

Abstract
Purpose: Retinitis pigmentosa (RP) is a heterogenous group of inherited retinal degenerations caused by mutations in at least 45 genes. Recently, the FAM161A gene was identified as the causative gene for RP28, an autosomal recessive form of RP.
Methods: We performed a clinical and molecular genetic study of a consanguineous Palestinian family with two three siblings affected with retinitis pigmentosa. DNA samples were collected from the index patient, his father, his affected sister, and two non-affected brothers. DNA sample from the index was subjected to high resolution genome-wide SNP array. Assuming identity-by-descent in this consanguineous family we applied homozygosity mapping to identify disease causing genes.
Results: The index patient reported night blindness since the age of 20 years, followed by moderate disease progression with decrease of peripheral vision, the development of photophobia and later on reduced central vision. At the age of 40 his visual acuity was counting fingers (CF) for both eyes, color discrimination was not possible and his visual fields were severely constricted. Funduscopic examination revealed a typical appearance of advanced RP with optic disc pallor, narrowed retinal vessels, bone-spicule like pigmentary changes in the mid-periphery and atrophic changes in the macula. His younger affected brother (37 years) was reported with overall milder symptoms, while the youngest sister (21 years) reported problems only with night vision. Applying high-density SNP arrays we identified several homozygous genomic regions one of which included the recently identified FAM161A gene mutated in RP28-linked autosomal recessive RP. Sequencing analysis revealed the presence of a novel homozygous nonsense mutation, c.1003C>T/p.R335X in the index patient and the affected sister.
Conclusion: We identified an RP28-linked RP family in the Palestinian population caused by a novel nonsense mutation in FAM161A. RP in this family shows a typical disease onset with moderate to rapid progression into severe visual impairment including central vision in the index and overall milder symptoms in the younger brother and sister.
INTRODUCTION
Retinitis pigmentosa (RP) is a clinically and genetically heterogeneous group of hereditary retinal disorders, being one of the most common types of retinal degenerations worldwide with a prevalence of 1:4000. So far, 53 genes have been associated with RP, whose defects cause a progressive loss of rod photoreceptor function, followed by cone photoreceptor dysfunction often leading to complete blindness [1]. At present, 36 genetic loci have been implicated in nonsyndromic autosomal recessive RP (arRP), each of them being responsible for a few percent of arRP cases [2].
Enormous efforts have been made in research in recent years. With the help of improved molecular genetic and functional diagnostic tools an early recognition and differentiation of retinal degenerations has become possible. Homozygosity mapping has been proven to be the most efficient tool for identifying novel arRP genes [3]. In the Israeli and Palestinian population this approach is highly effective, because of relatively high levels of consanguinity, large number of siblings in families and presence of isolated subpopulations [4-7].
As a part of an international cooperation, we used homozygosity mapping to identify the disease causing genes in families from Israel and the Palestinian Authority with retinal disorders. Here we report the identification of a novel mutation in the FAM161A gene in a consanguineous Palestinian family with three siblings suffering from RP.
METHODS
Clinical examination
Informed consent was obtained and examinations were carried out respecting the Code of Ethics of the World Medical Association (Declaration of Helsinki). Approval of the local Ethics Committee was obtained. A detailed patient history of the index patient and his affected siblings was recorded regarding disease onset, symptoms and progression. Visual acuity, visual field and morphological examinations of the anterior and posterior segments were carried out in the index patient. Furthermore, medical records of prior ophthalmological visits were used to complete the clinical aspect.
Genetic analysis
Genomic DNA was prepared from peripheral venous blood using the MasterPure™ DNA Purification Kit (Epicentre, Madison, WI) and stored in TE buffer. Whole genome SNP genotyping was done using Affymetrix 250k_NspI SNP arrays (Affymetrix, Santa Clara, CA) as outlined previously [8]. SNP chip genotypes were used for mapping of autozygous regions applying the online version of the Homozygosity Mapper software [9].
Mutation screening of the FAM161A gene was done by complete DNA sequencing of all coding exons and flanking intron and UTR sequences from PCR amplified genomic DNA. PCR fragments were treated with ExoSAP (GE Healthcare, Freiburg, Germany) and subsequently subjected to DNA sequencing applying BigDye Terminator 1.1 chemistry (Applied Biosystems, Darmstadt, Germany). All sequences were run on an ABI 3100 DNA sequencer, processed with the Sequence Analysis 5.1 software (Applied Biosystems) and aligned with the reference sequence applying SeqMan (Lasergene, Madison WI, USA). Assessment of the c.1003C>T/p.R335X mutation in controls was performed by pyrosequencing on a PyroMark Q96 instrument (Qiagen, Hilden, Germany) applying a custom developed SNP assay (PCR primers: 5’-AAT TTA TAG CAA GGG AGG AAC AGA-3’ and 5’-biotin-GAG GAA TGG GTC TGG CTT TAA-3’; sequencing primer: 5’-CAA GGG AGG AAC AGA AG-3’). Purification of PCR products and pyrosequencing reactions were done following the manufacturer’s protocol (Qiagen).
RESULTS
Clinical findings
One female and two male siblings of a consanguineous Palestinian family presented with typical symptoms and clinical findings of RP. The older brother (40 years) had noticed night blindness at the age of 20 years, as first symptom of his disease. This has been accompanied by photophobia and decreased peripheral vision, and his visual function showed a moderate worsening over the next years. At time of our examination his visual acuity was counting fingers (CF) for both eyes, color discrimination was not possible and his visual field showed a severe constriction disabling the patient`s self-orientation. He additionally reported a history of high myopia with radial keratotomy performed in both eyes at the age of 20 years. Current glass correction was -9.0 Dpt sphere for both eyes, however, glasses did not improve his visual function. Pupillary responses and intraocular pressure were normal. Examination of the anterior segments revealed numerous radial keratotomy scars on both eyes, with the central cornea appearing clear. There was no evidence of inflammation in the anterior segments and only a mild cataract on the right eye was observed. Funduscopic examination revealed slight optic disc pallor, narrowed retinal vessels and atrophic appearing maculae on both eyes. In the mid-periphery, widespread bone-spicule like pigmentary changes could be observed, typical for advanced retinitis pigmentosa (Figure 1). His younger brother (37 years) presented with overall milder symptoms, disease onset was reported at the age of 27 years. The youngest sister (21 years) did not report any disturbing visual symptoms, only problems with night vision were noticed recently. Unfortunately, a detailed clinical examination could not be performed.
Figure 1.
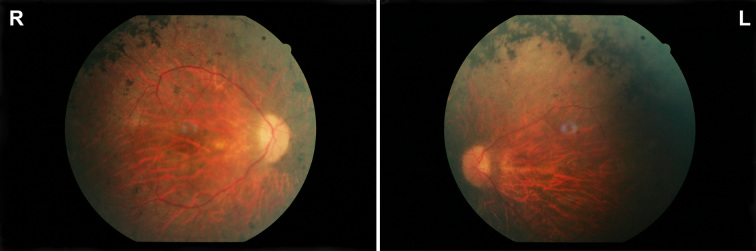
Morphological findings. Fundus pictures of the right (R) and left eye (L) of the index patient showing pallor optic discs, attenuated vessels, and characteristic pigment changes in the mid-periphery.
Genetic findings
DNA samples were collected from the index patient, his father, his affected sister, and two non-affected brothers after informed consent has been obtained. A DNA sample from the affected younger brother was not available. A DNA sample from the index was subjected to high resolution genome-wide SNP analysis using the Affymetrix 250K NspI SNP array. Assuming identity-by-descent in this consanguineous family we applied the program Homozygosity Mapper [9] for the identification of autozygous region in the genome. We observed several larger autozygous regions on chromosomes 1, 2, 3, 6, 11 and 16 including a ~ 25 Mb region between 47.2 M and 71.4M on chromosome 2. This region contains FAM161A, a gene that has been recently found to be mutated in arRP families linked to the RP28 locus [10,11]. We therefore performed Sanger sequencing of all coding exons and flanking intron and UTR sequences of the FAM161A gene which led to the identification of a homozygous nonsense mutation, c.1003C>T/p.R335X (RefSeq: NM_001201543), in the index patient (Figure 2). Segregation analysis within the family revealed that both the father and one of the unaffected brothers are heterozygous for the mutation whereas the other unaffected brother does not carry the mutation. (Figure 2).The youngest sister was found to be homozygous for the mutation c.1003C>T/p.R335X as well (Figure 2). Applying a custom pyrosequencing assay we further excluded the mutation in 87 ethnically matched controls (= 174 chromosomes) indicating that it is a rare variant in this population.
Figure 2.
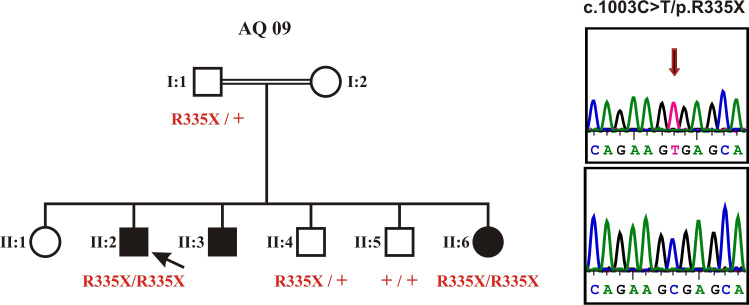
Pedigree and genetic findings. The index patient and his youngest sister show a homozygous nonsense mutation, c.1003C>T/p.R335X, while both the father and one of the unaffected brothers are heterozygous for the mutation and the other unaffected brother does not carry the mutation. The sequencing electropherograms show the homozygous mutant sequence (top) in comparison with a wildtype control sequence (bottom).
The p.R335X mutation is a novel mutation in FAM161A. It is predicted to result in a truncated protein that lacks – depending on the splice variant - 305 or 361 amino acid residues of the full length protein (Figure 3). However due to the location of the nonsense mutation in the central exon 3, mutant transcripts are expected to undergo nonsense-mediated mRNA decay. Moreover, truncated proteins are mostly unstable and rapidly degraded in the cell. We therefore reason, that the p.R335X mutation represent a virtual null allele of the FAM161A gene.
Figure 3.
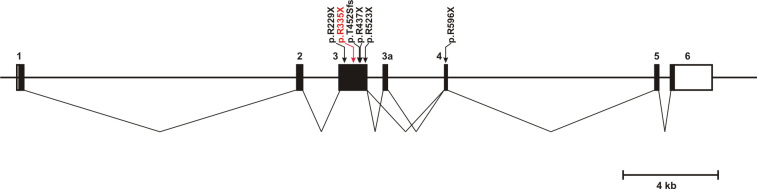
Structure of the FAM161A gene and localization of a currently known RP-associated mutation. Black bars indicate coding exons or exon segments whereas white bars represent the 5′ and 3′ untranslated region (UTR), respectively. The transcript splicing pattern is depicted below showing the two alternatively spliced isoforms including or lacking exon 3a. Most currently known mutations localize to exon 3; the novel p.R335X mutation found in this study is shown in red.
DISCUSSION
Retinitis pigmentosa is a genetically highly heterogeneous disorder with more than 50 genes currently known to be implicated in this condition. For the mapping of new loci and subsequent identification of new genes for autosomal recessive forms of RP, homozygosity mapping has been shown to be a highly effective method [3]. Homozygosity mapping relies on identity-by-descent of the mutant alleles and thus is most successful in consanguineous families. Consangineous marriages are rather common in the Palestinian population, therefore it provides a rich resource of families suitable for this approach. In fact, we were able to pick the correct candidate gene in this family, although our homozygosity mapping solely based on genotype data from a single affected individual.
The novel c.1003C>T/p.R335X mutation found in this study is just the fifth mutation reported in FAM161A. One of the initial gene identification reports relied on a study of RP patients from Israel, most of them of Jewish origin. In the 20 families reported in this study, only three different FAM161A mutations were found with the p.T452Sfs mutation being by far the most abundant allele [11]. The identification of the novel c.1003C>T/p.R335X mutation, different from those reported by Bandah-Rozenfeld and colleagues contrasts prior reports on genetic relatedness and shared mutations between the Jewish and the Palestian/Arab people in the near East [12,13]. However we cannot exclude a rather recent origin and thus limited spread of the c.1003C>T/p.R335X mutation in the Near East populations.
Most currently known mutations [10,11] cluster in exon 3 of the FAM161A gene (Figure 3), which is by far the largest coding exon of the whole gene. The mutations are predicted to result in a truncated polypeptide that lacks the carboxyterminus of the genuine protein. However, it is likely that the mutant transcripts undergo nonsense-mediated mRNA decay and thus only little of the truncated protein is expected to be synthesized. Interestingly, all four mutations that represent single nucleotide substitutions in exon 3 affect one of a total of 7 arginine –CGA- codons in this exon. CpG dinucleotides are prone to deamination of the cytosine and thus to result in the conversion into a –TGA- stop codon, however its abundance in this case is unusual. On average exon 3 of FAM161A has a rather normal GC content of 42%. However at least some of the mutated –CGA- codons are part of local segments with considerably higher GC content which might form DNA structures that a more exposed to mutating agents (Figure 4).
Figure 4.
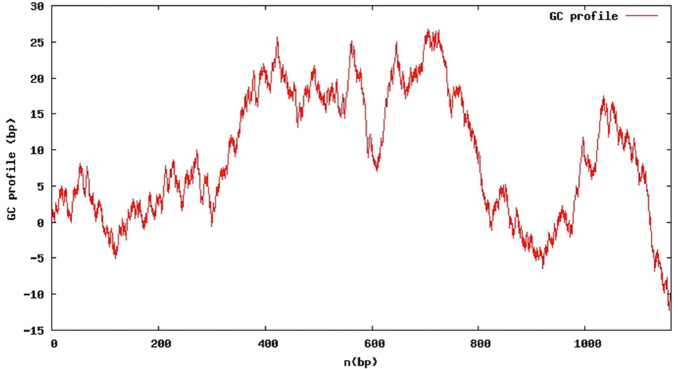
GC profile of exon 3 of FAM161A. The GC profile shows local segments of significantly elevated GC content.
Recent studies have shown that FAM161A localizes to the base of the connecting cilium in photoreceptors and is also associated with the basal body in ciliated mammalian cells [14,15]. Moreover, FAM161A interacts with other known components of sensory cilia including a number of ciliopathy-associated proteins [15]. Thus FAM161A is another member of the growing list of ciliary proteins implicated in human disease and FAM161A-associated RP must be considered as a ciliopathy. Phenotypes of ciliopathies are quite diverse and may involve impairment of multiple organs or functions such as vision, kidney, brain, skeletal abnormalities and obesity [16]. However, in the patients investigated in this study we did not observe any other obvious features or anamnestic history of extraocular symptoms typically related to ciliopathies. The clinical picture features all typical signs of retinitis pigmentosa including a history of night blindness as first symptom, progressive loss of visual function and characteristic morphological changes. Compared to the patients described by Langmann et al [10] and Bandah-Rozenfeld et al [11] it seems, that FAM161A-related arRP does not show unique clinical features, however, phenotype can be variable regarding disease onset and progression. Interestingly, high myopia was present in our case, similar to the findings of Bandah-Rozenfeld et al in their patient cohort [11].
In conclusion, we report here a novel c.1003C>T/p.R335X mutation in the FAM161A gene causing autosomal recessive retinitis pigmentosa in a family in the Palestinian Arabic population, which further accentuates its presence and relevance in Israeli and Palestinian populations.
Acknowledgments
We thank the family members for participation in this study. We also thank the Microarray Facility at the Medical Faculty of the Tübingen University for SNP chip processing. This work was supported by a Trilateral German-Israel-Palestinian Authority program grant of the Deutsche Forschungsgemeinschaft (SCHO 754/5–1 and WI1189/8–1).
REFERENCES
- 1.Zobor D, Zrenner E. Retinitis pigmentosa - a review. Pathogenesis, guidelines for diagnostics and perspectives. Ophthalmologe. 2012;109:501–14. doi: 10.1007/s00347-012-2555-6. [DOI] [PubMed] [Google Scholar]
- 2.Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29:335–75. doi: 10.1016/j.preteyeres.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Janecke AR, Thompson DA, Utermann G, Becker C, Hübner CA, Schmid E, McHenry CL, Nair AR, Rüschendorf F, Heckenlively J, Wissinger B, Nürnberg P, Gal A. Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat Genet. 2004;36:850–4. doi: 10.1038/ng1394. [DOI] [PubMed] [Google Scholar]
- 4.Zlotogora J. Genetic disorders among Palestinian Arabs: 1. Effects of consanguinity. Am J Med Genet. 1997;68:472–5. doi: 10.1002/(sici)1096-8628(19970211)68:4<472::aid-ajmg20>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 5.Collin RW, Safieh C, Littink KW, Shalev SA, Garzozi HJ, Rizel L, Abbasi AH, Cremers FP, den Hollander AI, Klevering BJ, Ben-Yosef T. Mutations in C2ORF71 cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2010;86:783–8. doi: 10.1016/j.ajhg.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zelinger L, Banin E, Obolensky A, Mizrahi-Meissonnier L, Beryozkin A, Bandah-Rozenfeld D, Frenkel S, Ben-Yosef T, Merin S, Schwartz SB, Cideciyan AV, Jacobson SG, Sharon D. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am J Hum Genet. 2011;88:207–15. doi: 10.1016/j.ajhg.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bandah-Rozenfeld D, Collin RW, Banin E, van den Born LI, Coene KL, Siemiatkowska AM, Zelinger L, Khan MI, Lefeber DJ, Erdinest I, Testa F, Simonelli F, Voesenek K, Blokland EA, Strom TM, Klaver CC, Qamar R, Banfi S, Cremers FP, Sharon D, den Hollander AI. Mutations in IMPG2, encoding interphotoreceptor matrix proteoglycan 2, cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2010;87:199–208. doi: 10.1016/j.ajhg.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wissinger B, Schaich S, Baumann B, Bonin M, Jägle H, Friedburg C, Varsanyi B, Hoyng CB, Dollfus H, Heckenlively JR, Rosenberg T, Rudolph G, Kellner U, Salati R, Plomp A, de Baere E, Andrassi-Darida M, Sauer A, Wolf C, Zobor D, Bernd A, Leroy B, Enyedi P, Cremers FPM, Lorenz B, Zrenner E, Kohl S. Large Deletions of the KCNV2 Gene are Common in Patients with Cone Dystrophy and Supernormal Rod Response. Hum Mutat. 2011;32:1398–406. doi: 10.1002/humu.21580. [DOI] [PubMed] [Google Scholar]
- 9.Seelow D, Schuelke M, Hildebrandt F, Nürnberg P. HomozygosityMapper--an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37:W593-9. doi: 10.1093/nar/gkp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langmann T, Di Gioia SA, Rau I, Stöhr H, Maksimovic NS, Corbo JC, Renner AB, Zrenner E, Kumaramanickavel G, Karlstetter M, Arsenijevic Y, Weber BH, Gal A, Rivolta C. Nonsense mutations in FAM161A cause RP28-associated recessive retinitis pigmentosa. Am J Hum Genet. 2010;87:376–81. doi: 10.1016/j.ajhg.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bandah-Rozenfeld D, Mizrahi-Meissonnier L, Farhy C, Obolensky A, Chowers I, Pe'er J, Merin S, Ben-Yosef T, Ashery-Padan R, Banin E, Sharon D. Homozygosity mapping reveals null mutations in FAM161A as a cause of autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2010;87:382–91. doi: 10.1016/j.ajhg.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nebel A, Filon D, Brinkmann B, Majumder PP, Faerman M, Oppenheim A. The Y chromosome pool of Jews as part of the genetic landscape of the Middle East. Am J Hum Genet. 2001;69:1095–112. doi: 10.1086/324070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zelinger L, Greenberg A, Kohl S, Banin E, Sharon D. An ancient autosomal haplotype bearing a rare achromatopsia-causing founder mutation is shared among Arab Muslims and Oriental Jews. Hum Genet. 2010;128:261–7. doi: 10.1007/s00439-010-0846-z. [DOI] [PubMed] [Google Scholar]
- 14.Zach F, Grassmann F, Langmann T, Sorusch N, Wolfrum U, Stöhr H. The retinitis pigmentosa 28 protein FAM161A is a novel ciliary protein involved in intermolecular protein interaction and microtubule association. Hum Mol Genet. 2012;21:4573–86. doi: 10.1093/hmg/dds268. [DOI] [PubMed] [Google Scholar]
- 15.Di Gioia SA, Letteboer SJ, Kostic C, Bandah-Rozenfeld D, Hetterschijt L, Sharon D, Arsenijevic Y, Roepman R, Rivolta C. FAM161A, associated with retinitis pigmentosa, is a component of the cilia-basal body complex and interacts with proteins involved in ciliopathies. Hum Mol Genet. 2012;21:5174–84. doi: 10.1093/hmg/dds368. [DOI] [PubMed] [Google Scholar]
- 16.Mockel A, Perdomo Y, Stutzmann F, Letsch J, Marion V, Dollfus H. Retinal dystrophy in Bardet-Biedl syndrome and related syndromic ciliopathies. Prog Retin Eye Res. 2011;30:258–74. doi: 10.1016/j.preteyeres.2011.03.001. [DOI] [PubMed] [Google Scholar]