplemented diet (1 mg/kg Se) and received 1000 mg/kg, i.g. DEHP for the last 10 days; (DEHP/SeD) Se-deficient diet (≤0.05 mg/kg Se) and received 1000 mg/kg, i.g. DEHP for the last 10 days.
Discussion
Several studies suggest an association of ROS with the mechanism of tumorigenesis by PPARα agonists, including phthalates (Reddy & Rao 1989; Erkekoglu et al. 2010a,b). Reactive oxygen species play a signalling role in the rapid increase in parenchymal cell proliferation caused by PPs, and Kupffer cells are also suggested to be a potential source of oxidants in rodent liver after treatment with DEHP or other PPs (Rose et al. 2000; Rusyn et al. 2001). Thus, in rodent liver, oxidant-related molecular events seem to be implicated in with other several pathways activated by PPARα agonists in vivo (Rusyn et al. 2000). The main goal of the present study was to investigate the hepatic toxicity of subacute DEHP exposure with special emphasis on its oxidant stress potential. To examine the possible protective effect of Se, the study was conducted with Se-adequate, Se-deficient and Se-supplemented rats.
Previous reports show that PPs elicit a classic pleiotropic response in the liver of rodents that includes increase in liver size (Moody et al. 1991; Reddy & Rao 1992), rapid increase in hepatocyte proliferation (Rusyn et al. 2000), marked increase in the number and size of peroxisomes, and enhanced activity of peroxisomal enzymes (Conway et al. 1989; Klaunig et al. 2003). The overall data of the DEHP group in the present study were in accordance with those data. Briefly, body weight gain was significantly less than control along with significant increase in relative liver weight; PPARα activation in the hepatocytes was evident with significant increase in the number of peroxisomes (100%) and increased immunohistochemistry and activity of the peroxisomal marker enzyme, CAT; ultrastructural examination showed marked histological alterations including cytoplasmic eosinophilia, presence of lipid droplets and degeneration of mitochondria (Rusyn et al. 2001). Increase in plasma ALT and AST levels also indicated DEHP-induced hepatocellular injury. Moreover, glycogen depletion in hepatocytes was noted, and this was consistent with earlier observations indicating the metabolic effects of DEHP-inducing glucose intolerance and alterations in hepatic glycogen content in rats (Mushtaq et al. 1980). Gayathri et al. (2004) reported that in utero DEHP exposure alters postnatal liver development, delaying the programming of glycogen metabolism in rats.
Studies have shown that agonists of PPARα, including DEHP, suppressed hepatic apoptosis (James et al. 1998; Roberts et al. 2000). However, conflicting results have also been reported. A study showed low concentrations of MEHP-decreased apoptosis, while higher concentrations elevated the frequency of apoptotic hepatocytes (Hasmall et al. 2000). More importantly, an in vivo study showed that the prototype PPARα agonist Wy-14643 caused a 40–60% increase in the number of apoptotic hepatocytes in mice (Isenberg et al. 1997). Reasons for these discrepancies are not clear. However, experimental models, as well as age of animals, may be the contributing factors (Youssef et al. 2003). In fact, a study that quantified the age-dependent effect of PPARα agonists on liver apoptosis revealed that only livers of middle age (50 week–old) and senescent (100 week–old) rats were sensitive, while no effect was observed in young (4 and 10 week–old) animals (Youssef et al. 2003). In the present study, we did not observe apoptosis suppression by DEHP treatment, suggesting either our animals were young as in the latter study or the dose and duration of DEHP treatment did not allow to interfere the hepatic apoptosis processes.
Indicating a decrease in the H2O2 detoxification ability, we observed significant decreases in the hepatic activities of GPx1, GPx4 and GST, and in the content of tGSH in DEHP-treated animals. Peroxisome proliferators were shown to increase hepatic peroxisomal fatty acid β-oxidation and the H2O2-generating enzyme acyl-CoA oxidase markedly in rats (Yeldandi et al. 2000). The subject is contentious that whether the depletion of GPx1 activity by DEHP exposure is the primary cause of intracellular H2O2 elevation or whether the increase in H2O2 levels cannot be compensated by the decreased GPx1 and even by the increased CAT activity as seen in this study. After all, excess H2O2 can potentially escape the peroxisomes and react with cellular macromolecules. On the other hand, decrease in hepatic GST activity was shown earlier with different PPs (O'Brein et al. 2001; Seo et al. 2004) and in Leydig cell cultures with DEHP and MEHP treatments (Erkekoglu et al. 2010b). It is clear that DEHP interferes with several components of GSH system and alters the oxidant–antioxidant balance of the cells.
The marked increases in the activities of TrxR1, CAT and SOD (particularly in Mn-SOD form) also showed DEHP-induced further alterations in the antioxidant defence of hepatocytes. A notable feature of these alterations was the marked enhancement of the activities of TrxR1 (∼75%) and Mn-SOD (∼two-fold) as seen in tumorogenic tissues. Thioredoxin reductase is considered as one of the major redox regulators in mammalian cells (Gromer et al. 2005) with roles in cell proliferation, transcription, DNA repair and angiogenesis (Arnér & Holmgren 2006). Thioredoxin reductase is also involved in a multitude of pathological conditions with special emphasis on cancer development and drug resistance due to its anti-apoptotic functions (Urig & Becker 2006). Thioredoxin reductase is overexpressed in many cancers (Arnér & Holmgren 2006; Arnér 2009). As targeting its deactivation often leads to a reversal in numerous malignant characteristics, it has been marked as a prime target for cancer therapy (Yoo et al. 2010). In the present study, we might postulate that the marked increase in TrxR1 activity might cause an increase in the reduction in thioredoxin (Trx) which induces cell growth, affects transcription factors, inhibits apoptosis and reduces ascorbate (Mustacich & Powis 2000). This high reduction rate might lead to higher growth of cancer cells induced by several environmental agents, including phthalates.
The induction of hepatic total SOD and Cu,Zn-SOD activities by DEHP might be as a cause of high cytoplasmic superoxide production, while high Mn-SOD activity might be an indicator of elevated mitochondrial superoxide production. Thiobarbituric acid reactive substance levels were ∼40% higher than control in DEHP-treated animals. Thus, all these results indicated the disturbance of cellular redox systems and the incidence of oxidative stress by DEHP exposure in rat liver. In fact, in vivo exposure of rodents to DEHP leads to an increased oxidative stress in liver, and the induction of peroxisomal and microsomal enzymes, a pathway largely dependent on activation of PPARα, contributes to the increased formation of ROS in hepatocytes (Rusyn et al. 2006). In vivo evidence for the increase in free radicals in liver before peroxisomal oxidases are induced was also reported (Rusyn et al. 2001).
Oxidative stress may play a pathogenetic role in several liver diseases, including HCC (Medina & Moreno-Otero 2005; Sasaki 2006). Antioxidants, including Se, appeared promising in prevention and treatment of hepatic damage (Medina & Moreno-Otero 2005). Se in supranutritional doses was shown to be tumour preventive in animal models (Darvesh & Bishayee 2010) and considered as a candidate treatment for hepatocellular carcinoma (HCC) (Erkhembayar et al. 2012). The potentially anti-carcinogenic mechanisms of Se include its effects on DNA stability, cell proliferation, necrotic and apoptotic cell death in healthy and malignant cells (Whanger 2004). Hepatocellular carcinoma is one of the most lethal cancers in the world, and both oxidative stress and inflammatory mechanisms have been implicated in its pathophysiology. An epidemiological study among chronic carriers of hepatitis B virus (HBV) and/or hepatitis C demonstrated an inverse correlation between plasma Se levels and HCC (Yu et al. 1999), and Se supplementation showed a protective effect against HBV infection (Costantini et al. 2011). The optimum level for the prevention and retardation of carcinogenesis in human cells is higher than the level commonly achieved under a diet not deficient in Se (Whanger 2004).
The data presented herein showed normal morphology of liver with slightly increased glycogen granules in the hepatocytes of Se-supplemented group. None of the measured parameters were different than the control, with the exception of TrxR1, the activity of which increased significantly (∼30%). A Se-dependent increase in the activity of TrxR1 in parallel with the increase in liver Se levels was also reported earlier (Erkhembayar et al. 2012).
In Se-deficient animals, except for a slight decrease in glycogen granules, decreased number of peroxisomes and a decline in CAT immunohistochemistry and activity, we did not observe any significant changes in the morphology and structure of hepatocytes. Consistent with these observations, Olsson et al. (1992) reported much less pronounced PP in Se-deficient rat livers. Moreover, we observed statistically significant increase in the number of apoptotic cells in SeD group vs. control group. In line with our results, Irmak et al. (2003) observed marked apoptosis and increased in oxidative stress in Se-deficient human HCC-derived cell lines. Aggravation of apoptotic response in Se deficiency is probably due to impaired capacity of GPx1 as well as CAT to degrade H2O2 (Demelash et al. 2004).
It is well known that GPx1 activity is highly regulated by dietary Se and decreases down to less than 1% of its normal value in Se deficiency in rat liver (Hafeman et al. 1974). 0.1 μg/g Se diet fulfils the requirement of Se in rats providing 100% of GPx1 activity in rats (National Research Council 1995). The Se dose for control group was chosen as 0.15 mg/kg which can fulfil the whole requirement of rats even they eat less than the animals in the same group. If the rats were fed with 0.05 mg/kg Se, this would provide 50% of GPx1 activity in the animals. With diet that contains <0.05 mg/kg Se, the animals will express lower amounts GPx1 protein and will show lower (<50%) GPx1 activity (Sundae 2003). In addition to its activity, rat liver mRNA level of GPx1 is also down-regulated by Se deficiency (Saedi et al. 1988). One milligram per kilogram Se containing diet was chosen as this amount will provide supraphysiological doses of Se (Sundae 2003). However, as also observed in the study, 1 mg/kg Se containing diet does not provide significant increase in the GPx1 expression and activity. We observed a drastic decline (93%) in hepatic GPx1 activity in SeD group, confirming that the Se deficiency was induced by the feeding schedule applied. Marked decreases in TrxR1 activity (2.2-fold) and GSH levels and an enhancement of LP were observed, also by the others (Turan et al. 2001; Erkhembayar et al. 2012). Reduced Se levels result in accumulation of lipid peroxides which might accelerate growth of HCC (Rohr-Udilova et al. 2012). Total SOD and Cu,Zn-SOD activities of SeD group markedly increased. The increase in Mn-SOD was as high as 2.5-fold compared with control affirming the induction of oxidative stress as also shown earlier in animals fed on Se-deficient diet (Giray et al. 2004).
Di(2-ethylhexyl)phthalate treatment caused the same level of liver enlargement in all DEHP-exposed groups regardless of Se status. Body weight gain was low in DEHP/SeD group compared with control, but DEHP/SeS animals gained higher weights than DEHP group indicating protection by Se. The overall data of DEHP/SeS group indicated further protective effects of Se supplementation against DEHP, evidenced by maintained normal morphology and presence of glycogen granules; decreased mitochondrial damage and AST levels, and markedly lowered Mn-SOD activity compared with DEHP group; maintained GPx1 and GPx4 activities and tGSH levels compared with control; and decreased LP down to the control and SeS levels. Se supplementation, however, did not have an effect on the PP activity of DEHP treatment. On the contrary, when Se deficiency accompanied by DEHP exposure, severe histological alterations, marked alterations in antioxidant enzyme activities and increase in LP were observed, indicating that this particular treatment causes oxidative stress, which can further augment to liver damage. Among the DEHP-exposed groups, the most severe histopathological lesions were observed in DEPH/SeD animals. Marked focal necrosis and inflammation observed by EM examination in opposition to normal morphology seen in SeD animals indicated the augmenting effect of DEHP on Se-deficient liver. Thus, the results presented herein implicated the important roles of Se in the structure, antioxidant defence and redox status of hepatocytes and also showed that the hepatic effects of DEHP changed for the worse in Se deficiency. Our previous studies similarly demonstrated that the testicular, renal and thyroidal toxicity of DEHP exposure in pubertal rats are modified by Se status (Erkekoglu et al. 2011a,b, 2012a,b). The same line of data was produced in both MA-10 Leydig cells and LNCaP cells where Se supplementation was highly protective against the cytotoxicity, genotoxicity and oxidative potential of DEHP and its main metabolite MEHP (Erkekoglu et al. 2010a,b). On the other hand, there are limited number of studies in literature determining the protective effect of other antioxidants against the hepatotoxicity of DEHP. In a study by El-Shinnawy (2013), the researcher showed that Apium graveolens (celery) oil seed extract was protective against the oxidative stress-producing potential of DEHP in rat liver. However, resveratrol and vitamin C did not provide any protection against the hepatoxicity of DEHP in rats (Botelho et al. 2009).
Conclusion
The present study, conducted to investigate the hepatic toxicity of subacute DEHP exposure at different Se status, demonstrated that DEHP exposure increased oxidative stress by disturbing the antioxidant balance in the hepatocytes, in addition to its PP effect and that Se status plays a crucial role as ameliorating or augmenting the effects of DEHP in liver. Protective effect of Se supplementation in DEHP-exposed rats was evidenced by improvement of histopathology and cellular antioxidant balance, and complete prevention of LP without interfering PP. Thus, the effect of Se seems to be not involved with peroxisomal action. These findings also suggest that the oxidative stress-inducing effect of DEHP is independent of its PP action and/or it occurs before peroxisomal oxidases induced as reported by Rusyn et al. (2001). In conclusion, our findings emphasize the critical role of Se as an effective redox regulator and the importance of Se status in protecting the liver structure and functions from oxidant stressor activity of DEHP. This effect of Se is in accordance with its suggested protective potential in liver disease including its promising effects in liver malignancies.
Acknowledgments
The work was supported by Hacettepe University Research Fund [Project No: 0701301001].
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Determination of SOD activity in liver homogenate samples.
References
- Aebi H. Catalase. In: Bergmeyer HU, editor. Methods of Enzymatic Analysis. New York: Academic press 3; 1974. p. 673. [Google Scholar]
- Akerboom TP, Sies H. Assay of glutathione, glutathione disulfide, and glutathione mixed disulfides in biological samples. Meth. Enzymol. 1981;77:373–382. doi: 10.1016/s0076-6879(81)77050-2. [DOI] [PubMed] [Google Scholar]
- Akingbemi BT, Ge R, Klinefelter GR, Zikrin BR, Hardy MP. Phthalate induced Leydig cell hyperplasia is associated with multiple endocrine disturbances. Proc. Natl Acad. Sci. USA. 2004;101:775–780. doi: 10.1073/pnas.0305977101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnér ES, Zhong L, Holmgren A. Preparation and assay of mammalian thioredoxin and thioredoxin reductase. Methods Enzymol. 1999;300:226–239. doi: 10.1016/s0076-6879(99)00129-9. [DOI] [PubMed] [Google Scholar]
- Arnér ES. Focus on mammalian thioredoxin reductases–important selenoproteins with versatile functions. Biochim. Biophys. Acta. 2009;1790:495–526. doi: 10.1016/j.bbagen.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Arnér ES, Holmgren A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006;16:420–426. doi: 10.1016/j.semcancer.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Botelho GG, Bufalo AC, Boareto AC, et al. Vitamin C and resveratrol supplementation to rat dams treated with di(2-ethylhexyl)phthalate: impact on reproductive and oxidative stress end points in male offspring. Arch. Environ. Contam. Toxicol. 2009;57:785–793. doi: 10.1007/s00244-009-9385-9. [DOI] [PubMed] [Google Scholar]
- Clark LC, Cantor KP, Allaway WH. Selenium in forage crops and cancer mortality in US counties. Arch. Environ. Health. 1991;46:37–42. doi: 10.1080/00039896.1991.9937427. [DOI] [PubMed] [Google Scholar]
- Conway JG, Cattley RC, Popp JA, Butterworth BE. Possible mechanisms in hepatocarcinogenesis by the peroxisome proliferator di(2-ethylhexyl)phthalate. Drug Metab. Rev. 1989;21:65–102. doi: 10.3109/03602538909029956. [DOI] [PubMed] [Google Scholar]
- Costantini S, Lepore MG, Castello G, Colonna G. Has selenium a chemopreventive effect on hepatocellular carcinoma? Mini. Rev. Med. Chem. 2011;1:599–610. doi: 10.2174/138955711795906950. [DOI] [PubMed] [Google Scholar]
- Darvesh AS, Bishayee A. Selenium in the prevention and treatment of hepatocellular carcinoma. Anticancer Agents Med. Chem. 2010;10:338–345. doi: 10.2174/187152010791162252. [DOI] [PubMed] [Google Scholar]
- Demelash A, Karlsson JO, Nilsson M, Björkman U. Selenium has a protective role in caspase-3-dependent apoptosis induced by H2O2 in primary cultured pig thyrocytes. Eur. J. Endocrinol. 2004;50:841–849. doi: 10.1530/eje.0.1500841. [DOI] [PubMed] [Google Scholar]
- El-Shinnawy NA. The therapeutic applications of celery oil seed extract on the plasticizer di(2-ethylhexyl) phthalate toxicity. Toxicol. Ind. Health. 2013 doi: 10.1177/0748233713475515. doi: 10.1177/0748233713475515 (Ahead of print) [DOI] [PubMed] [Google Scholar]
- Erkekoglu P, Rachidi W, De Rosa V, Giray B, Favier A, Hincal F. Protective effect of selenium supplementation on the genotoxicity of di(2-ethylhexyl)phthalate and mono(2-ethylhexyl)phthalate treatment in LNCaP cells. Free Radic. Biol. Med. 2010a;49:559–566. doi: 10.1016/j.freeradbiomed.2010.04.038. [DOI] [PubMed] [Google Scholar]
- Erkekoglu P, Rachidi W, Yuzugullu OG, et al. Evaluation of cytotoxicity and oxidative DNA damaging effects of di(2-ethylhexyl)-phthalate (DEHP) and mono(2-ethylhexyl)-phthalate (MEHP) on MA-10 Leydig cells and protection by selenium. Toxicol. Appl. Pharmacol. 2010b;248:52–62. doi: 10.1016/j.taap.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Erkekoglu P, Zeybek ND, Giray B, Asan E, Arnaud J, Hincal F. Reproductive toxicity of di(2-ethylhexyl) phthalate in selenium-supplemented and selenium-deficient rats. Drug Chem. Toxicol. 2011a;34:379–389. doi: 10.3109/01480545.2010.547499. [DOI] [PubMed] [Google Scholar]
- Erkekoglu P, Giray B, Rachidi W, et al. Effects of di(2-ethylhexyl)phthalate on testicular oxidant/antioxidant status in selenium-deficient and selenium-supplemented rats. Environ. Toxicol. 2011b doi: 10.1002/tox.20776. doi: 10.1002/tox.20776. [DOI] [PubMed] [Google Scholar]
- Erkekoglu P, Giray BK, Kızilgün M, et al. Di(2-ethylhexyl)phthalate-induced renal oxidative stress in rats and protective effect of selenium. Toxicol. Mech. Methods. 2012a;22:415–423. doi: 10.3109/15376516.2012.666652. [DOI] [PubMed] [Google Scholar]
- Erkekoglu P, Giray BK, Kızilgün M, et al. Thyroidal effects of di-(2-Ethylhexyl) phthalate in rats at different selenium status. J. Environ. Pathol. Toxicol. Oncol. 2012b;31:143–153. doi: 10.1615/jenvironpatholtoxicoloncol.v31.i2.60. [DOI] [PubMed] [Google Scholar]
- Erkhembayar S, Mollbrink A, Eriksson LC. The effect of sodium selenite on liver growth and thioredoxin reductase expression in regenerative and neoplastic liver cell proliferation. Biochem. Pharmacol. 2012;83:687–693. doi: 10.1016/j.bcp.2011.12.004. [DOI] [PubMed] [Google Scholar]
- Flohé L, Günzler WA. Assays of glutathione peroxidase. Meth. Enzymol. 1984;105:114–121. doi: 10.1016/s0076-6879(84)05015-1. [DOI] [PubMed] [Google Scholar]
- Fromme H, Gruber L, Schlummer M, et al. Intake of phthalates and di(2-ethylhexyl)adipate: results of the Integrated Exposure Assessment Survey based on duplicate diet samples and biomonitoring data. Environ. Int. 2007;33:1012–1020. doi: 10.1016/j.envint.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Ganther DE. Selenium metabolism, selenoproteins and mechanisms of cancer prevention: complexities with thioredoxin reductase. Carcinogenesis. 1999;20:1657–1666. doi: 10.1093/carcin/20.9.1657. [DOI] [PubMed] [Google Scholar]
- Gayathri NS, Dhanya CR, Indu AR, Kurup PA. Changes in some hormones by low doses of di (2-ethyl hexyl) phthalate (DEHP), a commonly used plasticizer in PVC blood storage bags & medical tubing. Indian J. Med. Res. 2004;119:139–144. [PubMed] [Google Scholar]
- Gazouli M, Yao ZX, Boujrad N, Corton JC, Culty M, Papadopoulos V. Effect of peroxisome proliferators on Leydig cell peripheral-type benzodiazepine receptor gene expression, hormone-stimulated cholesterol transport, and steroidogenesis: role of the peroxisome proliferator-activator receptor alpha. Endocrinology. 2002;143:2571–2583. doi: 10.1210/endo.143.7.8895. [DOI] [PubMed] [Google Scholar]
- Giray B, Riondel J, Richard MJ, Favier A, Hıncal F. Oxidant/antioxidant status in relation to thyroid hormone metabolism in selenium-and/or iodine-deficient rats. J. Trace Elem. Exp. Med. 2004;17:109–121. [Google Scholar]
- Gromer S, Eubel JK, Lee BL, Jacob J. Human selenoproteins at a glance. Cell. Mol. Life Sci. 2005;62:2414–2437. doi: 10.1007/s00018-005-5143-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- Hafeman DG, Sunde RA, Hoekstra WG. Effect of dietary selenium on erythrocyte and liver glutathione peroxidase in the rat. J. Nutr. 1974;104:580–587. doi: 10.1093/jn/104.5.580. [DOI] [PubMed] [Google Scholar]
- Hasmall SC, James NH, Macdonald N, Soames AR, Roberts RA. Species differences in response to diethylhexylphthalate: suppression of apoptosis, induction of DNA synthesis and peroxisome proliferator activated receptor alpha-mediated gene expression. Arch. Toxicol. 2000;74:85–91. doi: 10.1007/s002040050657. [DOI] [PubMed] [Google Scholar]
- Hernández-Díaz S, Mitchell AA, Kelley KE, Calafat AM, Hauser R. Medications as a potential source of exposure to phthalates in the U.S. population. Environ. Health Perspect. 2009;117:185–189. doi: 10.1289/ehp.11766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmak MB, Ince G, Ozturk M, Cetin-Atalay R. Acquired tolerance of hepatocellular carcinoma cells to selenium deficiency: a selective survival mechanism? Cancer Res. 2003;63:6707–6715. [PubMed] [Google Scholar]
- Isenberg JS, Kolaja KL, Ayoubi SA, Watkins JB, 3rd, Klaunig JE. Inhibition of WY-14,643 induced hepatic lesion growth in mice by rotenone. Carcinogenesis. 1997;18:1511–1519. doi: 10.1093/carcin/18.8.1511. [DOI] [PubMed] [Google Scholar]
- Jablonska E, Gromadzinska J, Reszka E, et al. Association between GPx1 Pro198Leu polymorphism, GPx1 activity and plasma selenium concentration in humans. Eur. J. Nutr. 2009;48:383–386. doi: 10.1007/s00394-009-0023-0. [DOI] [PubMed] [Google Scholar]
- James NH, Soames AR, Roberts RA. Suppression of hepatocyte apoptosis and induction of DNA synthesis by the rat and mouse hepatocarcinogen diethylhexylphlathate (DEHP) and the mouse hepatocarcinogen 1,4-dichlorobenzene (DCB) Arch. Toxicol. 1998;72:784–790. doi: 10.1007/s002040050574. [DOI] [PubMed] [Google Scholar]
- Klaunig JE, Babich MA, Baetcke KP, et al. PPARalpha agonist-induced rodent tumors: modes of action and human relevance. Crit. Rev. Toxicol. 2003;33:655–780. doi: 10.1080/713608372. [DOI] [PubMed] [Google Scholar]
- Kluwe WM, Huff JE, Matthews HB, Irwin R, Haseman JK. Comparative chronic toxicities and carcinogenic potentials of 2-ethylhexyl-containing compounds in rats and mice. Carcinogenesis. 1985;6:1577–1583. doi: 10.1093/carcin/6.11.1577. [DOI] [PubMed] [Google Scholar]
- Latini G. Monitoring phthalate exposure in humans. Clin. Chim. Acta. 2005;361:20–29. doi: 10.1016/j.cccn.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Maiorino M, Gregolin C, Ursini F. Phospholipid hydroperoxide glutathione peroxidase. Meth. Enzymol. 1990;186:448–457. doi: 10.1016/0076-6879(90)86139-m. [DOI] [PubMed] [Google Scholar]
- Marklund S, Marklund G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur. J. Biochem. 1974;47:469–474. doi: 10.1111/j.1432-1033.1974.tb03714.x. [DOI] [PubMed] [Google Scholar]
- Medina J, Moreno-Otero R. Pathophysiological basis for antioxidant therapy in chronic liver disease. Drugs. 2005;65:2445–2461. doi: 10.2165/00003495-200565170-00003. [DOI] [PubMed] [Google Scholar]
- Melnick RL. Is peroxisome proliferation an obligatory precursor step in the carcinogenicity of di(2-ethylhexyl)phthalate (DEHP)? Environ. Health Perspect. 2001;109:437–442. doi: 10.1289/ehp.01109437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody DE, Reddy JK. The hepatic effects of hypolipidemic drugs (clofibrate, nafenopin, tibric acid, and Wy-14,643) on hepatic peroxisomes and peroxisome-associated enzymes. Am. J. Pathol. 1978;90:435–446. [PMC free article] [PubMed] [Google Scholar]
- Moody DE, Reddy JK, Lake BG, Popp JA, Reese DH. Peroxisome proliferation and nongenotoxic carcinogenesis: commentary on a symposium. Fundam. Appl. Toxicol. 1991;16:233–248. doi: 10.1016/0272-0590(91)90108-g. [DOI] [PubMed] [Google Scholar]
- Mushtaq M, Srivastava SP, Seth PK. Effect of di-2-ethylhexyl phthalate (DEHP)on glycogen metabolism in rat liver. Toxicology. 1980;16:153–161. doi: 10.1016/0300-483x(80)90045-1. [DOI] [PubMed] [Google Scholar]
- Mustacich D, Powis G. Thioredoxin reductase. Biochem. J. 2000;346:1–8. [PMC free article] [PubMed] [Google Scholar]
- National Research Council (NRC) Nutrient Requirements of Laboratory Animals. 4th revised edn. Washington, DC: National Academy Press; 1995. [Google Scholar]
- Nose K. Role of reactive oxygen species in the regulation of physiological functions. Biol. Pharm. Bull. 2000;23:897–903. doi: 10.1248/bpb.23.897. [DOI] [PubMed] [Google Scholar]
- Oberley TD, Zhong W, Szweda LI, Oberley LW. Localization of antioxidant enzymes and oxidative damage products in normal and malignant prostate epithelium. Prostate. 2000;44:144–155. doi: 10.1002/1097-0045(20000701)44:2<144::aid-pros7>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- O'Brien ML, Spear BT, Glauert HP. Role of oxidative stress in peroxisome proliferator-mediated carcinogenesis. Crit. Rev. Toxicol. 2005;35:61–88. doi: 10.1080/10408440590905957. [DOI] [PubMed] [Google Scholar]
- O'Brien ML, Twaroski TP, Cunningham ML, Glauert HP, Spear BT. Effects of peroxisome proliferators on antioxidant enzymes and antioxidant vitamins in rats and hamsters. Toxicol Sci. 2001;60:271–278. doi: 10.1093/toxsci/60.2.271. [DOI] [PubMed] [Google Scholar]
- Olsson U, Garberg P, Lundgren B, et al. The involvement of selenium in peroxisome proliferation caused by dietary administration of clofibrate to rats. Chem. Biol. Interact. 1992;5:49–67. doi: 10.1016/0009-2797(92)90052-m. [DOI] [PubMed] [Google Scholar]
- Reddy JK, Rao MS. Oxidative DNA damage caused by persistent peroxisome proliferation: its role in hepatocarcinogenesis. Mutat. Res. 1989;214:63–68. doi: 10.1016/0027-5107(89)90198-x. [DOI] [PubMed] [Google Scholar]
- Reddy JK, Rao MS. Peroxisome proliferation and hepatocarcinogenesis. IARC Sci. Publ. 1992;116:225–235. [PubMed] [Google Scholar]
- Richard MJ, Portal B, Meo J, Coudray C, Hadjian A, Favier A. Malondialdehyde kit evaluated for determining plasma and lipoprotein fractions that react with thiobarbituric acid. Clin. Chem. 1992;38:704–709. [PubMed] [Google Scholar]
- Roberts RA, James NH, Hasmall SC, et al. Apoptosis and proliferation in nongenotoxic carcinogenesis: species differences and role of PPARalpha. Toxicol. Lett. 2000;112–113:49–57. doi: 10.1016/s0378-4274(99)00243-x. [DOI] [PubMed] [Google Scholar]
- Rohr-Udilova N, Sieghart W, Eferl R, et al. Antagonistic effects of selenium and lipid peroxides on growth control in early hepatocellular carcinoma. Hepatology. 2012;55:1112–1121. doi: 10.1002/hep.24808. [DOI] [PubMed] [Google Scholar]
- Rose ML, Rusyn I, Bojes HK, Belyea J, Cattley RC, Thurman RG. Role of Kupffer cells and oxidants in signaling peroxisome proliferator induced hepatocyte proliferation. Mutat. Res. 2000;448:179–192. doi: 10.1016/s0027-5107(99)00235-3. [DOI] [PubMed] [Google Scholar]
- Rusyn I, Rose ML, Bojes HK, Thurman RG. Novel role of oxidants in the molecular mechanism of action of peroxisome proliferators. Antioxid. Redox Signal. 2000;2:607–621. doi: 10.1089/15230860050192350. [DOI] [PubMed] [Google Scholar]
- Rusyn I, Kadiiska MB, Dikalova A, et al. Phthalates rapidly increase production of reactive oxygen species in vivo: role of Kupffer cells. Mol. Pharmacol. 2001;59:744–750. doi: 10.1124/mol.59.4.744. [DOI] [PubMed] [Google Scholar]
- Rusyn I, Peters JM, Cunningham ML. Modes of action and species-specific effects of di-(2-ethylhexyl)phthalate in the liver. Crit. Rev. Toxicol. 2006;36:459–479. doi: 10.1080/10408440600779065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saedi MS, Smith CG, Frampton J, Chambers I, Harrison PR, Sunde RA. Effect of selenium status on mRNA levels for glutathione peroxidase in rat liver. Biochem. Biophys. Res. Commun. 1988;153:855–861. doi: 10.1016/s0006-291x(88)81174-4. [DOI] [PubMed] [Google Scholar]
- Sasaki Y. Does oxidative stress participate in the development of hepatocellular carcinoma? J. Gastroenterol. 2006;41:1135–1148. doi: 10.1007/s00535-006-1982-z. [DOI] [PubMed] [Google Scholar]
- Seo KW, Kim KB, Kim YJ, Choi JY, Lee KT, Choi KS. Comparison of oxidative stress and changes of xenobiotic metabolizing enzymes induced by phthalates in rats. Food Chem. Toxicol. 2004;42:107–114. doi: 10.1016/j.fct.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Steinbrenner H, Sies H. Protection against reactive oxygen species by selenoproteins. Biochim. Biophys. Acta. 2009;1790:1478–1485. doi: 10.1016/j.bbagen.2009.02.014. [DOI] [PubMed] [Google Scholar]
- Sundae RA. Chapter 8. Regulation of selenoprotein expression. In: D Hatfield., editor. Selenium: Its Molecular Biology and Role in Human Health. Massachusetts: Kluwer Academic Publishers; 2003. pp. 81–98. [Google Scholar]
- Turan B, Acan NL, Ulusu NN, Tezcan EF. A comparative study on effect of dietary selenium and vitamin E on some antioxidant enzyme activities of liver and brain tissues. Biol. Trace Elem. Res. 2001;81:141–152. doi: 10.1385/BTER:81:2:141. [DOI] [PubMed] [Google Scholar]
- Urig S, Becker K. On the potential of thioredoxin reductase inhibitors for cancer therapy. Semin. Cancer Biol. 2006;16:452–465. doi: 10.1016/j.semcancer.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Weitzel F, Ursini F, Wendel A. Phospholipid hydroperoxide glutathione peroxidase in various mouse organs during selenium deficiency and repletion. Biochim. Biophys. Acta. 1990;1036:88–94. doi: 10.1016/0304-4165(90)90018-r. [DOI] [PubMed] [Google Scholar]
- Whanger PD. Selenium and its relationship to cancer: an update. Br. J. Nutr. 2004;91:11–28. doi: 10.1079/bjn20031015. [DOI] [PubMed] [Google Scholar]
- Willhite CC. Weight-of-evidence versus strength-of-evidence in toxicologic hazard identification: di(2-ethylhexyl)phthalate (DEHP) Toxicology. 2001;160:219–226. doi: 10.1016/s0300-483x(00)00451-0. [DOI] [PubMed] [Google Scholar]
- Wormuth M, Scheringer M, Vollenweider M, Hungerbühler K. What are the sources of exposure to eight frequently used phthalic acid esters in Europeans? Risk Anal. 2006;26:803–824. doi: 10.1111/j.1539-6924.2006.00770.x. [DOI] [PubMed] [Google Scholar]
- Yeldandi AV, Rao MS, Reddy JK. Hydrogen peroxide generation in peroxisome proliferator-induced oncogenesis. Mutat. Res. 2000;448:159–177. doi: 10.1016/s0027-5107(99)00234-1. [DOI] [PubMed] [Google Scholar]
- Yoo MH, Carlson BA, Tsuji P, Irons R, Gladyshev VN, Hatfield DL. Alteration of thioredoxin reductase 1 levels in elucidating cancer etiology. Methods Enzymol. 2010;474:255–275. doi: 10.1016/S0076-6879(10)74015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef JA, Bouziane M, Badr MZ. Age-dependent effects of nongenotoxic hepatocarcinogens on liver apoptosis in vivo. Mech. Ageing Dev. 2003;124:333–340. doi: 10.1016/s0047-6374(02)00189-6. [DOI] [PubMed] [Google Scholar]
- Yu MW, Horng IS, Hsu KH, Chiang YC, Liaw YF, Chen CJ. Plasma selenium levels and risk of hepatocellular carcinoma among men wit chronic hepatitis virus infection. Am. J. Epidemiol. 1999;150:367–374. doi: 10.1093/oxfordjournals.aje.a010016. [DOI] [PubMed] [Google Scholar]
- Zhu H, Jia Z, Misra H, Li YR. Oxidative stress and redox signaling mechanisms of alcoholic liver disease: updated experimental and clinical evidence. J. Dig. Dis. 2012;13:133–142. doi: 10.1111/j.1751-2980.2011.00569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwolak I, Zaporowska H. Selenium interactions and toxicity: a review. Selenium interactions and toxicity. Cell Biol. Toxicol. 2012;28:31–46. doi: 10.1007/s10565-011-9203-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Determination of SOD activity in liver homogenate samples.