

Abstract
Background
Previous genetic studies demonstrated association between the transcription factor ENGRAILED2 (EN2) and Autism Spectrum Disorder (ASD). Subsequent molecular analysis determined that the EN2 ASD-associated haplotype (rs1861972-rs1861973 A-C) functions as a transcriptional activator to increase gene expression. EN2 is flanked by 5 genes, SEROTONIN RECEPTOR5A (HTR5A), INSULIN INDUCED GENE1 (INSIG1), CANOPY1 HOMOLOG (CNPY1), RNA BINDING MOTIF PROTEIN33 (RBM33), and SONIC HEDGEHOG (SHH). These flanking genes are co-expressed with EN2 during development and coordinate similar developmental processes. To investigate if mRNA levels for these genes are altered in individuals with autism, post-mortem analysis was performed.
Methods
qRT-PCR quantified mRNA levels for EN2 and the 5 flanking genes in 78 post-mortem cerebellar samples. mRNA levels were correlated with both affection status and rs1861972-rs1861973 genotype. Molecular analysis investigated whether EN2 regulates flanking gene expression.
Results
EN2 levels are increased in affected A-C/G-T individuals (p = .0077). Affected individuals also display a significant increase in SHH and a decrease in INSIG1 levels. Rs1861972-rs1861973 genotype is correlated with significant increases for SHH (A-C/G-T) and CNPY1 (G-T/G-T) levels. Human cell line over-expression and knock-down as well as mouse knock-out analysis are consistent with EN2 and SHH being co-regulated, which provides a possible mechanism for increased SHH post-mortem levels.
Conclusions
EN2 levels are increased in affected individuals with an A-C/G-T genotype, supporting EN2 as an ASD susceptibility gene. SHH, CNPY1, and INSIG1 levels are also significantly altered depending upon affection status or rs1861972-rs1861973 genotype. Increased EN2 levels likely contribute to elevated SHH expression observed in the post-mortem samples
Introduction
Autism Spectrum Disorder (ASD) causes deficits in language and social skills along with increased repetitive interests and behaviors. ASD includes autism, Asperger Syndrome (AS), and Pervasive Developmental Disorder-Not Otherwise Specified (PDD-NOS). Individuals with autism display the most severe phenotypes while individuals with AS and PDD-NOS typically exhibit milder symptoms.
Genetic studies have implicated the homeobox transcription factor ENGRAILED2 (EN2) as an ASD susceptibility gene. We determined previously that the EN2 intronic rs1861972-rs1861973 haplotype is associated with ASD, with the A-C haplotype being over-transmitted to affected individuals while the other common haplotype (G-T) is over-represented in unaffected siblings [1], [2]. Six other groups have also reported EN2 association with ASD [3]–[8].
Our molecular genetic and biochemical studies demonstrated that the ASD-associated A-C haplotype functions as a transcriptional activator [9]. Luciferase assays determined that ASD-associated A-C haplotype is both sufficient and necessary for this activator function. A protein complex specifically binds to the A-C haplotype. These proteins were partially purified and two transcription factors, CUX1 and NFIB, were identified. Subsequent electromobility shift assays (EMSAs), over-expression and knock-down studies indicate that CUX1 and NFIB bind the A-C haplotype at the same time and both proteins are required to mediate the activator function. These data demonstrate that the ASD-associated A-C haplotype is a functional cis-regulatory element, and suggest that increased EN2 levels contribute to ASD risk [9]. Consistent with this possibility, recent study demonstrated that human EN2 is epigenetically regulated and increased levels are observed in individuals with autism but these results were not correlated with the rs1861972-rs1861973 haplotype [10].
EN2 also regulates developmental processes relevant to ASD. Numerous groups have demonstrated that En2 is essential for the topographic mapping of axons in both the tectum and cerebellum [11]–[15]. A recent study suggests that En2 is also important for maintaining excitatory/inhibitory balance [16]. Finally several groups have demonstrated that En2 is required for the development of ventral mid-hindbrain neurotransmitter systems [17]–[23].
EN2 maps to a gene poor region of the genome but the five genes within 1Mb of EN2 are co-expressed during brain development and perform similar biological functions. These genes are: SONIC HEDGEHOG (SHH), INSULIN INDUCED GENE1 (INSIG1), CANOPY1 HOMOLOG (CNPY1), SEROTONIN RECEPTOR5A (HTR5A), and RNA BINDING MOTIF PROTEIN33 (RBM33).
SHH is a morphogen that coordinates many aspects of CNS development including patterning, proliferation, and connectivity. Shh and En2 are co-expressed in the embryonic mid-hindbrain as well as the post-natal and adult cerebellum. During embryogenesis Shh and En2 play important roles in the development of ventral mid-hindbrain neurotransmitter systems [24]–[26]. In the cerebellum Shh promotes granule cell neurogenesis while En2 inhibits proliferation [27], [28].
INSIG1 regulates cholesterol biosynthesis, and cholesterol modification is needed for Shh activity [29], [30]. Insig1, Shh, and En2 are co-expressed in the embryonic mid-hindbrain as well as the adult cerebellum. Interestingly, Smith-Lemli-Opitz Syndrome (SLOS) is a mono-genic disorder with defects in cholesterol biosynthesis. 50-86% of individuals with SLOS are diagnosed with ASD, suggesting an involvement of cholesterol metabolism in the disorder [31].
CNPY1 is a positive regulator of Fgf signaling and Cnpy1 induces En2 expression in the developing mid-hindbrain [32], [33]. The Fgf pathway is necessary for neurogenesis, survival, and connectivity, all of which have been implicated in ASD [34]. Cnpy1 and En2 are also co-expressed in the adult cerebellum.
HTR5A is a 5HT receptor and abnormalities in the serotonin pathway have been consistently implicated in ASD [35]. Htr5a is co-expressed with En2 throughout development and in the adult cerebellum. Finally, RBM33 contains homology to a predicted RNA binding domain. RNA binding proteins regulate multiple aspects of RNA processing including splicing, translation, and in neurons transport mRNAs to the synapse for local translation [36]. Rbm33 expression has not been analyzed developmentally but is detected with the other flanking genes in the adult cerebellum. Together these results indicate En2 and the flanking genes regulate similar developmental processes and are co-expressed during development, suggesting they may be co-regulated.
Given the ASD-associated A–C haplotype functions as a transcriptional activator in cultured mouse neurons and human cell lines [9], the next important question is whether the A–C haplotype and affection status are correlated with altered EN2 expression in individuals with autism. To investigate this possibility, post-mortem analysis was performed. We asked if affection status and rs1861972-rs1861973 genotype are correlated with EN2 levels. Because the flanking genes regulate ASD relevant developmental functions and are co-expressed with EN2, their levels were also examined and correlated with affection status and rs1861972-rs1861973 genotype.
Results
EN2 levels are elevated in post-mortem cerebellum
To investigate if EN2 levels are altered in individuals affected with autism, 90 age and sex matched cerebellar post-mortem samples (35 autism and 55 control) were obtained from NICHD Brain and Tissue Bank for Developmental Disorders, and Harvard Brain Tissue Resource Center (Tables S1 and S2 in File S1). The adult cerebellum was chosen as a structure to analyze because EN2 and all the flanking genes are expressed in the cerebellum, and our previous molecular analysis demonstrated that the ASD-associated A–C haplotype is functional in mouse cerebellar neurons. To assess genotype effects on EN2 levels, genomic DNA was isolated and each sample was genotyped for rs1861972 and rs1861973. The three genotypes (A-C/A-C, A-C/G-T, and G-T/G-T) were equally distributed between autism and control groups (Table S3 in File S1). To measure EN2 mRNA levels, total RNA was also isolated and quality was assessed using RNA integrity number (RIN). 78 samples (29 autism and 49 control) with RIN values greater than 3 were used for the final analysis (Table S1 in File S1). cDNA was then generated and Taqman qRT-PCR was performed in triplicate for each sample and normalized to GAPDH.
Post-mortem analysis results can be significantly affected by covariates including: affection, genotype, age, sex, post-mortem interval (PMI), RNA integrity number (RIN), as well as interaction between affection, genotype, and sex (affection*genotype, affection*sex, genotype*sex, and affection*genotype*sex). To take this into account all statistical analysis was adjusted for these covariates. For each gene expression level, insignificant covariates were removed and then a statistical model was established to correct for any significant covariates (see methods for details). Prescription drug use and comorbidity information is also available for a subset of affected individuals but matching information is mostly absent for control individuals rendering covariate analysis impossible. If no drug use or comorbidity in control group is assumed, autism affection status was positively correlated with both prescription drug use and comorbid disorders like epilepsy (P<.0001).
While EN2 was expressed at higher levels in the post-mortem samples from individuals with autism (Figure S1 in File S1), analysis of covariance for EN2 levels demonstrated a significant interaction between affection and genotype (p = 0.0006) (Table S4 in File S1). This result indicates the effect of the rs1861972-rs1861973 haplotype is significantly different between the autism and control groups. Based on this finding, rs1861972-rs1861973 genotype effects on EN2 levels were compared separately for the affected and control groups.
For individuals with autism, EN2 levels exhibited significant differences among three genotypes (p = 0.0225)(Fig 1). Affected A-C/G-T heterozygotes displayed significantly higher EN2 levels than two other genotypes (54% higher than the A-C/A-C, p = 0.0077 and 45% higher than the G-T/G-T, p = 0.0243) (Table 1 and Fig 1). For control individuals, EN2 levels also exhibited significant differences among three genotypes (p<0.0001) (data not shown). However, G-T/G-T homozygotes displayed the highest EN2 levels in control individuals (29% higher than the A-C/A-C, p<.0001, and 47% higher than the A-C/G-T, p = 0.0001) while A-C/G-T is the lowest (Table 1 and Fig 1). In other words, individuals with autism display 74% higher EN2 levels than controls within A-C/G-T group (p = 0.0005) (Table 1 and Figure S2 in File S1). Although the effect of RIN is already taken into account in our statistical model, we performed the same analyses using samples of RIN>5(N = 59). The results displayed a similar pattern of changes albeit with slightly compromised significance due to the decreased number of samples.
Figure 1. EN2 levels are elevated in individuals with autism and an A-C/G-T genotype.

EN2 mRNA levels were measured in 29 autism and 49 control cerebellar samples using Taqman qRT-PCR. Based on the interaction between genotype and affection status, EN2 levels were compared between genotypes (A-C/A-C, A-C/G-T, G-T/G-T) in control and autism groups separately. In each group EN2 levels are normalized to 1 for A-C/A-C genotype and presented as fold change for the other genotypes (EN2 levels are similar between control A-C/A-C and autism A-C/A-C group (<0.5% difference). Fold difference was calculated based on ΔΔCt values. Type 3 tests of fixed effects, *P<.05, **P<.01, ***P<.001, ****P<.0001. AC/AC – individuals homozygous for the rs1861972-rs1861973 A-C haplotype, GT/GT – individuals homozygous for the G-T haplotype, AC/GT – individuals heterozygous for the A-C/G-T haplotype.
Table 1. EN2 levels comparisons between affection and genotype groups.
| Genotype | Affection | Least squared meana | Standard errorsb | P-valuec | Fold changed | Standard errorse |
| AC/AC | Autism vs. Control | 0.007615 | 0.1462 | 0.9586 | 0.9947 | 0.1015 |
| AC/GT | −0.7963 | 0.2188 | 0.0005 | 1.7366 | 0.2678 | |
| GT/GT | 0.2897 | 0.1599 | 0.0744 | 0.8180 | 0.0915 | |
| Affection | Genotype | Least squared meana | Standard errorsb | P-valuec | Fold changed | Standard errorse |
| Autism | AC/AC vs. AC/GT | 0.6236 | 0.2273 | 0.0077 | 0.6490 | 0.1041 |
| AC/AC vs. GT/GT | 0.09040 | 0.1940 | 0.6426 | 0.9393 | 0.1280 | |
| AC/GT vs. GT/GT | −0.5332 | 0.2315 | 0.0243 | 1.4471 | 0.2366 | |
| Control | AC/AC vs. AC/GT | −0.1803 | 0.1300 | 0.1699 | 1.1331 | 0.1027 |
| AC/AC vs. GT/GT | 0.3725 | 0.08992 | <.0001 | 0.7724 | 0.0483 | |
| AC/GT vs. GT/GT | 0.5527 | 0.1373 | 0.0001 | 0.6817 | 0.0653 |
Least squared means were calculated from ΔΔCt values (ΔCtautism–ΔCtcontrol or ΔCt1st genotype–ΔCt2nd genotype) after adjusting for significant covariates.
Standard error for least squared mean.
Type 3 tests of fixed effects were performed considering all the significant covariates. Significant values (5% cut-off) are in bold.
Fold change of each first group versus the second is calculated as 2−(least squared mean).
Standard error of fold change.
These data demonstrate that EN2 levels are elevated in A-C/G-T affected individuals. In addition, different rs1861972-rs1861973 genotypes are correlated with increased EN2 levels in affected (A-C/G-T) and control (G-T/G-T) individuals.
Mediators of A-C haplotype function are not increased in post-mortem cerebellum
Since the transcription factors, CUX1 and NFIB bind the A-C haplotype and mediate its function, their mRNA levels were also measured in the post-mortem samples. No significant interaction between affection status and genotype was observed for either gene. In addition, no significant difference in mRNA level was observed between affected and control groups or between the three genotype groups for either gene (Figure S3, Tables S5 and S6 in File S1). These data determined CUX1 and NFIB transcript levels are not altered by affection status or rs1861972-rs1861973 genotype in post-mortem cerebellum.
Post-mortem expression analysis for five flanking genes of EN2
Five genes flank EN2 in a 1Mb region: HTR5A and INSIG1 5′ of EN2 while CNPY1, RBM33, and SHH map 3′ of EN2 (Fig 2). Animal studies indicate that En2 and these 5 flanking genes are co-expressed (www.brain-map.org; genome.ucsc.edu/cgi-bin/hgVisiGene) and coordinate similar neurodevelopmental processes. These data led us to hypothesize that levels of the flanking genes may also be altered in the cerebellar post-mortem samples.
Figure 2. Genomic map of EN2 and five flanking genes.
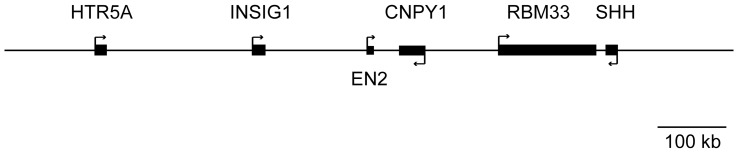
Genomic region encompassing 500 kb upstream and 500 kb downstream of EN2 gene is illustrated and drawn to scale. Solid boxes represent each gene and arrows indicate the transcribed DNA strand. Gene symbols for each gene are shown.
To examine this possibility, normalized mRNA levels were measured in the same 29 affected and 49 control individuals by Taqman qRT-PCR. Analysis of covariance resulted in slightly different final models for each gene (Tables S7-9 in File S1). Unlike EN2, no significant interaction between affection and genotype was observed for any of the five genes. Thus EN2 is the only gene in the 1Mb area, whose expression levels display an interaction between affection status and rs1861972-rs1861973 genotype. Based on this finding, comparisons for the five flanking genes were made separately for affection status and genotype.
SHH and INSIG1 levels are altered in autism
After adjusting for covariates, SHH levels exhibited a significant increase in the affected group compared to control (26% increase, p = 0.0327) (Fig 3A, Table S10 in File S1). INSIG1, on the other hand, displayed decreased levels in affected individuals compared to control (13% decrease, p = 0.0352) (Fig 3A, Table S11 in File S1). None of the other genes displayed a significant difference. These data demonstrate SHH and INSIG1 levels are altered in these individuals with autism (Table 2 for summary).
Figure 3. SHH,INSIG1, and CNPY1 levels are correlated with affection status or rs1861972-rs1861973 genotype.
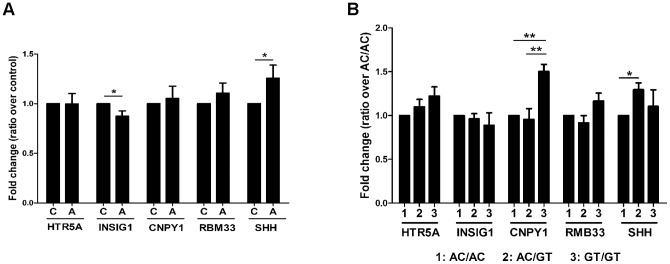
mRNA levels of the five flanking genes were measured using Taqman qRT-PCR. No interaction between affection status and genotype was observed, so comparisions between autism and control group were made regardless of genotype (A) and between the three genotypes regardless of affection status (B). (A) For each gene control levels are normalized to 1,and transcript levels are presented as fold change in autism using ΔΔCt values. Type 3 tests of fixed effects, *p<0.05. Note that SHH and INSIG1 levels are significantly altered in affected individuals. C – control, A – autism group. (B) Each transcript level is normalized to 1 for the genotype with the lowest quantity (AC/AC for HTR5A; GT/GT for INSIG1; AC/GT for CNPY1; AC/GT for RBM33; AC/AC for SHH). Levels for other genotypes are presented as fold change using ΔΔCt values. Type 3 tests of fixed effects, *p<0.05, **p<0.01. CNPY1 and SHH levels are correlated with rs1861972-rs1861973 genotype.
Table 2. Summary of flanking gene results.
| Post-mortem | EN2 over-expression | EN2 knock-down | CUX1-NFIB knock-down | En2 knock-out | |
| SHH | ↑a | ↑ | ↓ | ↓ | ↓ |
| CNPY1 | ↑b | NSd | ↓ | ↓ | ↓ |
| INSIG1 | ↓c | ↑ | NS | ↓ | NS |
| RBM33 | NS | NS | ↓ | NS | NDe |
| HTR5A | NS | ↑ | ↓ | ↓ | ND |
increased in A-C/G-T individuals and affected individuals.
increased in G-T/G-T individuals.
decreased in affected individuals.
NS-not significant.
ND-not determined.
SHH and CNPY1 levels are correlated with rs1861972-rs1861973 genotypes
Differences between the three genotypes were then compared regardless of affection status. SHH levels displayed a significant increase in A-C/G-T heterozygotes compared to A-C/A-C homozygotes (30% increase, p = 0.0297) (Fig 3B, Table S10 in File S1). CNPY1 exhibited increased levels in G-T/G-T homozygotes compared to A-C/A-C and A-C/G-T individuals (50%, p = 0.0037 and 57% increase, p = 0.0013, respectively) (Fig 3B, Table S12 in File S1). None of the other genes displayed a significant difference. These data demonstrate that the rs1861972-rs1861973 genotype is correlated with altered SHH and CNPY1 levels (Table 2 for summary).
EN2 and SHH co-regulation
To investigate if increased levels of EN2 could contribute to the altered expression of these flanking genes, EN2 was over-expressed in non-neuronal (HEK293T) and neuronal (PFSK-1) cell lines, Flanking mRNA levels were measured by Taqman qRT-PCR, normalized to GAPDH, and compared to cells transfected with a non-silencing or empty vector. For EN2 over-expression, average SHH mRNA levels are increased 5.72 fold in HEK293T cells and 5.5 fold in PFSK-1 cells (Fig 4a, b). We then examined the effect of EN2 knock-down on flanking gene expression in both cell lines. 72% and 78% knock-down of EN2 mRNA was observed in HEK293T and PFSK-1 cells respectivelly. Average SHH levels are significantly decreased by ∼45% in both HEK293T and PFSK-1 cells (Fig 4c, d).
Figure 4. SHH expression is regulated by EN2 levels.

To investigate if EN2 regulates flanking gene expression, over-expression and knock-down analyses were performed. (A, B) For over-expression analysis human EN2 cDNA (EN2) or empty pCMV-Tag3B vectors (Ctrl) were transfected transiently into HEK293T (A) and PFSK1 (B) cells. SHH is expressed in both cell lines. mRNA levels were measured by Taqman qRT-PCR and normalized to GAPDH. Relative SHH mRNA levels are presented as fold difference of EN2 over-expression versus control condition. qRTPCR was performed in triplicate and average ΔCt values were used for statistical analyses. N = 8–9, (C, D) EN2 knock-down (KD) was achieved by transfecting shRNAmir constructs (Open Biosystems) into HEK293T cells (C) and PFSK1 (D) cells. A nonsilencing construct was used as a control (Ctrl). SHH mRNA levels were measured as described above. N = 3–6. (E) Both CUX1 and NFIB bind the EN2 A-C haplotype and mediate its transcriptional activator function. To investigate the effect of CUX1 and NFIB on the flanking gene levels, stable double knock-downs (KD) were established in HEK293T cells and analyzed. A non-silencing control cell line (Ctrl) was also generated. mRNA levels for each gene were measured using Taqman qRT-PCR. N = 3. In sum SHH expression consistently mirrors EN2 levels in all five experiments. Student T-test, two-tailed, paired, *P<.05, **P<.01, ***P<.001.
Next we examined the effect of stable CUX1 and NFIB single and double knock-downs generated in HEK293T cells. CUX1 and NFIB bind the ASD-associated A-C haplotype and mediate its activator function. Previous analysis demonstrated that endogenous EN2 expression is decreased significantly by 60% only in the double knock-downs. The single knock-downs had no effect on EN2 levels [9]. In the double knock-down, average SHH levels are significantly decreased (Fig 4e).
Finally we investigated the effect of the En2 knock-out on flanking gene expression. Dopaminergic, noradrenergic and serotonergic neurons are generated from the ventral mid-hindbrain at E10.5. Previous studies indicate that Shh signaling helps coordinate these developmental events [25], [37], [38]. Given the relevance to ASD etiology, mRNA levels were quantitated in micro-dissected mid-hindbrain junction from the En2 knock-out and wild type littermates. Average Shh levels are significantly decreased in the knock-out (Fig 5a). We then examined the spatial expression pattern of Shh by ISH. Shh is expressed in a ventral domain in the embryonic mid-hindbrain. In the En2ko/ko, the Shh expression domain is reduced (Fig 5c, d). To quantify this difference the entire mid-hindbrain was sectioned, all sections were then subjected to ISH and the average expression domain area was measured. A 24% decrease was observed which is consistent with the reduced mRNA levels (Fig 5a, b)
Figure 5. Shh levels are decreased in the ventral mid-hindbrain junction of En2ko/ko.

A) Ventral E10.5 mid-hind junction was dissected from En2 knock-out (KO) embryos and wild-type (WT) littermates. Shh mRNA levels were measured by SYBR Green qRT-PCR and reduced expression is observed in the En2ko/ko. N = 11 Student T-test, two-tailed, paired, **P<.01. B-D) Shh ISHs were performed at the same age and a reduced expression area was observed in the KO compared to WT. N = 5 Student T-test, two-tailed, paired, *P<.05.
Transcript levels for some of the flanking genes are also increased or decreased in the above experiments but unlike SHH they are never affected in a consistent manner (Figure S4 in File S1). Thus only SHH is consistently positively correlated with EN2 - with over-expression leading to increased SHH levels while knock-down/knock-out results in decreased SHH levels. In sum all of the above experiments indicate that SHH and EN2 are co-regulated, and suggest that increased levels of EN2 in the post-mortem samples likely contribute to elevated levels of SHH.
Discussion
The present study demonstrates that EN2 levels are elevated in affected A-C/G-T individuals, providing additional evidence that EN2 is an ASD susceptibility gene. These data are consistent with our in vitro molecular and biochemical data indicating that the A-C haplotype functions as a transcriptional activator to increase reporter activity and endogenous EN2 levels. Another recent post-mortem study also demonstrates increased EN2 levels in cerebellar post-mortem samples but did not correlate expression with the ASD-associated haplotype [10]. Together, these results support the model that increased EN2 levels contribute to ASD pathogenesis.
Our EN2 post-mortem analysis demonstrates an interaction between affection status and rs1861972-rs1861973 genotype. This result indicates that the rs1861972-rs1861973 genotype functions differently depending upon the affection status. In the affected group, individuals with an A-C/G-T genotype express EN2 at the highest levels. However, in the control group, G-T/G-T individuals display the highest EN2 levels. These data suggest that other ASD genetic, epigenetic, environmental or treatment factors work in concert with the rs1861972-rs1861973 haplotype to affect EN2 levels. This finding is consistent with ASD being an epistatic, multi-factorial disorder. The identification of these other ASD factors will aid our understanding of how rs1861972-rs1861973 function is regulated.
Interestingly, affected A-C/G-T individuals display higher EN2 levels than the affected A-C/A-C group. Given the A-C haplotype functions as a transcriptional activator in cell culture analysis, the simplest model is A-C/A-C individuals should exhibit the highest levels of EN2 levels. However previous in vitro molecular genetic analysis used standard over-expression and knock-down approaches that never examined the allelic dosage effect of rs1861972-rs1861973 genotype. Possible explanations for why the A-C/A-C genotype did not result in higher EN2 levels include the following. One, autism-specific environmental factors diminish the effect of the A-C haplotype, reducing levels more in A-C/A-C than A-C/G-T or G-T/G-T individuals. For example, prescription drug use and associated comorbid disorders like epilepsy in the autism co-hort could affect EN2 levels through the A-C haplotype. Some information on medication, comorbidity, and severity of symptoms is available for a subset of affected individuals. Unfortunately the number of individuals with relevant information was not sufficient to warrant a formal statistical test. In addition other unrecorded variables specific to the A-C/A-C autism group likely exist, and these could impact EN2 levels. Two, other ASD risk factors specifically interact with the A-C/G-T genotype to elevate EN2 levels. Three, because ASD is a heterogeneous disorder, increased EN2 levels might only be observed in a subset of affected A-C/A-C individuals, which might be detected if the number of samples was increased substantially. Four, it is possible that the A-C and G-T haplotypes function in trans to create a unique cis-regulatory element. Transvection is a phenomenon whereby cis-regulatory elements function in trans to control gene expression. Reports indicate that transvection regulates gene expression in both humans and Drosophila, suggesting this mechanism is possible [39], [40]
Nevertheless, our post-mortem and molecular analysis is consistent with increased EN2 levels contributing to ASD pathogenesis. Animal studies indicate that En2 regulates several important aspects of brain development relevant to ASD including connectivity, E/I balance, as well as serotonin and norepinephrine system development [15], [16], [24]-[26], [41], [42]. Over-expression and mis-expression studies in animal models have demonstrated that levels and proper spatial-temporal expression of En2 is necessary for normal brain development [11], [13]–[15], [43]–[45]. Thus altered levels or mis-expression at a critical time points could affect any number of developmental processes relevant to ASD.
Levels for some of the flanking genes were also significantly increased or decreased in the post-mortem samples (Table 2). SHH levels were increased in affected individuals as well as individuals with an A-C/G-T genotype. INSIG1 levels were decreased in affected individuals, while CNPY1 was increased in G-T/G-T individuals (Table 2). None of these genes displayed an interaction between genotype and affection status, indicating that ASD only affects rs1861972-rs1861973 regulation of EN2. Finally RBM33 and HTR5A did not display any difference in expression.
Because EN2, SHH, CNPY1 and INSIG1 are co-expressed during development and regulate similar developmental processes, we investigated whether elevated levels of EN2 may contribute to altered flanking gene expression. SHH levels were affected in all analyses. When EN2 was over-expressed, SHH levels were also increased as observed in the post-mortem samples. When EN2 levels were decreased by knock-down, the CUX1-NFIB double knock-down, or in the En2 knock-out, SHH levels were also decreased. These results are consistent with EN2 regulating SHH expression, which is supported by research in Drosophila [46]-[48]. Thus one likely down-stream effect of increased EN2 levels is elevated SHH.
While EN2 regulation of SHH likely contributes to increased levels, other factors may also play a role. SHH is increased in all affected post-mortem samples regardless of genotype so other ASD risk factors also likely contribute to the increased expression. SHH levels are also increased in A-C/G-T individuals, suggesting that the rs1861972-rs1861973 haplotype may regulate SHH at a distance. Consistent with this possibility the CUX1-NFIB double knock-down displays reduced SHH levels while the single CUX1 and NFIB knock-downs had no effect on SHH levels. It is well established that enhancers can function hundreds of kilobases away from transcriptional start sites. Interestingly, a classic example of long-range transcriptional regulation is Shh where spatially restricted expression in the developing limb, notochord, and floor plate is controlled by distant enhancers [49]-[51]. The cis-regulatory elements important for Shh cerebellar expression have not been identified.
Thus EN2 regulation, other ASD risk factors, and the rs1861972-rs1861973 haplotype all likely contribute to increased SHH levels. Shh functions as a morphogen during development to coordinate numerous developmental processes including proliferation, neuronal specification, connectivity and synaptogenesis [52], [53]. Shh also plays an essential role in the patterning of the mid-hindbrain to generate ventral dopaminergic, noradrenergic and serotonergic neurons [24], [25], [54], [55]. These neurons then innervate the vast majority of the brain and control behaviors relevant to ASD such as attention, sleep-wake states and anxiety/mood. We observed diminished levels of Shh in the E10.5 En2 knockout consistent with a possible effect on ventral neurotransmitter development. SHH is also potent mitogen [52]. While many studies point to synaptic defects in autism, macrocephaly and accelerated brain growth have also been consistently reported [56], [57]. Elevated levels of SHH during brain development could contribute to this phenotype. Thus altered SHH levels may contribute to some of the neurodevelopmental phenotypes observed in individuals with ASD.
Finally, CNPY1 and INSIG1 levels are also affected in the post-mortem samples but unlike SHH these post-mortem effects do not seem to be due to increased EN2 levels but instead other ASD factors likely play a more significant role.
In summary, our studies demonstrate that EN2, SHH, CNPY1 and INSIG1 levels are significantly altered in our post-mortem analysis. Increased EN2 levels are consistent with our previous molecular genetic data for the ASD-associated A-C haplotype. The altered levels of SHH is likely to be due to a combination of other ASD-trans acting factors, long range cis-regulatory effects of the rs1861972-rs1861973 haplotype, and regulation by EN2. The CNPY1 and INSIG1 effects are more likely due to other ASD risk factors. All four genes function in similar cell biological pathways relevant to ASD, and alterations in their levels could perturb CNS development and contribute to ASD pathogenesis.
Materials and Methods
All human post-mortem analysis and mouse studies were approved by the University of Medicine and Dentistry of New Jersey (UMDNJ) New Brunswick/Piscataway Institutional Review Board (IRB) and UMDNJ-Robert Wood Johnson Medical School (RWJMS) Institutional Animal Care and Use Committee (IACUC) (I12-013-3) committees. Written consent was obtained from both committees prior to initiating the analysis. All human post-mortem samples were obtained from NICHD Brain and Tissue Bank for Developmental Disorders and the Harvard Brain Tissue Resource Center. All samples are de-identified with no identifiable medical or personal information.
Post-mortem analysis
We obtained 90 frozen postmortem cerebellar tissue samples from 35 individuals with autism and 55 controls with no obvious diagnosable psychiatric disorder. Frozen cerebellar tissue was homogenized for isolation of RNA and DNA, and each post-mortem sample was genotyped for rs1861972-rs1861973. RNA integrity was assessed, cDNA was generated from 78 samples with RIN values greater than 3, and the QRTPCR was performed. See File S1 for details.
Post-mortem statistical analysis
Statistical analysis was performed in collaboration with biostatistician Yong Lin Ph.D. All results are adjusted for significant covariates. Only EN2 levels displayed an interaction between genotype and affection status so comparisons were made considering both variables together. For all the other genes, no interaction was observed so comparisons were made for each variable (affection or genotype) separately. Since variations were not the same between the various genotype and affection groups, analysis of covariance with heterogeneous variance model was used to test if affection status or genotype is correlated with significant changes in mRNA levels. The response variable is the average ΔCt, which was generated by performing qRTPCR in triplicate for each transcript in every post-mortem sample. The full model for analysis of covariance includes 10 variables: affection, genotype, sex, and interaction between affection, genotype and sex (affection*genotype, affection*sex, genotype*sex, and affection*genotype*sex), as well as age, post-mortem interval (PMI), and RIN. The Type 3 tests of fixed effects were performed, and insignificant covariates were removed one by one to obtain the final model, which includes only significant covariates. Pair-wise comparisons were conducted based on the final model so any difference in expression levels was adjusted for significant covariates. Since all analysis assumed normality, these assumptions were verified for the final model. Tests for normality of residuals were conducted, and QQ-plots as well as the residuals plots were generated for the final model. Based on these analyses, three potential outliers for INSIG1 levels (AN16641 and AN00764 - affected G-T/G-T; UMB4899- affected A-C/G-T), one for CNPY1 (AN16641- affected G-T/G-T), and one for CUX1 (AN16641- affected G-T/G-T) were detected. For these three genes, sensitivity analysis was performed by removing outliers. Pair-wise comparisons were conducted based on this final model and any difference in expression levels was adjusted for significant covariates. Differences in gene expression levels were estimated for each comparison by calculating least squared mean of ΔΔCt values. Standard error was calculated for each least squared mean and significance was assessed using the Type 3 tests of fixed effects for EN2 and all flanking genes. Tukey's adjustment was used for all pair-wise comparisons. Fold increase and decrease for each pair was calculated using the formula 2-least squared mean. A significance level of 5% was used for all the tests.
Over-expression, knock-down analysis and knockout analysis
Standard over-expression and knock-down analysis was performed. For the En2 knock-out analysis, the En2tm1Alj knockout allele was maintained on a 12∶12 light:dark cycle. Heterozygous matings were performed, impregnated females were identified and sacrificed. The mid-hindbrain junction was dissected underneath a Nikon stereo microscope in 1xPBS using forebrain-midbrain junction and rhombomere 4 as landmarks. The tissue was flash frozen, total RNA was isolated as described above and yolk sac DNA was used to determine genotype. For the over-expression, knock-down and knock-out analysis, flanking mRNA levels were measured by standard SYBR green QRTPCR. See File S1 for details.
Supporting Information
Supporting figures and tables.
(PDF)
Acknowledgments
We thank NICHD Brain and Tissue Bank for Developmental Disorders, the Harvard Brain Tissue Resource Center for the post-mortem samples, and all participating families for the post-mortem samples. We thank the Autism Tissue Program and especially Jane Pickett for all their help.
Funding Statement
This work was supported by National Institutes of Health (R01 MH076624), New Jersey Governor's Council on Autism Research, National Alliance for Autism Research, Autism Speaks, and National Alliance for Research on Schizophrenia and Depression Young Investigator Award to JHM; National Institutes of Health (R01 MH076435, RC1 MH088288) to LMB. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Benayed R, Gharani N, Rossman I, Mancuso V, Lazar G, et al. (2005) Support for the homeobox transcription factor gene ENGRAILED 2 as an autism spectrum disorder susceptibility locus. Am J Hum Genet 77: 851–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gharani N, Benayed R, Mancuso V, Brzustowicz LM, Millonig JH (2004) Association of the homeobox transcription factor, ENGRAILED 2, 3, with autism spectrum disorder. Molecular psychiatry 9: 474–484. [DOI] [PubMed] [Google Scholar]
- 3. Yang P, Lung FW, Jong YJ, Hsieh HY, Liang CL, et al. (2008) Association of the homeobox transcription factor gene ENGRAILED 2 with autistic disorder in Chinese children. Neuropsychobiology 57: 3–8. [DOI] [PubMed] [Google Scholar]
- 4. Yang P, Shu BC, Hallmayer JF, Lung FW (2010) Intronic single nucleotide polymorphisms of engrailed homeobox 2 modulate the disease vulnerability of autism in a han chinese population. Neuropsychobiology 62: 104–115. [DOI] [PubMed] [Google Scholar]
- 5. Sen B, Singh AS, Sinha S, Chatterjee A, Ahmed S, et al. (2010) Family-based studies indicate association of Engrailed 2 gene with autism in an Indian population. Genes Brain Behav 9: 248–255. [DOI] [PubMed] [Google Scholar]
- 6. Petit E, Herault J, Martineau J, Perrot A, Barthelemy C, et al. (1995) Association study with two markers of a human homeogene in infantile autism. Journal of medical genetics 32: 269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brune CW, Korvatska E, Allen-Brady K, Cook EH, Jr., Dawson G, et al.. (2007) Heterogeneous association between engrailed-2 and autism in the CPEA network. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics. [DOI] [PubMed]
- 8. Wang L, Jia M, Yue W, Tang F, Qu M, et al. (2008) Association of the ENGRAILED 2 (EN2) gene with autism in Chinese Han population. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics 147B: 434–438. [DOI] [PubMed] [Google Scholar]
- 9. Choi J, Ababon MR, Matteson PG, Millonig JH (2012) Cut-like homeobox 1 and nuclear factor I/B mediate ENGRAILED2 autism spectrum disorder-associated haplotype function. Human molecular genetics 21: 1566–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. James SJ, Shpyleva S, Melnyk S, Pavliv O, Pogribny IP (2013) Complex epigenetic regulation of engrailed-2 (EN-2) homeobox gene in the autism cerebellum. Transl Psychiatry 3: e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baader SL, Vogel MW, Sanlioglu S, Zhang X, Oberdick J (1999) Selective disruption of "late onset" sagittal banding patterns by ectopic expression of engrailed-2 in cerebellar Purkinje cells. J Neurosci 19: 5370–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Herrup K, Kuemerle B (1997) The compartmentalization of the cerebellum. Annual review of neuroscience 20: 61–90. [DOI] [PubMed] [Google Scholar]
- 13. Sillitoe RV, Gopal N, Joyner AL (2009) Embryonic origins of ZebrinII parasagittal stripes and establishment of topographic Purkinje cell projections. Neuroscience 162: 574–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sillitoe RV, Stephen D, Lao Z, Joyner AL (2008) Engrailed homeobox genes determine the organization of Purkinje cell sagittal stripe gene expression in the adult cerebellum. J Neurosci 28: 12150–12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sillitoe RV, Vogel MW, Joyner AL (2010) Engrailed homeobox genes regulate establishment of the cerebellar afferent circuit map. The Journal of neuroscience : the official journal of the Society for Neuroscience 30: 10015–10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tripathi PP, Sgado P, Scali M, Viaggi C, Casarosa S, et al. (2009) Increased susceptibility to kainic acid-induced seizures in Engrailed-2 knockout mice. Neuroscience 159: 842–849. [DOI] [PubMed] [Google Scholar]
- 17. Ritvo ER, Yuwiler A, Geller E, Ornitz EM, Saeger K, et al. (1970) Increased blood serotonin and platelets in early infantile autism. Archives of general psychiatry 23: 566–572. [DOI] [PubMed] [Google Scholar]
- 18. Campbell M, Friedman E, Green WH, Collins PJ, Small AM, et al. (1975) Blood serotonin in schizophrenic children. A preliminary study. International pharmacopsychiatry 10: 213–221. [DOI] [PubMed] [Google Scholar]
- 19. Cook EH Jr, Rowlett R, Jaselskis C, Leventhal BL (1992) Fluoxetine treatment of children and adults with autistic disorder and mental retardation. J Am Acad Child Adolesc Psychiatry 31: 739–745. [DOI] [PubMed] [Google Scholar]
- 20. McDougle CJ, Naylor ST, Cohen DJ, Aghajanian GK, Heninger GR, et al. (1996) Effects of tryptophan depletion in drug-free adults with autistic disorder. Archives of general psychiatry 53: 993–1000. [DOI] [PubMed] [Google Scholar]
- 21. Wass S (2011) Distortions and disconnections: disrupted brain connectivity in autism. Brain and cognition 75: 18–28. [DOI] [PubMed] [Google Scholar]
- 22. Minshew NJ, Keller TA (2010) The nature of brain dysfunction in autism: functional brain imaging studies. Current opinion in neurology 23: 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rubenstein JL (2010) Three hypotheses for developmental defects that may underlie some forms of autism spectrum disorder. Current opinion in neurology 23: 118–123. [DOI] [PubMed] [Google Scholar]
- 24. Alenina N, Bashammakh S, Bader M (2006) Specification and differentiation of serotonergic neurons. Stem cell reviews 2: 5–10. [DOI] [PubMed] [Google Scholar]
- 25. Ye W, Shimamura K, Rubenstein JL, Hynes MA, Rosenthal A (1998) FGF and Shh signals control dopaminergic and serotonergic cell fate in the anterior neural plate. Cell 93: 755–766. [DOI] [PubMed] [Google Scholar]
- 26. Simon HH, Scholz C, O′Leary DD (2005) Engrailed genes control developmental fate of serotonergic and noradrenergic neurons in mid- and hindbrain in a gene dose-dependent manner. Molecular and cellular neurosciences 28: 96–105. [DOI] [PubMed] [Google Scholar]
- 27. Wechsler-Reya RJ, Scott MP (1999) Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22: 103–114. [DOI] [PubMed] [Google Scholar]
- 28.Rossman ITaD-B, E (2008) ENGRAILED 2 and Cerebellar Development in the Pathogenesis of Autism Spectrum Disorders. Autism: Current Theories and Evidence Ed A W Zimmerman, Humana Press: 3–40.
- 29. Gong Y, Lee JN, Lee PC, Goldstein JL, Brown MS, et al. (2006) Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell metabolism 3: 15–24. [DOI] [PubMed] [Google Scholar]
- 30. Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, et al. (2002) Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110: 489–500. [DOI] [PubMed] [Google Scholar]
- 31. Porter FD (2008) Smith-Lemli-Opitz syndrome: pathogenesis, diagnosis and management. European journal of human genetics EJHG 16: 535–541. [DOI] [PubMed] [Google Scholar]
- 32. Hirate Y, Okamoto H (2006) Canopy1, a novel regulator of FGF signaling around the midbrain-hindbrain boundary in zebrafish. Current biology : CB 16: 421–427. [DOI] [PubMed] [Google Scholar]
- 33. Matsui T, Thitamadee S, Murata T, Kakinuma H, Nabetani T, et al. (2011) Canopy1, a positive feedback regulator of FGF signaling, controls progenitor cell clustering during Kupffer's vesicle organogenesis. Proceedings of the National Academy of Sciences of the United States of America 108: 9881–9886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vaccarino FM, Grigorenko EL, Smith KM, Stevens HE (2009) Regulation of cerebral cortical size and neuron number by fibroblast growth factors: implications for autism. Journal of autism and developmental disorders 39: 511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lam KS, Aman MG, Arnold LE (2006) Neurochemical correlates of autistic disorder: a review of the literature. Res Dev Disabil 27: 254–289. [DOI] [PubMed] [Google Scholar]
- 36. Bramham CR, Wells DG (2007) Dendritic mRNA: transport, translation and function. Nature reviews Neuroscience 8: 776–789. [DOI] [PubMed] [Google Scholar]
- 37. Lam CS, Sleptsova-Friedrich I, Munro AD, Korzh V (2003) SHH and FGF8 play distinct roles during development of noradrenergic neurons in the locus coeruleus of the zebrafish. Mol Cell Neurosci 22: 501–515. [DOI] [PubMed] [Google Scholar]
- 38. Hirate Y, Okamoto H (2006) Canopy1, a novel regulator of FGF signaling around the midbrain-hindbrain boundary in zebrafish. Curr Biol 16: 421–427. [DOI] [PubMed] [Google Scholar]
- 39. Sandhu KS, Shi C, Sjolinder M, Zhao Z, Gondor A, et al. (2009) Nonallelic transvection of multiple imprinted loci is organized by the H19 imprinting control region during germline development. Genes & development 23: 2598–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu H, Huang J, Wang J, Jiang S, Bailey AS, et al. (2008) Transvection mediated by the translocated cyclin D1 locus in mantle cell lymphoma. The Journal of experimental medicine 205: 1843–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brunet I, Weinl C, Piper M, Trembleau A, Volovitch M, et al. (2005) The transcription factor Engrailed-2 guides retinal axons. Nature 438: 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wizenmann A, Brunet I, Lam JS, Sonnier L, Beurdeley M, et al. (2009) Extracellular Engrailed participates in the topographic guidance of retinal axons in vivo. Neuron 64: 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Millen KJ, Hui CC, Joyner AL (1995) A role for En-2 and other murine homologues of Drosophila segment polarity genes in regulating positional information in the developing cerebellum. Development 121: 3935–3945. [DOI] [PubMed] [Google Scholar]
- 44. Millen KJ, Wurst W, Herrup K, Joyner AL (1994) Abnormal embryonic cerebellar development and patterning of postnatal foliation in two mouse Engrailed-2 mutants. Development 120: 695–706. [DOI] [PubMed] [Google Scholar]
- 45. Baader SL, Sanlioglu S, Berrebi AS, Parker-Thornburg J, Oberdick J (1998) Ectopic overexpression of engrailed-2 in cerebellar Purkinje cells causes restricted cell loss and retarded external germinal layer development at lobule junctions. J Neurosci 18: 1763–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zecca M, Basler K, Struhl G (1995) Sequential organizing activities of engrailed, hedgehog and decapentaplegic in the Drosophila wing. Development 121: 2265–2278. [DOI] [PubMed] [Google Scholar]
- 47. Guillen I, Mullor JL, Capdevila J, Sanchez-Herrero E, Morata G, et al. (1995) The function of engrailed and the specification of Drosophila wing pattern. Development 121: 3447–3456. [DOI] [PubMed] [Google Scholar]
- 48. de Celis JF, Ruiz-Gomez M (1995) groucho and hedgehog regulate engrailed expression in the anterior compartment of the Drosophila wing. Development 121: 3467–3476. [DOI] [PubMed] [Google Scholar]
- 49. Jeong Y, El-Jaick K, Roessler E, Muenke M, Epstein DJ (2006) A functional screen for sonic hedgehog regulatory elements across a 1 Mb interval identifies long-range ventral forebrain enhancers. Development 133: 761–772. [DOI] [PubMed] [Google Scholar]
- 50. Jeong Y, Leskow FC, El-Jaick K, Roessler E, Muenke M, et al. (2008) Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nature genetics 40: 1348–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, et al. (2003) A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Human molecular genetics 12: 1725–1735. [DOI] [PubMed] [Google Scholar]
- 52. Traiffort E, Angot E, Ruat M (2010) Sonic Hedgehog signaling in the mammalian brain. J Neurochem 113: 576–590. [DOI] [PubMed] [Google Scholar]
- 53. Harwell CC, Parker PR, Gee SM, Okada A, McConnell SK, et al. (2012) Sonic hedgehog expression in corticofugal projection neurons directs cortical microcircuit formation. Neuron 73: 1116–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cordes SP (2005) Molecular genetics of the early development of hindbrain serotonergic neurons. Clinical genetics 68: 487–494. [DOI] [PubMed] [Google Scholar]
- 55. Lam CS, Sleptsova-Friedrich I, Munro AD, Korzh V (2003) SHH and FGF8 play distinct roles during development of noradrenergic neurons in the locus coeruleus of the zebrafish. Molecular and cellular neurosciences 22: 501–515. [DOI] [PubMed] [Google Scholar]
- 56. DiCicco-Bloom E, Lord C, Zwaigenbaum L, Courchesne E, Dager SR, et al. (2006) The developmental neurobiology of autism spectrum disorder. J Neurosci 26: 6897–6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Redcay E, Courchesne E (2005) When is the brain enlarged in autism? A meta-analysis of all brain size reports. Biol Psychiatry 58: 1–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting figures and tables.
(PDF)