

Abstract
Estrogens are known to play a role in modulating metabolic processes within the body. The Aromatase knockout (ArKO) mice have been shown to harbor factors of Metabolic syndrome with central adiposity, hyperinsulinemia and male-specific hepatic steatosis. To determine the effects of estrogen ablation and subsequent replacement in males on whole body glucose metabolism, three- and six-month-old male ArKO mice were subjected to whole body glucose, insulin and pyruvate tolerance tests and analyzed for ensuing metabolic changes in liver, adipose tissue, and skeletal muscle. Estrogen-deficient male ArKO mice showed increased gonadal adiposity which was significantly reduced upon 17β-estradiol (E2) treatment. Concurrently, elevated ArKO serum leptin levels were significantly reduced upon E2 treatment and lowered serum adiponectin levels were restored to wild type levels. Three-month-old male ArKO mice were hyperglycemic, and both glucose and pyruvate intolerant. These phenotypes continued through to 6 months of age, highlighting a loss of glycemic control. ArKO livers displayed changes in gluconeogenic enzyme expression, and in insulin signaling pathways upon E2 treatment. Liver triglycerides were increased in the ArKO males only after 6 months of age, which could be reversed by E2 treatment. No differences were observed in insulin-stimulated ex vivo muscle glucose uptake nor changes in ArKO adipose tissue and muscle insulin signaling pathways. Therefore, we conclude that male ArKO mice develop hepatic glucose intolerance by the age of 3 months which precedes the sex-specific development of hepatic steatosis. This can be reversed upon the administration of exogenous E2.
Introduction
The vital role of estrogen balance in male metabolic regulation has been highlighted as both men with aromatase deficiency and aromatase knockout (ArKO) mice develop metabolic syndrome (MetS) [1]–[5]. The ArKO mouse, with targeted deletion of exon 9 of the Cyp19 gene, is an estrogen-deficiency due to its inability to convert androgens into estrogens. The ArKO mouse presents with some of the key factors of MetS - central obesity, hypertriglyceridemia, hyperglycemia, hypo-HDL-cholesterolemia and male-only-hepatic steatosis [5].
When adipose tissue exceeds its storage capacity, as is witnessed in obesity, other peripheral tissues have the potential to begin to increase storage of excessive non-esterified fatty acids (NEFA) [6]. Increased plasma NEFA inhibit glucose utilization, stimulate gluconeogenesis [7], [8] and increase inflammation. Ectopic lipid accumulation in the liver (hepatic steatosis) has been proposed as a link between the development of obesity and insulin resistance [9].
A key element in maintaining glucose homeostasis is the control of liver gluconeogenesis by insulin and glucagon [10]. Gluconeogenesis is reliant on two key enzymes - glucose-6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK) [11]. In states of hepatic insulin resistance, these enzymes are not suppressed by insulin and result in elevated endogenous glucose production which contributes to hyperglycemia. Sexually dimorphic differences in the prevalence of hepatic steatosis exist, with significantly higher rates displayed in boys (51.2%) than age matched girls (12.2%) [12] and in white men (42%) versus matched women (24%) [13]. Fat accumulation in the liver in both genders has been associated with insulin resistance and visceral depot size, but only males showed a direct relationship with the steroid hormone profile (free androgen and estrogen index) [12] thereby suggesting that hepatic steatosis may be regulated by sex hormones.
In agreement with human data, we have previously demonstrated that the male ArKO mice suffer from hepatic steatosis which can be rectified upon estrogens treatment [14], [15]. However, the mechanisms behind this have not been studied, emphasizing the need for further investigation into the MetS phenotype of the ArKO mouse.
In this study we provide direct evidence that using the ArKO mouse model, which has no aromatase and hence essentially zero levels of estrogens, allowed us to investigate the effects of exogenous estrogens on the management of glucose metabolism without the interference of endogenous estrogen production and determined that estrogen deficiency leads to hepatic glucose intolerance which precedes the development of male-specific hepatic steatosis.
Materials and Methods
All efforts were made to minimize animal suffering and procedures were approved by the Monash Medical Centre Animal Ethics Committee (Permit Number: MMCB2008/08)
Mice
Aromatase Knockout (ArKO) mice (C57Black6 X J129) were generated by disruption of the Cyp19 gene [16]. Homozygous null or wild-type (WT) offspring were bred by crossing heterozygous ArKO and were genotyped by PCR [17]. Mice were housed in groups under pathogenic free conditions, fed a soy-free mouse chow (Glen Forest Stock feeders, Perth, Australia) and water ad libitum as previously described [17].
Treatment and tissue collection
Estrogen and placebo treatment
Male mice of both ArKO and WT at 3 and 6 months of age were used in these studies, groups included were: untreated WT, untreated ArKO and ArKO implanted 17β-estradiol pellet (E2). Three month-old ArKO mice were implanted with E2 pellet (E2) (0.05 mg in 21 days i.e. 2.5 µg/day, Innovative Research of America, Toledo, OH, USA) for 3 weeks. Six month-old ArKO mice were implanted with E2 pellet (0.15 mg in 60 days, i.e. 2.5 µg/day; Innovative Research of America, Toledo, OH, USA) 6 weeks. In preliminary studies no difference in body mass or glucose tolerance were detected between untreated and placebo pellet (saline 60 day slow release, Innovative Research of America, Toledo, OH, USA) treated six month-old ArKO male mice (Table S1 and Figure S1). A total number of 5–15 mice were used per group and age, details specified on related graphs. After treatment, mice were killed using a lethal dose of anesthetic (Ketamine (300 mg/kg)/Xylazine (30 mg/kg)). Blood was collected by cardiac puncture and serum was separated, and stored at −20°C. Adipose, liver and muscle tissues were removed, weighed, snap frozen and stored at −80°C.
Insulin stimulated tissue collection
After overnight fasting, mice were anaesthetized (6 mg pentobarbital sodium/100 g body weight; Sigma, St Louis USA), and injected with a bolus of insulin (150 U/kg, Actrapid; Novo Nordisk, Bagsvaerd, Denmark) via the descending branch of the inferior vena cava. After 5 min, skeletal muscles (gastrocnemius, extensor digitorum longus, tibialis anterior and soleus), liver and gonadal fat were excised and rapidly frozen in liquid nitrogen for storage at −80°C. Fasting blood samples were collected, serum separated and stored at −20°C.
Tolerance tests
All mice cohorts were subjected to glucose, insulin and pyruvate tolerance tests, with approximately five days of recovery between each test; glucose tolerance test (GTT – 1 g glucose/kg of body weight, after 8 h of fasting), insulin tolerance test (ITT – 0.5U insulin/kg of body weight, after 8 h of fasting) and pyruvate tolerance test (PTT- 1 g pyruvate/kg of body weight, after ∼16–20 h of fasting). Blood samples were obtained by tail bleeding and analyzed for glucose content (AccuChek Performer, Roche, Mannheim, Germany) immediately before and at 20, 40, 60, 90 and 120 min after an intraperitoneal injection.
Ex-vivo muscle glucose uptake assay
Soleus and extensor digitorum longus (EDL) muscles from WT and ArKO mice were dissected from anesthetized 6 month-old male mice and ex vivo glucose uptake was measured with or without insulin (2.8 nM Actrapid; Novo Nordisk, Bagsvaerd, Denmark) as previously described [18]. Muscle tracer uptake was measured by liquid scintillation counting (Tri-Carb 2000, Packard Instrument) of the muscle lysate/supernatant which was prepared by homogenization of the tissue sample in ice-cold protein lysis buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 10 mM NaF, 1 mM sodium pyrophosphate, 0.5 mM EDTA, 250 mM sucrose, 1 mM DTT, 1 mM Na3VO4, 1% Triton X-100 and 1 Roche protease inhibitor tablet per 50 ml) followed by centrifugation at 4°C, 13,000 rpm for 30 min and collected.
Insulin assay
Insulin was measured by ELISA (#EZRMI-13K, Linco Research, St. Charles, MO, USA) following manufacturers' protocol. Briefly, a 96-well microtiter plate pre-coated with monoclonal mouse anti-rat insulin antibody was incubated with 10 µl of control, standards or sample serum plus a biotinylated anti-insulin antibody horseradish peroxidase and a light sensitive 3,3′,5,5′ tetramethylbenzidine substrate allowing enzyme activity to be measured at 450 nm on the Envision Plate reader v 1.09 (Perkin Elmer, Waltham, MA, USA). HOMA-IR was calculated using the following formula: HOMA-IR = fasting glucose (mmol/l) × fasting insulin (µU/l)/22.5.
Protein isolation
Samples were homogenised in ice-cold protein lysis buffer (2.5 mM HEPES, 68.5 mM NaCl, 0.5 mM MgCl2, 0.5 mM CaCl2, 5 mM NaF, 1 mM EDTA, 5 mM Na pyrophosphate, 1 mM NaVO4, 0.5% Nonidet P40 and 5% glycerol) containing protease inhibitors (Complete Mini; Roche, Mannheim, Germany), then incubated on ice for 45 min. Samples were then centrifuged for 15 min at 14,000 g before assay of supernatants for protein content by the bicinchoninic acid (BCA) method (Pierce Biotechnology, Rockford, IL, USA)
Western analysis
Isolated protein (50 µg) together with 1X sample loading buffer (0.125 M Tris-Cl, 4% SDS, 20% v/v Glycerol, 0.2 M DTT, 0.02% Bromophenol Blue, pH 6.8) was boiled for 5 mins at 95°C before SDS-PAGE analysis (10% polyacrylamide resolving gel and 4% stacking gel). Samples were transferred to a nitrocellulose membrane (Amersham™ GE Healthcare, Piscataway USA). Primary antibodies diluted in 1% BSA in Tris Buffered Saline with 1% Tween were incubated overnight at 4°C (mouse anti-Phosphor-Akt (Ser473), rabbit anti-Akt, rabbit anti-phosphor-AMPK (Thr172), rabbit anti-AMP alpha (Cell Signaling Technologies, Beverly MA, USA). Appropriate Alexa Fluor secondary antibodies were incubated at room temperature for 1 h. Protein band intensities were quantified using the Odyssey infrared imaging system (Licor Biosciences, Lincoln, NE, USA). All protein quantifications were normalized to housekeeping protein β tubulin (mouse- anti- β tubulin; Millipore, California USA).
RNA isolation and real-time PCR
Total RNA was extracted from WT, ArKO and ArKO + E2 mouse frozen liver tissue using Ultraspec RNA isolation system (Fisher Biotec, WA, Australia) following manufacturer's protocol. All samples were DNase I treated (DNAfree™, Ambion, Foster City, CA, USA) and concentrations were quantified using the NanoDrop®1000 (Thermo, Willmington, USA). Total RNA (1 µg) was reverse transcribed (Roche, Mannheim, Germany) and amplified by real-time PCR using a SYBR ‘Master Mix’ (DMSO (Sigma-Aldrich, USA) 10X Gold PCR Buffer, AmpliTaqGold (Applied Biosystems, California, USA), 1 M Mg(oAC)2 (Sigma-Aldrich, USA), 100 mM dACT, dCCT, dGCT and dTCT (Bioline, Australia) 1 µg/µl 6-ROX and 10,000X con SYBR (Invitrogen, USA)) in the ABI7900H (Applied Biosystems, California, USA) with specific oligonucleotide pairs (see Table 1). Real-time PCR data were calculated using ΔΔCT method of analysis. All samples were normalized to housekeeping gene cyclophilin transcripts levels.
Table 1. Real-time PCR primers.
| Transcript | Oligonucleotide pairs (5′-3′) | Amplicon Size (bp) |
| Glucose-6-phosphatase (G6Pase) | For: TCTTGTGGTTGGGATACTGG | 110 |
| Rev: AGCAATGCCTGACAAGACTC | ||
| Phosphophenolpyruvate carboxykinase (PEPCK) | For: GTGTCCCCCTTGTCTATGAA | 163 |
| Rev: GGTATTTGCCGAAGTTGTAG | ||
| Insulin Receptor Substrate 1 (IRS1) | For;CAGCGATGGCGAAGGCACCA | 272 |
| Rev;GCGAACCGGACACGGAAGCA | ||
| Insulin Receptor Substrate 2 (IRS2) | For: GCGGCCTCATCTTCTTCACT | 129 |
| Rev: AACTGAAGTCCAGGTTCATATA | ||
| Glycogen Substrate Kinase 3 beta (GSK3β) | For: ACCAATATTTCCTGGGGACA | 130 |
| Rev: GTACCTTGATTTGAGGGAAT | ||
| Fatty Acid Synthase (Fasn) | For: CACAGATGATGACAGGAGATGGA | 205 |
| Rev: TCGGAGTGAGGCTGGGTTGATA | ||
| Stearoyl CoA desaturase 1 (Scd-1) | For: CATCATTCTCATGGTCCTGCT | 236 |
| Rev: CCCAGTCGTACACGTCATT | ||
| Acetyl-CoA Carboxylas (ACCα) | For: TGTTGGGGTTATTTCAGTGTTGC | 236 |
| Rev: TGTCCAGCCAGCCAG GTCG | ||
| Cyclophilin | For: CTTGGGCCGCGTCTCCTTC | 180 |
| Rev: TGCCGCCAGTGCCATTAT |
List of primer sequences (and expected amplicon sizes) used for Real-Time PCR.
Liver triglyceride (TG) content
Saponified extracts were prepared from frozen liver samples, and the triglyceride content was quantified by comparing to a glycerol standard curve, as previously described [19], [20]. Briefly, hepatic triglycerides (TG) were extracted from 100–300 mg liver tissues overnight at 55°C in ethanolic potassium hydroxide, and neutralized with water:ethanol (1∶1 vol/vol). Saponification proceeded with 1 M MgCl2 by vortexing and incubating at 4°C. Saponified liver extracts (the upper phase) were separated by centrifugation, and the glycerol content was quantified with Free Glycerol Reagent (Sigma-Aldrich, Missouri, USA), which generate a dye with an absorbance at 540 nm (measured using the Wallac 1420 Victor Plate Reader, LabX, Midland ON Canada). All blanks, glycerol standards and samples were quantified in duplicates.
Adipokine panels
Adipokine levels in mouse serum were analyzed using the adipokine LINCOplex Kit 96 well assay according to manufacturer's protocol (Mouse Serum single-plex adiponectin panel #MADPK-71-ADPN; Mouse Serum Adipokine kit #MADPK-71K – Leptin; Millipore, California USA). Briefly, standards, quality controls and serum samples were pipetted in duplicate in a 96-well microtiter filter assay plate and incubated overnight at 4°C with antibody immobilized beads. Mouse adipokine detection antibodies were added and incubated. Unhybridized antibodies were removed and Streptavidin-Phycoerythrine incubated. Luminex sheath fluid was added; plates were scanned and analyzed on the Luminex® 100 instrument and software (Luminex Corporation, Texas, USA).
Statistical analysis
All statistical analyses were performed using GraphPad Prism® version 5 for windows (GraphPad Software Inc). WT untreated, ArKO untreated and ArKO + E2 treatment groups were analyzed using a two-tailed Mann-Whitney test. Data are expressed as mean ± standard deviation (SD).
Results
Effects of estrogens on body and gonadal adipose tissue weights
There were no differences between male WT and ArKO mice in body weight at both ages (3 and 6 month-old; Table 2). Placebo treatment in ArKO mice did not induce any changes in body weight or gonadal adipose tissue weight (Table S1). However upon 17β-estradiol (E2) treatment, body weights of ArKO mice at 3 and 6 months were reduced (p<0.05 and p<0.0001 vs untreated KO respectively; Table 2), and beyond that of the WT at 6 months (p<0.01 vs WT). In a consistent pattern, gonadal adipose tissue weights were increased in the 3 and 6 month-old untreated ArKO compared to that of WT (p<0.01 and p<0.05 respectively), and again reduced upon E2 treatment (p<0.001 and p<0.0001 vs untreated KO respectively).
Table 2. Body and adipose weights, adipokines levels.
| WT | KO | KOE | |
| Body weight (g) | |||
| 3mth | 25.50±2.71 | 30.03±6.66 | 25.21±2.73# |
| 6mth | 36.71±6.14 | 37.12±3.99 | 29.45±3.72**### |
| Gonadal adipose (g) | |||
| 3 mth | 0.37±0.11 | 0.91±0.47** | 0.31±0.10### |
| 6 mth | 1.05±0.52 | 1.69±0.33* | 0.69±0.32### |
| Leptin (ng/ml) | |||
| 6 mth | 5.93±0.78 | 11.79±7.9* | 3.85±1.4*## |
| Adiponectin (µg/ml) | |||
| 6 mth | 9.87±1.04 | 7.38±1.53* | 9.61±3.55 |
Body and gonadal adipose tissue weights in grams (g) and serum leptin in micrograms per milliliter (µg/ml) and adiponectin levels in nanograms per milliliter (ng/ml) of 3 and 6 month-old male wild type (WT), aromatase knockout (KO) and 2.5 µg/day 17β-estradiol-treated KO (KOE). Data are expressed as mean ± SD; n = 7–8 per group,*p<0.05 and **p<0.01 versus WT mice. # p<0.05, ## p<0.01 ### p<0.001 versus KO.
Serum adiponectin and leptin levels
Six month-old ArKO mice had reduced levels of serum adiponectin (p<0.05; Table 2), which returned to WT levels upon E2 treatment. Conversely, serum leptin levels in the ArKO mice were higher than WT counterparts (p<0.03). E2 administration dramatically reduced serum leptin levels in ArKO mice to below untreated WT controls (p<0.01, Table 2).
The effects of estrogens on serum insulin and glucose levels
Fasted basal serum insulin and glucose levels of the 3 month-old male ArKO mice were elevated, (n/s and p<0.05 respectively) compared to WT counterparts, a difference which was improved by E2 (2.5 µg/day) treatment (p<0.05 and n/s, Figure 1a, b). HOMA-IR showed a trend for increase between WT and untreated ArKO (p = 0.051) but was significantly reduced in ArKO with E2 treatment (p<0.05). In 6 month-old animals, there were no differences between WT and ArKO untreated fasted basal serum insulin, glucose or HOMA-IR, additionally no changes were seen upon E2 treatment (2.5 µg/day) in the ArKO's (Figure 1a, b, c)
Figure 1. Serum glucose, insulin and HOMA-IR levels.

(a) Fasted and 20 min post glucose infused serum glucose (b) Fasted serum insulin and (c) HOMA-IR (fasted serum glucose (mmol/l) × fasted serum insulin (µU/l)/22.5) from 3 and 6 month-old fasted male wildtype (WT) and aromatase knockout (KO) and KO treated with 2.5 µg/day estrogen (KOE). Expression data from samples (n = shown on corresponding bar) per genotype are shown, and are presented from replicate analysis as the mean ± SD. *p<0.05 versus WT mice. # p<0.05 versus KO.
Whole body glucose, pyruvate and insulin tolerance in the ArKO mice
Whole body tolerance tests were performed on WT, ArKO and E2 administrated mice at 3 and 6 months of age. No differences were seen in glucose tolerance between untreated and six-week placebo treated ArKO mice (Figure S1).
Three month-old animals
Three month-old ArKO mice exhibited lower glucose and pyruvate tolerance compared to WT littermates (p<0.05 and p<0.01 respectively, Area Under Curve (AUC), Figure 2a, b), highlighting a loss of glycemic control. After 3 weeks of E2 treatment (2.5 µg/day), glucose tolerance and insulin sensitivity were improved (p<0.01 and p<0.01 respectively, AUC) as compared to the untreated ArKO. Further, insulin sensitivity with E2 treatment in the ArKO lowered below that of the WT littermates (p<0.05; Figure 2a, c).
Figure 2. Three month-old male whole body glucose, insulin and pyruvate tolerance.

Whole body tolerance tests were completed on fasted three month-old male wildtype (WT) and aromatase knockout (KO) and 2.5 µg/day 17β-estradiol-treated KO (KOE) (a) glucose tolerance test and corresponding area under curve; (b) insulin tolerance test and corresponding area under curve; (c) pyruvate tolerance test and corresponding area under curve Data are presented from replicate analysis (n = shown on corresponding bar) as the mean ± SD. *p<0.05, **p<0.01, ***p<0.001, versus expression in age-matched WT samples and #p<0.05 and ##p<0.01 versus KO samples.
Six month-old animals
Six month-old ArKO mice displayed impaired glucose tolerance (p<0.01, AUC) and signs of pyruvate intolerance (p = 0.052, AUC) as compared to WT littermates (Figure 3a, b). The administration of E2 (2.5 µg/day) for 6 weeks to ArKO mice markedly improved glucose tolerance (p<0.05, AUC) and insulin sensitivity (p<0.05, AUC) As seen in the 3-month old mice, E2 treatment in the ArKO mouse was able to reduce insulin sensitivity below levels observed in WT littermates (Figure 3a, c).
Figure 3. Six month-old male whole body glucose, insulin and pyruvate tolerance.

Whole body tolerance tests were completed on fasted 6 month-old male wildtype (WT), aromatase knockout (KO) and 2.5 µg/kg 17β-estradiol-treated KO (KOE). (a) Glucose tolerance test and corresponding area under curve; (b) insulin tolerance test and corresponding area under curve; (c) pyruvate tolerance test and corresponding area under curve. Data are presented from replicate analysis (n = shown on corresponding bar) as the mean ± SD. *p<0.05, **p<0.01, versus expression in age-matched WT samples and #p<0.05 and ##p<0.01 versus KO samples
Liver triglycerides in the male ArKO
In 3 month old animals, there were no differences in liver triglycerides levels between WT, ArKO and ArKO+E2 mice. In 6 month old animals, liver triglycerides were substantially increased in ArKO compared with WT animals (p<0.01) and E2 treatment reduced triglycerides to below WT levels (p<0.001; Figure 4). These 6 month-old ArKO mice also had significantly (p<0.001) higher triglyceride levels compare to the 3 month-old ArKO male mice (Figure 4).
Figure 4. Liver Triglyceride levels.
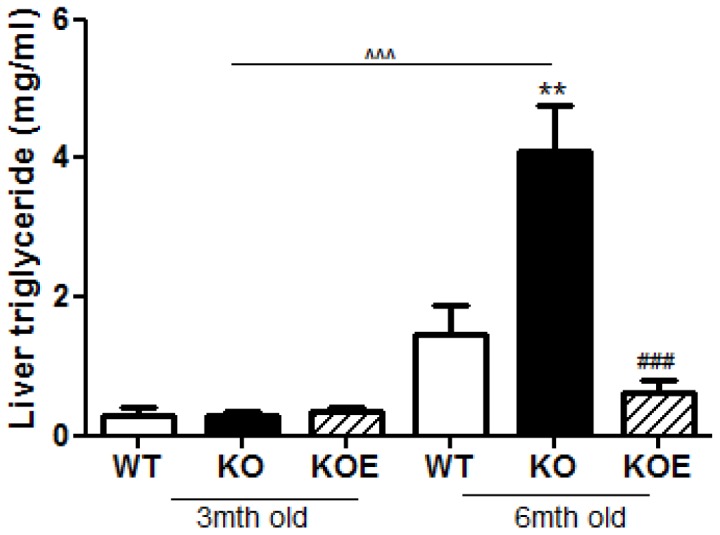
Liver triglyceride assay were performed on 3 and 6 month-old fasted male wildtype (WT), aromatase knockout (KO) and 17β-estradiol-treated KO (KOE) mice. Expression data from 7–8 samples per genotype are shown following and presented from replicate analysis as the mean ± SD. **p<0.01, versus expression in age-matched WT samples and ###p<0.001 versus age-matched KO samples and ∧∧p<0.01, ∧∧∧p<0.001, versus 3mth old KO samples.
Ex-vivo muscle glucose uptake
After observing signs of glucose dysregulation in the untreated ArKO male mice, we investigated ex vivo insulin stimulated muscle glucose uptake in 6 month-old mice. Surprisingly, there were no differences in insulin-stimulated glucose uptake in EDL or soleus muscle between the untreated ArKO mice and their WT littermates (Figure S2a, b). These data suggest that whole-body glucose intolerance in ArKO mice is not due to defects in skeletal muscle insulin-sensitivity.
Gonadal adipose tissue, liver and muscle protein phosphorylation
To determine the potential mechanism responsible for differences in glucose intolerance between WT and ArKO mice, the expression levels and phosphorylation of Akt and AMPK, which plays a role in insulin mediated glucose uptake, in gonadal adipose tissue, skeletal muscle and liver were analyzed. In 6 month-old untreated ArKO mice, there were no changes in the phosphorylation of Akt or AMPK, normalized to total protein, in investigated adipose, muscle and liver tissues compared to WT mice (Figure 5a, b, c). However, with 6 weeks of E2 treatment (2.5 µg/day), there was a clear increase in liver Akt phosphorylation and decrease in AMPK phosphorylation compared to both the untreated ArKO (p<0.001 and p<0.001 respectively) and WT counterparts (p<0.01 and p<0.01 respectively; Figure 5c).
Figure 5. Western blot analyses of insulin stimulated and basal tissue.

Protein phosphorylation analyses of Akt and AMPK levels were performed on protein extracted from insulin stimulated (a) gonadal adipose tissue, (b) gastrocnemius muscle, (c) liver in insulin stimulated liver of 6 month-old male wildtype (WT) and aromatase knockout (KO) and 2.5 µg/day estrogen-replaced KO (KOE) mice. Expression data from 6–8 samples per genotype are shown, and presented from replicate analysis as the mean ± SD. *p<0.05, **p<0.01, ***p<0.001 versus expression in age-matched WT samples and #p<0.05, ###p<0.001 versus KO samples.
Insulin signaling pathway mRNA expression levels in the liver
After witnessing significant changes in the liver phenotype but not gonadal adipose tissue or skeletal muscle, the regulation of hepatic glucose homeostasis was investigated.
In the 3 month-old mice both GSK3 β and IRS1 mRNA levels were significantly increased (p<0.01 and p<0.01, respectively) in the ArKO compared to WT counterparts. Upon E2 treatment GSK3β mRNA expression was subsequently decreased (p<0.001 vs KO and p<0.01 vs WT), whereas IRS1 mRNA levels remained elevated with no improvement compared to untreated ArKO. Estrogen treatment was also able to significantly reduce G6Pase and PEPCK mRNA levels (p<0.001 and p<0.05 respectively) in the ArKO mouse. There was no difference in the expression levels of IRS2 (Figure 6a).
Figure 6. Real-time PCR analysis of gluconeogenesis mRNA expression in the liver.
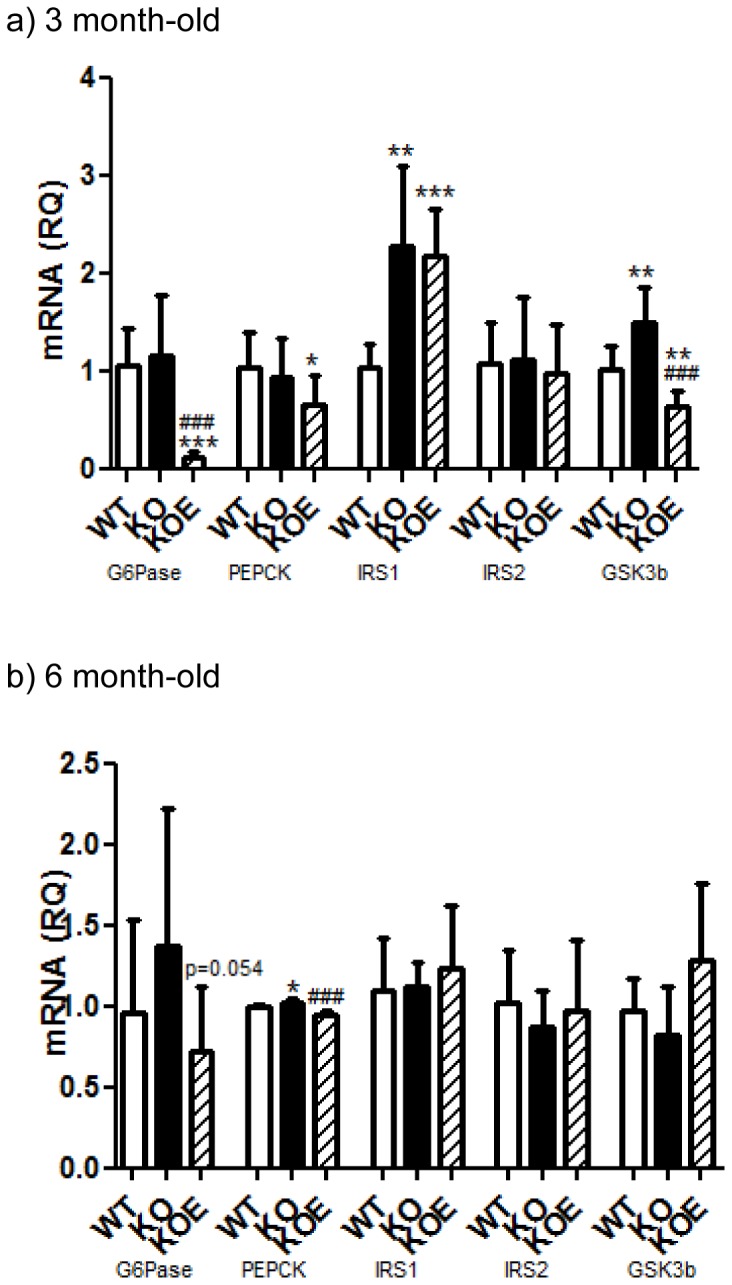
Real-time-PCR analyses of G6Pase, PEPCK, IRS1, IRS2 and GSK3β mRNA expression were performed on cDNA derived from total RNA prepared from fasted liver tissue of (a) 3 and (b) 6 month-old male wildtype (WT, n = 8), aromatase knockout (KO, n = 8) and 2.5 µg/day 17β-estradiol-treated KO (KOE, n = 7) mice. Expression data from 7–8 samples per genotype are shown following normalization for cyclophilin mRNA expression, and presented from replicate analysis as the mean ± SD. *p<0.05,**p<0.01 and ***p<0.001 versus expression in age-matched WT samples and ##p<0.01, ###p<0.05 versus KO samples.
Consistent with differences in pyruvate tolerance, fasted 6 month-old ArKO male mice liver transcript levels of PEPCK (Figure 6b) were significantly increased (p<0.05) compared to WT. Upon E2 treatment, G6Pase and PEPCK gene expressions are both decreased (p = 0.054 and p<0.001 respectively) compared to untreated ArKO. However, neither ArKO untreated nor E2 treatment had any effects on GSK3β, IRS1 and IRS2 expression levels (Figure 6b).
Lipid profile mRNA expression levels in the liver
Fasn, and Scd-1 mRNA levels were increased in the ArKO liver compare to the WT counterparts in the 3month-old (p<0.05 and p<0.01 respectively) and 6month-old (n/s and p<0.05 respectively) mice. Estrogen treatment was able to reverse these increases in all cases. ACCα transcript levels were significantly increased (p<0.05) in the 6 month-old ArKO only and not 3 month-old ArKO. However, both 6 and 3 month-old mice had reduced ACCα mRNA when administered E2 (p<0.001vs both WT and KO respectively) (Figure 7a, b).
Figure 7. Real-time PCR analysis of mRNA lipid profile in the liver.
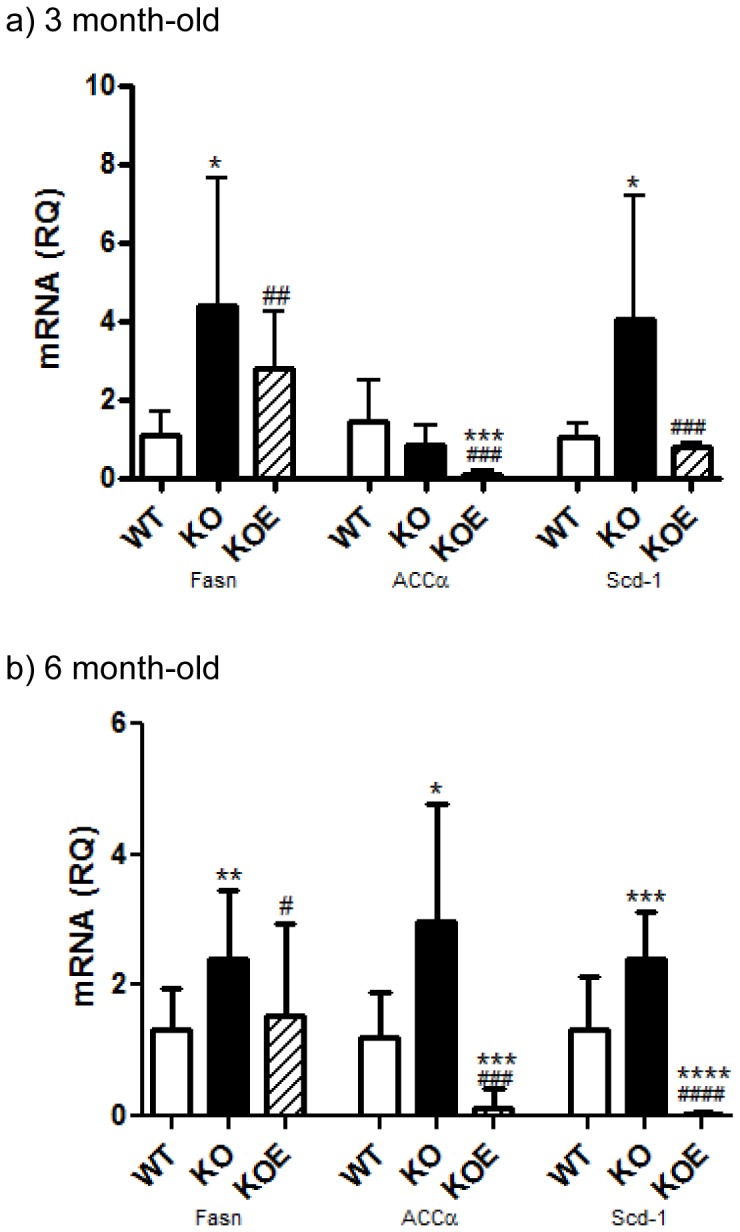
Real-time-PCR analyses of Fasn, ACCα and Scd-1 mRNA expression were performed on cDNA derived from total RNA prepared from fasted liver tissue of (a) 3 and (b) 6 month-old male wildtype (WT, n = 8), aromatase knockout (KO, n = 8) and 2.5 µg/day 17β-estradiol-treated KO (KOE, n = 7) mice. Expression data from 7–8 samples per genotype are shown following normalization for cyclophilin mRNA expression, and presented from replicate analysis as the mean ± SD. *p<0.05, **p<0.01 and **p<0.001, versus expression in age-matched WT samples and ##p<0.01, ###p<0.001 versus KO samples.
Discussion
This study presents data on whole body glucose tolerance in the estrogen deficient male ArKO which is consistent with other studies [21], and provides additional and new insight into the specific actions of key peripheral tissue (liver, adipose and muscle) involved in insulin signalling. Male ArKO mice displayed glucose intolerance at 3-and 6-months of age which could be primarily attributed to elevated hepatic gluconeogenesis as indicated by the results of pyruvate tolerance tests. In agreement with this conclusion, livers from ArKO mice had impaired insulin-stimulated Akt phosphorylation and increased expression of gluconeogenic and lipid biosynthesis enzymes. In contrast, in the absence of estrogens, adipose tissue and muscle insulin sensitivity were unchanged in terms of insulin stimulated Akt and AMPK phosphorylation and ex-vivo glucose uptake, despite the expected changes in adipokine levels (lowered adiponectin and elevated leptin levels) with increased body weight and fat deposits in ArKO mice. Interestingly, male ArKO mice did not display a significant increase in liver triglycerides until 6 months of age, thus demonstrating that hepatic glucose intolerance precedes hepatic steatosis in these mice. Importantly, we demonstrated that E2 replacement can improve most aspects of this phenotype.
The intricate ‘cause and effect’ relationship between hepatic steatosis and insulin resistance remains a subject of controversy and research. It has been reported that men have higher rates of insulin resistance which may be related to greater visceral and hepatic adiposity in conjunction with the lower levels of estrogens [22]. The hepatic steatosis phenotype in ArKO mice has also been partially attributed to increases in de novo lipogenesis [14] and impaired β-oxidation [23], which can be ameliorated upon estrogen treatment [14]. Accumulation of lipids within the liver may lead to reduced insulin signalling, impaired GLUT translocation and inflammation and is independently linked to type II diabetes [24]–[28]. However, it is also suggested that hepatic steatosis is secondary to an insulin resistant state [9] due to a lack of insulin dependent suppression of lipid synthesis. Our current detection of glucose intolerance without concomitant signs of hepatic steatosis in 3 month-old ArKO mice, followed by hepatic steatosis presentation at 6 months of age, supports this hypothesis.
Our investigation of the lipid profile in the male ArKO mouse liver revealed that mRNA expression of genes involved in fatty acid (FA) synthesis; Fasn (an enzyme involved in the synthesis of palmitate) and Scd-1 (a catalyst of palmitate to unsaturated FA) were already increased by 3 months of age continuing into 6 months. However, ACCα (which provides the malonyl-CoA substrate for FA synthesis) mRNA levels were significantly increased in the 6 month old animals but not in the 3 month old animals. Thus, the resultant translational and physiological effects of increased liver triglycerides and hepatic steatosis do not occur at 3 months of age but at a later age.
Other models of reduced estrogen activity such as the estrogen receptor α knockout (ERαKO) mouse have also confirmed the role of estrogens in disrupted hepatic glucose homeostasis [29], [30], although these experiments have only been published in female mice to date [31], [32]. Furthermore, estrogen administration to ob/ob mice reduced hepatic insulin resistance and decreased expression of genes involved in hepatic lipid biosynthesis [33] and improved liver dysfunction parameters [34].
In ArKO mice, estrogens' role in hepatic glucose intolerance may be explained by its ability to significantly reduce key hepatic insulin signaling genes G6Pase and PEPCK upon E2 administration, which we observed by 3 months of age and continued through to 6 months of age. These findings support previous studies which demonstrate direct inhibitory effects of E2 on PEPCK expression via PGC1α (peroxisome proliferator activated receptor gamma co-activator 1 alpha) [35], [36] and studies showing estrogens administration to ob/ob mice reduces G6Pase expression [37]. GSK3β was elevated in the ArKO compared to WT and reduced upon E2 treatment at 3months, but not evident at 6 months of age. GSK-3β impairs the ability of insulin to activate glucose disposal and over-expression is associated with insulin resistance. Additionally, the increases seen in IRS1 at 3 months of age (not seen at 6 months) and the lack of recovery upon estrogen treatment at this age also correlated with the lack of improvement in pyruvate tolerance. Furthermore, Kim et.al recently defined the timed dosing effects of estrogen and concluded that short term E2 treatment improved glucose homeostasis via hepatic gluconeogenesis rather than insulin sensitivity in the muscles [38].
Glucose uptake and production is regulated by the insulin signaling pathway of which Akt plays a fundamental role. Akt is known to be expressed in insulin sensitive tissue and Akt2 knockout mice display severe impairment of insulin stimulated whole body glucose disposal [39]. Increased Akt phosphorylation with improved glucose tolerance was observed in the liver of the E2 treated male ArKO mice. Recent studies highlighting specific E2 effects on Akt demonstrated that in ochidectomized male mice with resultant insulin resistance phenotype, showed E2 alone could increase Akt phosphorylation without effecting other parameters whereas combined T+E2 replacement returned whole body insulin sensitivity to normal levels [40]. Akt is also involved in glycogen synthesis, an insulin stimulated system of glucose storage through the phosphorylation and inactivation of GSK-3β. A vital constituent in both cellular and whole-body energy balance preservation is the adenosine monophosphate-activated protein kinase (AMPK). AMPK also plays a role in increasing glucose uptake and β-oxidation in peripheral tissues, as well as decreasing liver glucose production [41]. Unexpectedly, a steep decrease in levels of phosphorylated AMPK was also present in our estrogen-treated animals. Previous studies have identified that insulin stimulated Akt in the heart can induce Ser485/491 phosphorylation of AMPK which inhibits AMPK Thr172 phosphorylation [42], [43]. This may explain the decrease in AMPK Thr172 phosphorylation seen in the insulin stimulated male ArKO mice treated with E2. In summary, our data indicate that the hepatic steatosis in the 6 month old mice is not a result of decreased phosphor-AMPK and the E2 recovery of hepatic steatosis is not mediated through activation of AMPK.
The ArKO mouse model allowed us to observe the effects of estrogen absence on the relationship between the progression and development of glucose intolerance and hepatic steatosis. Additionally, as the ArKO mouse remains estrogen responsive this allows the effects of administered exogenous E2 to be investigated. The absence of estrogens in the male ArKO mice leads to hepatic glucose intolerance from 3 to 6 months of age with consistent improvement upon E2 treatment. This highlights the potential of combined roles of both estrogens and glucose homeostasis in the development of hepatic steatosis in the male ArKO mice.
Conclusion
This study shows that the absence of aromatase and hence ablation of estrogens causes defects in hepatic glucose homeostasis at a young age preceding the development of hepatic steatosis which is only witnessed in aged male ArKO mice. Exogenous E2 treatment is able to improve both glucose homeostasis and the pursuant hepatic steatosis phenotype.
Supporting Information
Untreated vs. placebo treated ArKO mice glucose tolerance test. Whole body glucose tolerance tests were completed on fasted six month-old male aromatase knockout (KO) and 2.5 µg/day 17β-estradiol-treated KO (KOE) (a) glucose tolerance test and (b) corresponding area under curve; Data are presented from replicate analysis (n = shown on corresponding bar) as the mean ± SD.
(PPT)
Ex-vivo muscle glucose uptake. Ex-vivo muscle glucose uptake assays were performed on 6 month-old fasted male wildtype (WT), or aromatase knockout (KO) mice, treated with insulin or without (basal). Tracer uptake was measured in dissected (a) soleus and (b) extensor digitorum longus (EDL) muscles. Expression data from n = 8 samples per genotype as the mean ± SD. *p<0.05, **p<0.01 and ***p<0.01.
(PPT)
Body and gonadal tissue weight of untreated versus placebo treated KO mice. Body and gonadal adipose tissue weights in grams (g) of 6 month-old male aromatase knockout untreated (KO) and placebo-treated KO (KOP). Data are expressed as mean ± SD; n = 3–8 per group.
(PPT)
Funding Statement
This research was supported by Australian National Health and Medical Research Council project grants ID 338510, 494813, and 628553 and program grant 441100. MLS was supported by Monash Postgraduate Scholarship; WCB was supported by a Fred P Archer Fellowship. The authors acknowledge the support of the Victorian Government through the Operational Infrastructure Scheme. SQT was supported by the Cancer Council of Victoria Postdoctoral Cancer Research Fellowship and PHI audit #13–33. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Maffei L, Murata Y, Rochira V, Tubert G, Aranda C, et al. (2004) Dysmetabolic Syndrome in a Man with a Novel Mutation of the Aromatase Gene: Effects of Testosterone, Alendronate, and Estradiol Treatment. J Clin Endocrinol Metab 89: 61–70. [DOI] [PubMed] [Google Scholar]
- 2. Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K (1995) Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab 80: 3689–3698. [DOI] [PubMed] [Google Scholar]
- 3. Carani C, Qin K, Simoni M, Faustini-Fustini M, Serpente S, et al. (1997) Effect of testosterone and estradiol in a man with aromatase deficiency. N Engl J Med 337: 91–95. [DOI] [PubMed] [Google Scholar]
- 4. Herrmann BL, Janssen OE, Hahn S, Broecker-Preuss M, Mann K (2005) Effects of estrogen replacement therapy on bone and glucose metabolism in a male with congenital aromatase deficiency. Horm Metab Res 37: 178–183. [DOI] [PubMed] [Google Scholar]
- 5. Jones ME, Boon WC, Proietto J, Simpson ER (2006) Of mice and men: the evolving phenotype of aromatase deficiency. Trends Endocrinol Metab 17: 55–64. [DOI] [PubMed] [Google Scholar]
- 6.Galic S, Oakhill JS, Steinberg GR (2009) Adipose tissue as an endocrine organ. Mol Cell Endocrinol. [DOI] [PubMed]
- 7. Muller G, Jordan H, Jung C, Kleine H, Petry S (2003) Analysis of lipolysis in adipocytes using a fluorescent fatty acid derivative. biochimie 85: 1245–1256. [DOI] [PubMed] [Google Scholar]
- 8. Frayn KN (2002) Adipose tissue as a buffer for daily lipid flux. Diabetologia 45: 1201–1210. [DOI] [PubMed] [Google Scholar]
- 9. Lonardo A, Lombardini S, Ricchi M, Scaglioni F, Loria P (2005) Review article: hepatic steatosis and insulin resistance. Aliment Pharmacol Ther 22 Suppl 264–70. [DOI] [PubMed] [Google Scholar]
- 10. Nuttall FQ, Ngo A, Gannon MC (2008) Regulation of hepatic glucose production and the role of gluconeogenesis in humans: is the rate of gluconeogenesis constant? Diabetes Metab Res Rev 24: 438–458. [DOI] [PubMed] [Google Scholar]
- 11. Barthel A, Schmoll D (2003) Novel concepts in insulin regulation of hepatic gluconeogenesis. American Journal of Physiology - Endocrinology & Metabolism 285: E685–692. [DOI] [PubMed] [Google Scholar]
- 12. Denzer C, Thiere D, Muche R, Koenig W, Mayer H, et al. (2009) Gender-specific prevalences of fatty liver in obese children and adolescents: roles of body fat distribution, sex steroids, and insulin resistance. J Clin Endocrinol Metab 94: 3872–3881. [DOI] [PubMed] [Google Scholar]
- 13. Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, et al. (2004) Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 40: 1387–1395. [DOI] [PubMed] [Google Scholar]
- 14. Hewitt KN, Pratis K, Jones ME, Simpson ER (2004) Estrogen replacement reverses the hepatic steatosis phenotype in the male aromatase knockout mouse. Endocrinology 145: 1842–1848. [DOI] [PubMed] [Google Scholar]
- 15. Jones ME, Thorburn AW, Britt KL, Hewitt KN, Misso ML, et al. (2001) Aromatase-deficient (ArKO) mice accumulate excess adipose tissue. J Steroid Biochem Mol Biol 79: 3–9. [DOI] [PubMed] [Google Scholar]
- 16. Fisher C, Graves K, Parlow A, Simpson E (1998) Characterization of mice deficient in aromatase (ArKO) because of targeted disruption of the cyp19 gene. Proc Natl Acad Sci U S A 95: 6965–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Robertson KM, O'Donnell L, Jones ME, Meachem SJ, Boon WC, et al. (1999) Impairment of spermatogenesis in mice lacking a functional aromatase (cyp 19) gene. Proc Natl Acad Sci U S A 96: 7986–7991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jorgensen SB, Honeyman J, Oakhill JS, Fazakerley D, Stockli J, et al. (2009) Oligomeric resistin impairs insulin and AICAR-stimulated glucose uptake in mouse skeletal muscle by inhibiting GLUT4 translocation. Am J Physiol Endocrinol Metab 297: E57–66. [DOI] [PubMed] [Google Scholar]
- 19. Chow JD, Jones ME, Prelle K, Simpson ER, Boon WC (2011) A selective estrogen receptor alpha agonist ameliorates hepatic steatosis in the male aromatase knockout mouse. J Endocrinol 210: 323–334. [DOI] [PubMed] [Google Scholar]
- 20. Norris AW, Chen L, Fisher SJ, Szanto I, Ristow M, et al. (2003) Muscle-specific PPAR{gamma}-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J Clin Invest 112: 608–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takeda K, Toda K, Saibara T, Nakagawa M, Saika K, et al. (2003) Progressive development of insulin resistance phenotype in male mice with complete aromatase (CYP19) deficiency. J Endocrinol 176: 237–246. [DOI] [PubMed] [Google Scholar]
- 22. Geer EB, Shen W (2009) Gender differences in insulin resistance, body composition, and energy balance. Gend Med 6 Suppl 160–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nemoto Y, Toda K, Ono M, Fujikawa-Adachi K, Saibara T, et al. (2000) Altered expression of fatty acid-metabolizing enzymes in aromatase-deficient mice. J Clin Invest 105: 1819–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boren J, Taskinen MR, Olofsson SO, Levin M (2013) Ectopic lipid storage and insulin resistance: a harmful relationship. J Intern Med 274: 25–40. [DOI] [PubMed] [Google Scholar]
- 25. Yki-Jarvinen H (2010) Liver fat in the pathogenesis of insulin resistance and type 2 diabetes. Dig Dis 28: 203–209. [DOI] [PubMed] [Google Scholar]
- 26.Ruhl CE, Everhart JE (2004) Epidemiology of nonalcoholic fatty liver. Clin Liver Dis 8: : 501–519, vii. [DOI] [PubMed] [Google Scholar]
- 27.Harrison SA, Neuschwander-Tetri BA (2004) Pharmacologic management of nonalcoholic fatty liver disease. Clin Liver Dis 8: : 715–728, xii. [DOI] [PubMed] [Google Scholar]
- 28. Loria P, Lonardo A, Carulli L, Verrone AM, Ricchi M, et al. (2005) Review article: the metabolic syndrome and non-alcoholic fatty liver disease. Aliment Pharmacol Ther 22 Suppl 231–36. [DOI] [PubMed] [Google Scholar]
- 29. Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS (2000) Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci U S A 97: 12729–12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Foryst-Ludwig A, Clemenz M, Hohmann S, Hartge M, Sprang C, et al. (2008) Metabolic actions of estrogen receptor beta (ERbeta) are mediated by a negative cross-talk with PPARgamma. PLoS Genet 4: e1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bryzgalova G, Gao H, Ahren B, Zierath JR, Galuska D, et al. (2006) Evidence that oestrogen receptor-alpha plays an important role in the regulation of glucose homeostasis in mice: insulin sensitivity in the liver. Diabetologia 49: 588–597. [DOI] [PubMed] [Google Scholar]
- 32.Ribas V, Nguyen MT, Henstridge DC, Nguyen AK, Beaven SW, et al.. (2009) Impaired Oxidative Metabolism and Inflammation are Associated with Insulin Resistance in ER{alpha} Deficient Mice. Am J Physiol Endocrinol Metab. [DOI] [PMC free article] [PubMed]
- 33. Gao H, Bryzgalova G, Hedman E, Khan A, Efendic S, et al. (2006) Long-term administration of estradiol decreases expression of hepatic lipogenic genes and improves insulin sensitivity in ob/ob mice: a possible mechanism is through direct regulation of signal transducer and activator of transcription 3. Mol Endocrinol 20: 1287–1299. [DOI] [PubMed] [Google Scholar]
- 34. Hamden K, Carreau S, Boujbiha MA, Lajmi S, Aloulou D, et al. (2008) Hyperglycaemia, stress oxidant, liver dysfunction and histological changes in diabetic male rat pancreas and liver: protective effect of 17 beta-estradiol. Steroids 73: 495–501. [DOI] [PubMed] [Google Scholar]
- 35. Herzog B, Cardenas J, Hall RK, Villena JA, Budge PJ, et al. (2006) Estrogen-related receptor alpha is a repressor of phosphoenolpyruvate carboxykinase gene transcription. J Biol Chem 281: 99–106. [DOI] [PubMed] [Google Scholar]
- 36. Hu P, Kinyamu HK, Wang L, Martin J, Archer TK, et al. (2008) Estrogen induces estrogen-related receptor alpha gene expression and chromatin structural changes in estrogen receptor (ER)-positive and ER-negative breast cancer cells. J Biol Chem 283: 6752–6763. [DOI] [PubMed] [Google Scholar]
- 37. Borthwick EB, Houston MP, Coughtrie MW, Burchell A (2001) The antihyperglycemic effect of estrone sulfate in genetically obese-diabetic (ob/ob) mice is associated with reduced hepatic glucose-6-phosphatase. Horm Metab Res 33: 721–726. [DOI] [PubMed] [Google Scholar]
- 38. Kim JY, Jo KJ, Kim OS, Kim BJ, Kang DW, et al. (2010) Parenteral 17beta-estradiol decreases fasting blood glucose levels in non-obese mice with short-term ovariectomy. Life Sci 87: 358–366. [DOI] [PubMed] [Google Scholar]
- 39. Bjornholm M, Zierath JR (2005) Insulin signal transduction in human skeletal muscle: identifying the defects in Type II diabetes. Biochem Soc Trans 33: 354–357. [DOI] [PubMed] [Google Scholar]
- 40. Muthusamy T, Murugesan P, Balasubramanian K (2009) Sex steroids deficiency impairs glucose transporter 4 expression and its translocation through defective Akt phosphorylation in target tissues of adult male rat. Metabolism 58: 1581–1592. [DOI] [PubMed] [Google Scholar]
- 41. Hardie DG (2003) Mini-review: The AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology 144: 5179–5183. [DOI] [PubMed] [Google Scholar]
- 42. Steinberg GR, Kemp BE (2009) AMPK in Health and Disease. Physiol Rev 89: 1025–1078. [DOI] [PubMed] [Google Scholar]
- 43. Horman S, Vertommen D, Heath R, Neumann D, Mouton V, et al. (2006) Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem 281: 5335–5340. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Untreated vs. placebo treated ArKO mice glucose tolerance test. Whole body glucose tolerance tests were completed on fasted six month-old male aromatase knockout (KO) and 2.5 µg/day 17β-estradiol-treated KO (KOE) (a) glucose tolerance test and (b) corresponding area under curve; Data are presented from replicate analysis (n = shown on corresponding bar) as the mean ± SD.
(PPT)
Ex-vivo muscle glucose uptake. Ex-vivo muscle glucose uptake assays were performed on 6 month-old fasted male wildtype (WT), or aromatase knockout (KO) mice, treated with insulin or without (basal). Tracer uptake was measured in dissected (a) soleus and (b) extensor digitorum longus (EDL) muscles. Expression data from n = 8 samples per genotype as the mean ± SD. *p<0.05, **p<0.01 and ***p<0.01.
(PPT)
Body and gonadal tissue weight of untreated versus placebo treated KO mice. Body and gonadal adipose tissue weights in grams (g) of 6 month-old male aromatase knockout untreated (KO) and placebo-treated KO (KOP). Data are expressed as mean ± SD; n = 3–8 per group.
(PPT)