

Abstract
We previously found that vitamin K3 (menadione, 2-methyl-1,4-naphthoquinone) inhibits the activity of human mitochondrial DNA polymerase γ (pol γ). In this study, we focused on plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone), and chemically synthesized novel plumbagins conjugated with C2:0 to C22:6 fatty acids (5-O-acyl plumbagins). These chemically modified plumbagins enhanced mammalian pol inhibition and their cytotoxic activity. Plumbagin conjugated with chains consisting of more than C18-unsaturated fatty acids strongly inhibited the activities of calf pol α and human pol γ. Plumbagin conjugated with oleic acid (C18:1-acyl plumbagin) showed the strongest suppression of human colon carcinoma (HCT116) cell proliferation among the ten synthesized 5-O-acyl plumbagins. The inhibitory activity on pol α, a DNA replicative pol, by these compounds showed high correlation with their cancer cell proliferation suppressive activity. C18:1-Acyl plumbagin selectively inhibited the activities of mammalian pol species, but did not influence the activities of other pols and DNA metabolic enzymes tested. This compound inhibited the proliferation of various human cancer cell lines, and was the cytotoxic inhibitor showing strongest inhibition towards HT-29 colon cancer cells (LD50 = 2.9 µM) among the nine cell lines tested. In an in vivo anti-tumor assay conducted on nude mice bearing solid tumors of HT-29 cells, C18:1-acyl plumbagin was shown to be a promising tumor suppressor. These data indicate that novel 5-O-acyl plumbagins act as anti-cancer agents based on mammalian DNA replicative pol α inhibition. Moreover, the results suggest that acylation of plumbagin is an effective chemical modification to improve the anti-cancer activity of vitamin K3 derivatives, such as plumbagin.
Introduction
Cancer is a major global public health problem. Epidemiological and animal studies have indicated that chemopreventive natural products are associated with a reduced risk of cancer development [1], [2]. Furthermore, selective inhibitors of DNA polymerases (pols) are considered potentially useful anti-cancer, anti-viral, anti-parasitic, and anti-pregnancy agents because some are known to suppress human cancer and normal cell proliferation, and are cytotoxic [3], [4].
Pol (DNA-dependent DNA polymerase, E.C. 2.7.7.7) catalyzes deoxyribonucleotide addition to the 3′-hydroxyl terminus of primed double-stranded DNA (dsDNA) molecules [5]. The human genome encodes at least 15 DNA pols, which function in cellular DNA synthesis [6], [7]. Eukaryotic cells contain three replicative pols (α, δ, and ε), one mitochondrial pol (γ), and at least 11 non-replicative pols [β, ζ, η, θ, ι, κ, λ, μ, ν, terminal deoxynucleotidyl transferase (TdT), and REV1] [8], [9]. Pols have a highly conserved structure, with their overall catalytic subunits showing little variation among species; conserved enzyme structures are usually preserved over time as they perform important cellular functions that confer evolutionary advantages. Based on sequence homology, eukaryotic pols can be divided into four main families, termed A, B, X, and Y [8]. Family A includes mitochondrial pol γ as well as pols θ and ν; family B includes the three replicative pols α, δ, and ε and also pol ζ; family X comprises pols β, λ, and μ as well as TdT; and family Y includes pols η, ι, and κ in addition to REV1 [9]. We have studied selective inhibitors of each pol derived from natural products including food materials and nutrients for more than 18 years [10], [11]. We have found that vitamin K3, but not K1 or K2, is a potent inhibitor of human pol γ [12], [13].
Vitamin K3 (menadione, 2-methyl-1,4-naphthoquinone, 3 of Fig. 1) is a fat-soluble compound that contains quinone as its principal chemical feature. Quinones are a class of organic compounds that are derived from aromatic compounds via the exchange of an even number of –CH = groups for –C( = O)– groups and any necessary rearrangement of double bonds, resulting in a fully conjugated cyclic dione structure. The toxicological properties of quinones, which act as alkylating agents, have also been examined. For example, quinones are known to interact with flavoproteins to generate reactive oxygen species (ROS) that can induce biological injury [14]–[17]. In this study, we focused on 5-hydroxy-2-methyl-1,4-naphthoquinone (plumbagin, 1 of Fig. 1), which has the common naphthoquinone skeleton and a hydroxyl group and a methyl group at the C-5 and C-2 positions, respectively. Plumbagin (1) is found in the plants of the Plumbaginaceae, Droseraceae, Ancestrocladaceae, and Dioncophyllaceae families. The chief source of plumbagin (1) is the root of Plumbago zeylanica L. (also known as “Chitrak”). Plumbagin (1) has been shown to exert anti-carcinogenic, anti-atherosclerotic, and anti-microbial effects [18]–[21]. The root of P. zeylanica L. has been used in Indian medicine for approximately 2,750 years and its components possess anti-atherogenic, cardiotonic, hepatoprotective, and neuroprotective properties [19]. Plumbagin (1) has potent anti-proliferative and apoptotic activities in various types of human cancers, and its mechanism of cytotoxicity is by inhibition of a PI-5 kinase for ROS generation [22].
Figure 1. Structure of plumbagin (1), 5-O-acyl plumbagins (2), vitamin K3 (3), juglone (4), and 5-O-acyl juglones (5).
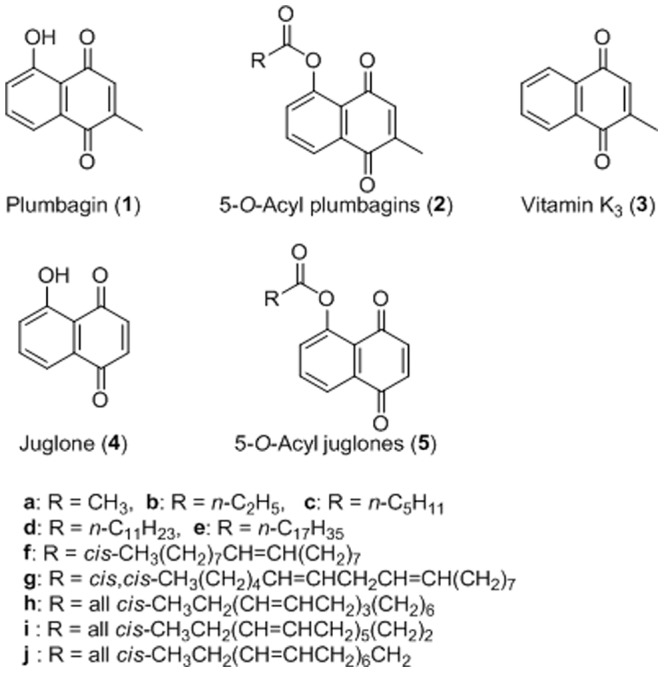
“R” represents a saturated or unsaturated alkyl group in 5-O-acyl plumbagins (2) and 5-O-acyl juglones (5).
We previously found that a vitamin K3 (3) derivative, juglone (5-hydroxy-1,4-naphthoquinone, 4 of Fig. 1), conjugated with fatty acids such as 5-O-acyl juglones (5a–j of Fig. 1) were stronger pol inhibitors than juglone alone (4) [23], therefore, ten 5-O-acylated derivatives of plumbagin (2a–j of Fig. 1) were chemically synthesized from plumbagin (1) and fatty acids to compare with 5-O-acyl juglones (5a–j). In this study, we first investigated the stability of 5-O-acyl plumbagins (2) and 5-O-acyl juglones (5). We also assayed the inhibitory effects of ten 5-O-acyl plumbagins (2a–j) on mammalian pol activity, cytotoxicity in human cancer cell lines and in vivo anti-tumor activity compared with plumbagin (1) and 5-O-acyl juglones (5). The relationship between the pol inhibitory and anti-tumor effects of vitamin K3-based acylated derivatives is discussed.
Materials and Methods
Materials
A chemically synthesized DNA template, poly(dA), was purchased from Sigma-Aldrich Inc. and a customized oligo(dT)18 DNA primer was produced by Sigma-Aldrich Japan K.K. (Hokkaido, Japan). Radioactive nucleotide [3H]-labeled 2′-deoxythymidine-5′-triphosphate (dTTP; 43 Ci/mmol) was obtained from Moravek Biochemicals Inc. (Brea, CA, USA). All other reagents were of analytical grade from Nacalai Tesque Inc. (Kyoto, Japan).
Enzymes
Pol α was purified from calf thymus by immunoaffinity column chromatography as described by Tamai et al. [24]. Recombinant rat pol β was purified from Escherichia coli JMpβ5 as described by Date et al. [25]. The human pol γ catalytic gene was cloned into pFastBac. Histidine-tagged enzyme was expressed using the BACTO-BAC HT Baculovirus Expression System according to the manufacturer's instructions (Life Technologies, Frederick, MD, USA) and purified using ProBound resin (Invitrogen Japan, Tokyo Japan) [26]. Human pols δ and ε were purified by nuclear fractionation of human peripheral blood cancer cells (Molt-4) using the second subunit of pol δ and ε-conjugated affinity column chromatography, respectively [27]. A truncated form of human pol η (residues 1–511) tagged with His6 at its C-terminal was expressed in E. coli cells and purified as described by Kusumoto et al. [28]. A recombinant mouse pol ι tagged with His6 at its C-terminal was expressed and purified by Ni–NTA column chromatography [29]. A truncated form of pol κ (residues 1–560) with 6× His-tags attached at the C-terminus was overexpressed in E. coli and purified as described by Ohashi et al. [30]. Recombinant human His-pol λ was overexpressed and purified according to a method described by Shimazaki et al. [31]. Recombinant human His-pol μ was overexpressed in E. coli BL21 and purified using Glutathione Sepharose 4B (GE Healthcare Bio-Science Corp., Piscataway, NJ, USA) column chromatography according to the method for pol λ preparation by Shimazaki et al. [31]. Pol α from a higher plant, cauliflower, was purified from the inflorescence structure according to the methods outlined by Sakaguchi et al. [32]. Recombinant rice (Oryza sativa L. cv. Nipponbare) pol λ tagged with His6 at the C-terminus was expressed in E. coli and purified as described by Uchiyama et al. [33]. Calf TdT, Taq pol, T4 pol, T7 RNA polymerase, and T4 polynucleotide kinase were purchased from Takara Bio Inc. (Kyoto, Japan). The Klenow fragment of pol I from E. coli was purchased from Worthington Biochemical Corp. (Freehold, NJ, USA). Bovine pancreas deoxyribonuclease I was obtained from Stratagene Cloning Systems (La Jolla, CA, USA).
Measurement of pol activity
The reaction mixtures for calf pol α, rat pol β, plant pol α, and prokaryotic pols have been described previously [34], [35]; those for pol γ as well as pols δ and ε were previously described by Umeda et al. [26] and Ogawa et al. [36], respectively. Those for pols η, ι, and κ were the same as for pol α, and those for pols λ and μ were the same as for pol β. For the pol reactions, poly(dA)/oligo(dT)18 (A/T, 2/1) and dTTP were used as the DNA template-primer substrate and nucleotide (dNTP; 2′-deoxynucleoside-5′-triphosphate) substrate, respectively. For the TdT reactions, oligo(dT)18 (3′-OH) and dTTP were used as the DNA primer substrate and nucleotide substrate, respectively.
The chemically synthesized 5-O-acyl plumbagins (2a–j) were dissolved in distilled dimethyl sulfoxide (DMSO) to various concentrations and sonicated for 30 s. Subsequently, 4 µL aliquots were mixed with 16 µL of each enzyme (0.05 units) in 50 mM Tris-HCl at pH 7.5, containing 1 mM dithiothreitol, 50% glycerol (by vol), and 0.1 mM ethylenediaminetetraacetic acid and held at 0°C for 10 min. These inhibitor-enzyme mixtures in 8 µL volumes were next added to 16 µL of enzyme standard reaction mixture and incubated at 37°C for 60 min, except for Taq pol, which was incubated at 74°C for 60 min. Activity without inhibitor was considered 100%, and the relative activity was determined for each inhibitor concentration. One unit of pol activity was defined as the amount of each enzyme that catalyzed the incorporation of 1 nmol dTTP into synthetic DNA template primers in 60 min, at 37°C, and under standard reaction conditions [34], [35].
Other enzyme assays
The activities of calf primase of pol α, T7 RNA polymerase, mouse inosine-5′-triphosphate (IMP) dehydrogenase (type II), T4 polynucleotide kinase, and bovine deoxyribonuclease I were measured in standard assays according to the manufacturer's specifications, as described by Tamiya-Koizumi et al. [37], Nakayama and Saneyoshi [38], Mizushina et al. [39], Soltis et al. [40], and Lu and Sakaguchi [41], respectively.
Thermal transition of DNA
Thermal transition profiles of dsDNA to single-stranded DNA with or without test compound were obtained with a spectrophotometer (UV-2500; Shimadzu Corp., Kyoto, Japan) equipped with a thermoelectric cell holder according to previous methods [42]. Calf thymus DNA (6 µg/mL) was dissolved in 0.1 M sodium phosphate buffer (pH 7.0) containing 1% DMSO. The solution temperature was equilibrated to 75°C for 10 min, and then increased by 1°C at 2-min intervals for each measurement point. Any change in the absorbance (260 nm) of the compound itself at each temperature point was automatically subtracted from that of DNA plus the compound in the spectrophotometer.
Cell culture and measurement of cancer cell viability
The following human cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA): lung (A549), prostate (DU145 and PC3), colon (HCT116 and HT-29), cervix (HeLa), hepatocellular liver (HepG2), breast (MCF-7), and pancreatic cancer (PANC-1). These cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, penicillin (100 units/mL), streptomycin (100 µg/mL), and 1.6 mg/mL NaHCO3 at 37°C in a humid atmosphere of 5% CO2/95% air. For the cell viability assay, cells were seeded at 1×103 cells/well in a 96-well microplate with various concentrations of the test compounds, and incubated for 48 h. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) solution was added to a final concentration 0.6 mg/mL in Milli-Q purified water for 2 h [43], after which time the medium was discarded and the cells lysed in DMSO. A540 was then measured in a microplate reader (Model 680, Bio-Rad Laboratories, Hercules, CA, USA).
In vivo assessment of anti-tumor assay
Male BALB/c nu/nu mice at 5 week of age (18 g) were purchased from Japan SLC, Inc. (Shizuoka, Japan). Mice receiving standard laboratory chow and water ad libitum were acclimatized for 1 week before inoculation with cancer cells. For in vivo experiments, human colon HT-29 cells (1×107 cells/mouse) were subcutaneously injected into nude mice. Mice bearing solid tumors that had grown to approximately 100 mm3 in volume (tumor volume = length×width×height) at 12 days after implantation were used for the assessment of anti-tumor effects. They were divided randomly into five groups (n = 6/group). One of the five groups was a control group injected with 0.1 mL of phosphate buffered saline (PBS) alone, and other groups were injected with vitamin K3 (3), juglone (4), C18:1-acyl juglone (5f), and C18:1-acyl plumbagin (2f) dissolved in PBS at a dose of 5 mg/kg. The above administrations all took place between days 12–39 subsequent to implantation. All mice were injected subcutaneously 14 times at 1-day intervals with the compound and PBS alone (control). Tumor growth was measured at 1-day intervals for 40 days after implantation. At the end of the in vivo anti-tumor assay, some mice treated with the test compounds and PBS were independently examined to observe major organs such as lung, heart, spleen, stomach, liver, pancreas, kidney, intestine, and brain.
This animal study was approved by the Institutional Animal Care and Use Committee of Kobe Gakuin University, and was performed according to the guidelines outlined in the Care and Use of Laboratory Animals of Kobe Gakuin University. The animals were anesthetized with pentobarbital before undergoing cervical dislocation. The mice that had been bred in-house with free access to food and water were used for all experiments. All of the mice were maintained under a 12-h light/dark cycle and housed at a room temperature of 25°C.
Analytical instruments for synthetic 5-O-acyl plumbagins (2a–j)
Melting points, determined on a Yanaco Micro Melting Point apparatus, are uncorrected. NMR spectra were recorded on a Bruker spectrometer (Avance 400). Chemical shifts are expressed in δ (ppm) relative to Me4Si or the residual solvent resonance, and coupling constants (J) are expressed in Hz. The following abbreviations are used for spin multiplicity: s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, m = multiplet, br = broad. Infrared spectra (IR) were recorded on a HORIBA FT-720, using NaCl (neat) or KBr pellets (solid) and are reported in wavenumbers (cm−1). High resolution mass spectra (HRMS) were obtained on a JEOL mass spectrometer (JMS-700 MStaion) using fast atom bombardment (FAB), or a Fourier transformation-ion cyclotron resonance-mass spectrometer, Bruker solariX (FT-ICR-MS) by using electrospray ionization (ESI) and laser desorption ionization (LDI) techniques. Analytical thin-layer chromatography (TLC) was performed on Silica Gel 60 F254 plates (Merck). Flash chromatography was carried out on SiliaFlash F60 (Silicycle).
Synthesis and characterization of 5-O-acetoxy-2-methyl-1,4-naphthoquinone (C2:0-Acyl plumbagin, 2a)
Acetic anhydride (0.5 mL) was added to a solution of plumbagin (1) (107 mg, 0.57 mmol) in pyridine (1.0 mL), and the mixture was stirred at room temperature for 4 h. After the solvent was removed, the residue was purified by silica gel chromatography (EtOAc/hexanes 1∶3, v/v) to yield 2a (118 mg, 90%) as a yellow solid [44]. Mp 118–124°C; IR (KBr) 3049, 2987, 2966, 2927, 1761, 1662, 1593, 1431, 1375, 1365, 1271, 1203, 1024, 910, 785 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.06 (dd, J = 8.0 Hz, 0.8 Hz, 1H), 7.73 (t, J = 8.0 Hz, 1H), 7.36 (dd, J = 8.0 Hz, 0.8 Hz, 1H), 6.71 (q, J = 1.2 Hz, 1H), 2.45 (s, 3H), 2.17 (d, J = 1.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 184.8, 183.6, 169.4, 149.2, 146.9, 136.8, 134.4, 133.8, 129.4, 125.1, 123.4, 21.1, 16.0; HRMS (FAB) calcd for C13H10O4Na ([M+Na]+) 253.0477, found 253.0482
General procedure for the preparation of 5-O-acyl plumbagins (2b–f) using MNBA
Et3N (2.5–2.6 equiv.), 2-methyl-6-nitrobenzoic anhydride (MNBA) (1.5 equiv.) and N,N-dimethyl-4-aminopyridine (DMAP) (0.1 equiv.) were added to a solution of plumbagin (1) (1.0 equiv.) and carboxylic acid (1.5 equiv.) in CH2Cl2 at room temperature. The mixture was stirred at room temperature under a N2 atmosphere until no further TLC changes were observed. The reaction was quenched by the addition of H2O, and the mixture was extracted with CHCl3. The combined extracts were washed with brine, dried over Na2SO4, and concentrated. The residue was purified by silica gel chromatography using EtOAc/hexanes as eluent.
Synthesis and characterization of 5-O-propanolyloxy-2-methyl-1,4-naphthoquinone (C3:0-Acyl plumbagin, 2b)
Following the general procedure, the reaction of plumbagin (1) (52 mg, 0.28 mmol) with propionic acid (30 µL, 0.41 mmol) using MNBA (143 mg, 0.41 mmol), Et3N (96 µL, 0.69 mmol), and DMAP (3.4 mg, 0.03 mmol) for 2 h gave the crude product. The crude product was purified by silica gel column chromatography (EtOAc/hexanes = 1/5, v/v) to give 2b (60 mg, 89%) as a yellow solid [44]. Mp = 108–112°C; IR (KBr) 3076, 3043, 2981, 2937, 1766, 1662, 1630, 1591, 1358, 1265, 1194, 1132, 1080, 1024, 904, 881, 781 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.06 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 7.73 (t, J = 8.0 Hz, 1H), 7.36 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 6.71 (q, J = 1.2 Hz, 1H), 2.77 (q, J = 7.6 Hz, 2H), 2.16 (d, J = 1.2 Hz, 3H), 1.32 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 184.8, 183.6, 172.8, 149.3, 146.8, 136.9, 134.4, 133.8, 129.4, 125.0, 123.5, 27.6, 16.0, 8.7; HRMS (FAB) calcd for C14H12O4Na ([M+Na]+) 267.0633, found 267.0639.
Synthesis and characterization of 5-O-hexanoyloxy-2-methyl-1,4-naphthoquinone (C6:0-Acyl plumbagin, 2c)
Following the general procedure, the reaction of plumbagin (1) (100 mg, 0.53 mmol) with caproic acid (100 µL, 0.80 mmol) using MNBA (274 mg, 0.80 mmol), Et3N (185 µL, 1.33 mmol), and DMAP (6.7 mg, 0.05 mmol) for 2 h gave the crude product. The crude product was purified by silica gel column chromatography (EtOAc/hexanes = 1/6, v/v) to give 2c (102 mg, 68%) as a yellow solid. Mp = 45–47°C; IR (KBr) 3086, 2958, 2929, 2871, 1766, 1664, 1631, 1595, 1454, 1362, 1267, 1234, 1138, 1105, 912, 891, 791 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.99 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 7.65 (t, J = 8.0 Hz, 1H), 7.28 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 6.64 (q, J = 1.2 Hz, 1H), 2.66 (t, J = 7.6 Hz, 2H), 2.09 (d, J = 1.2 Hz, 3H), 1.75 (quin, J = 7.6 Hz, 2H), 1.41-1.28 (m, 4H), 0.87 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 184.9, 183.6, 172.2, 149.4, 146.8, 136.9, 134.4, 133.8, 129.5, 125.0, 123.6, 34.2, 31.3, 24.1, 22.4, 16.1, 14.0; HRMS (FAB) calcd for C17H18O4Na ([M+Na]+) 309.1103, found 309.1099.
Synthesis and characterization of 5-O-dodecanoyloxy-2-methyl-1,4-naphthoquinone (C12:0-Acyl plumbagin, 2d)
Following the general procedure, the reaction of plumbagin (1) (53 mg, 0.28 mmol) with lauric acid (85 mg, 0.43 mmol) using MNBA (147 mg, 0.43 mmol), Et3N (100 µL, 0.72 mmol), and DMAP (3.5 mg, 0.03 mmol) for 19 h gave the crude product. The crude product was purified by silica gel column chromatography (EtOAc/hexanes = 1/9, v/v) to give 2d (90 mg, 86%) as a yellow crystal; Mp 41–42°C; IR (KBr) 2922, 2850, 1755, 1654, 1632, 1593, 1464, 1358, 1271, 1230, 1188, 1144, 939, 918, 893, 787 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.06 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 7.73 (t, J = 8.0 Hz, 1H), 7.35 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 6.71 (q, J = 1.2 Hz, 1H), 2.73 (t, J = 7.6 Hz, 2H), 2.16 (d, J = 1.2 Hz, 3H), 1.81 (quin, J = 7.6 Hz, 2H), 1.38-1.27 (brm, 16H), 0.88 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 184.9, 183.6, 172.2, 149.4, 146.8, 136.9, 134.4, 133.8, 129.5, 125.0, 123.6, 34.2, 31.9, 29.6 (2C), 29.5, 29.3, 29.3, 29.2, 24.4, 22.7, 16.1, 14.1; HRMS (ESI-LDI) calcd for C23H30O4Na ([M+Na]+) 393.2036, found 393.2032.
Synthesis and characterization of 5-O-octadecanoyloxy-2-methyl-1,4-naphthoquinone (C18:0-Acyl plumbagin, 2e)
Following the general procedure, the reaction of plumbagin (1) (102 mg, 0.54 mmol) with stearic acid (231 mg, 0.81 mmol) using MNBA (280 mg, 0.81 mmol), Et3N (189 µL, 1.36 mmol), and DMAP (6.6 mg, 0.05 mmol) for 19 h gave the crude product. The crude product was purified by silica gel column chromatography (EtOAc/hexanes = 1/10, v/v) to give 2e (139 mg, 56%) as a yellow solid. Mp = 67–70°C; IR (KBr) 2922, 2850, 1759, 1660, 1593, 1541, 1514, 1471, 1273, 1142, 1105, 893, 789 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.99 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 7.65 (t, J = 8.0 Hz, 1H), 7.27 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 6.63 (q, J = 1.2 Hz, 1H), 2.66 (t, J = 7.6 Hz, 2H), 2.09 (d, J = 1.2 Hz, 3H), 1.74 (quin, J = 7.6 Hz, 2H), 1.31-1.19 (brm, 28H), 0.81 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 184.9, 183,6, 172.2, 149.4, 146.8, 136.9, 134.4, 133.8, 129.5, 125.0, 123.6, 34.2, 31.9, 29.7 (5C), 29.6 (2C), 29.6, 29.5, 29.3, 29.2, 29.2, 24.4, 22.7, 16.0, 14.1; HRMS (FAB) calcd for C29H42O4 Na ([M+Na]+) 477.2981, found 447.2988.
Synthesis and characterization of 5-O-oleoyl-2-methyl-1,4-naphthoquinone (C18:1-Acyl plumbagin, 2f)
Following the general procedure, the reaction of plumbagin (1) (100 mg, 0.53 mmol) with oleic acid (0.25 mL, 0.79 mmol) using MNBA (275 mg, 0.80 mmol), Et3N (186 µL, 1.33 mmol), and DMAP (6.5 mg, 0.05 mmol) for 24 h gave the crude product. The crude product was purified by silica gel column chromatography (EtOAc/hexanes = 1/20, v/v) to give 2f (166 mg, 69%) as yellow oil. IR (neat) 3005, 2925, 2854, 1770, 1664, 1595, 1462, 1356, 1271, 1190, 1107, 895, 783 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.06 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 7.72 (t, J = 8.0 Hz, 1H), 7.34 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 6.70 (q, J = 1.2 Hz, 1H), 5.40-5.35 (m, 2H), 2.73 (t, J = 7.6 Hz, 2H), 2.16 (d, J = 1.2 Hz, 3H), 2.06-1.97 (brm, 4H), 1.81 (quin, J = 7.6 Hz, 2H), 1.47-1.27 (brm, 20H), 0.88 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 184.9, 183.6, 172.1, 149.3, 146.8, 136.9, 134.4, 133.8, 130.0, 129.7, 129.5, 125.0, 123.5, 34.2, 31.9, 29.8, 29.7, 29.6, 29.6, 29.5, 29.3, 29.2, 29.1, 27.2, 27.2, 24.4, 22.7, 16.1, 14.1; HRMS (FAB) calcd for C29H40O4Na ([M+Na]+) 475.2824, found 475.2821.
General procedure for the preparation of acyl plumbagins (2g–j) via acyl chlorides
Oxalyl chloride (3.0–3.6 equiv.) was added to a solution of carboxylic acid (1 equiv.) in CH2Cl2 at 0°C. The mixture was stirred at room temperature for 3–4.5 h. The solvent was removed to yield crude acyl chloride. A solution of the acyl chloride (2.2–5.8 equiv.), plumbagin (1) (1 equiv.) and DMAP (0.1 equiv.) in pyridine was stirred at room temperature. The mixture was stirred at room temperature under a N2 atmosphere until no further TLC changes were observed. The reaction was quenched by the addition of H2O, and the mixture was extracted with CHCl3. The extracts were washed with brine, dried over Na2SO4, and concentrated. The residue was purified by silica gel chromatography using hexanes/ethyl acetate as eluent.
Synthesis and characterization of 5-O-linoleoyloxy-2-methyl-1,4-naphthoquinone (C18:2-Acyl plumbagin, 2g)
Following the general procedure, linoleoyl chloride was prepared by treatment of linoleic acid (110 mg, 0.39 mmol) with oxalyl chloride (100 µL, 1.16 mmol) for 4 h. The reaction of plumbagin (1) (34 mg, 0.18 mmol) with the crude linoleoyl chloride using DMAP (2.2 mg, 0.02 mmol) in pyridine for 2 h gave the crude product. The crude product was purified by silica gel column chromatography (toluene/hexanes = 1/10, v/v) to give 2g (31.9 mg, 39%) as yellow oil. IR (neat) 3008, 2927, 2854, 1768, 1664, 1595, 1462, 1358, 1271, 1190, 1009, 1026, 982, 895, 783 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.06 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 7.73 (t, J = 8.0 Hz, 1H), 7.35 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 6.71 (q, J = 1.2 Hz, 1H), 5.42-5.33 (m, 4H), 2.78 (t, J = 7.6 Hz, 2H), 2.73 (t, J = 7.6 Hz, 2H), 2.16 (d, J = 1.2 Hz, 3H), 2.08-2.03 (m, 4H), 1.81 (quin, J = 7.6 Hz, 2H), 1.36-1.26 (m, 14H), 0.94-0.86 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 184.9, 183.6, 172.2, 149.3, 146.8, 136.9, 134.4, 133.8, 130.2, 130.1, 129.5, 128.0, 127.9, 125.0, 123.5, 34.2, 32.5, 31.5, 29.6, 29.5, 29.3, 29.2, 29.1, 27.2, 25.6, 24.4, 22.6, 16.1, 14.1; HRMS (FAB) calcd for C29H38O4Na ([M+Na]+) 473.2668, found 473.2666.
Synthesis and characterization of 5-O-linolenoyloxy-2-metyl-1,4-naphthoquinone (C18:3-Acyl plumbagin, 2h)
Following the general procedure, α-linolenoyl chloride was prepared by treatment of α-linolenic acid (200 mg, 0.72 mmol) with oxalyl chloride (200 µL, 2.33 mmol) for 4.5 h. The reaction of plumbagin (1) (25 mg, 0.13 mmol) with the crude α-linolenoyl chloride using DMAP (1.6 mg, 0.01 mmol) in pyridine for 18 h gave the crude product. The crude product was purified by silica gel column chromatography (EtOAc/hexanes = 1/20, v/v) to give 2h (32 mg, 54%) as yellow oil; IR (neat) 3010, 2927, 2856, 1768, 1664, 1595, 1462, 1358, 1271, 1107, 974, 912, 783 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.05 (dd, J = 7.6 Hz, 1.2 Hz, 1H), 7.71 (t, J = 7.6 Hz, 1H), 7.34 (dd, J = 7.6 Hz, 1.2 Hz, 1H), 6.70 (q, J = 1.2 Hz, 1H), 5.41-5.33 (m, 6H), 2.83-2.71 (m, 6H), 2.16 (d, J = 1.2 Hz, 3H), 2.12-2.00 (m, 4H), 1.81 (quin, 2H), 1.48-1.30 (brm, 8H), 0.98 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 184.9, 183.6, 172.2, 149.4, 146.8, 136.9, 134.4, 133.8, 132.0, 130.3, 130.0, 128.2 (2C), 127.7, 127.1, 125.0, 123.6, 34.2, 29.6, 29.2, 29.1, 27.2, 25.6, 25.5, 25.5, 24.4, 20.5, 16.1, 14.3; HRMS (FAB) calcd for C29H37O4 ([M+H]+) 449.2692, found 449.2694.
Synthesis and characterization of 5-O-eicosapentaenoyloxy-2-methyl-1,4-naphthoquinone (C20:5-Acyl plumbagin, 2i)
Following the general procedure, eicosapentaenoyl chloride was prepared by treatment of eicosapentaenoic acid (194 mg, 0.64 mmol) with oxalyl chloride (200 µL, 2.33 mmol) for 3 h. The reaction of plumbagin (1) (20 mg, 0.10 mmol) with the crude eicosapentaenoyl chloride using DMAP (1.3 mg, 0.01 mmol) in pyridine for 4 h gave the crude product. The crude product was purified by silica gel column chromatography (EtOAc/hexanes = 1/20, v/v) to give 4i (40 mg, 80%) as yellow oil. IR (neat) 3012, 2962, 1768, 1664, 1595, 1446, 1358, 1271, 1190, 1126, 897, 783, 717 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.06 (dd, J = 7.6 Hz, 1.2 Hz, 1H), 7.72 (t, J = 7.6 Hz, 1H), 7.34 (dd, J = 7.6 Hz, 1.2 Hz, 1H), 6.70 (q, J = 1.2 Hz, 1H), 5.50-5.35 (m, 10H), 2.90-2.72 (m, 10H), 2.27 (q, J = 7.6 Hz, 2H), 2.16 (d, J = 1.2 Hz, 3H), 2.08 (quin, J = 7.6 Hz, 2H), 1.90 (quin, J = 7.6 Hz, 2H), 0.97 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 184.8, 183.6, 171.9, 149.3, 146.8, 136.9, 134.4, 133.8, 132.0, 129.4, 129.0, 128.9, 128.5, 128.3, 128.2, 128.1, 128.1, 127.8, 127.0, 125.0, 123.5, 33.6, 26.6, 25.6, 25.6, 25.6, 25.5, 24.3, 20.5, 16.1, 14.3; HRMS (ESI-LDI) calcd for C31H36O4Na ([M+Na]+) 495.2506, found 495.2505.
Synthesis and characterization of 5-O-docosahexaenoyloxy-2-methyl-1,4-naphthoquinone (C22:6-Acyl plumbagin, 2j)
Following the general procedure, docosahexaenoyl chloride was prepared by treatment of docosahexaenoic acid (210 mg, 0.64 mmol) with oxalyl chloride (200 µL, 2.33 mmol) for 3 h. The reaction of plumbagin (1) (20 mg, 0.11 mmol) with the crude docosahexaenoyl chloride using DMAP (1.3 mg, 0.01 mmol) in pyridine for 3 h gave the crude product. The crude product was purified by silica gel column chromatography (EtOAc/hexanes = 1/20, v/v) to give 2j (39 mg, 70%) as yellow oil; IR (neat) 3012, 2964, 2927, 2875, 1768, 1662, 1595, 1358, 1271, 1190, 1128, 1026, 980, 897, 783, 714 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.06 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 7.73 (t, J = 8.0 Hz, 1H), 7.35 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 6.71 (q, J = 1.2 Hz, 1H), 5.34 (m, 12H), 2.92-2.73 (m, 12H), 2.60 (q, J = 7.2 Hz, 2H), 2.16 (d, J = 1.2 Hz, 3H), 2.07 (quin, J = 7.2 Hz, 2H), 0.97 (t, J = 0.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 184.8, 183.6, 171.5, 149.3, 146.8, 136.9, 134.4, 133.8, 132.0, 129.5, 129.4, 128.5, 128.2 (2C), 128.2, 128.1, 128.1, 128.1, 127.8, 127.8, 127.0, 125.1, 123.5, 34.1, 25.6, 25.6, 25.6, 25.6, 25.5, 22.4, 20.5, 16.1, 14.3; HRMS (ESI-LDI) calcd for C33H38O4Na ([M+Na]+) 521.2662, found 521.2661.
Stability of C18:1-acyl plumbagin (2f) and C18:1-acyl juglone (5f) under basic conditions
A 2.2×10−2 M solution of Triton B in MeOH was added to a 2.2×10−2 M solution of each sample in 1,4-dioxane. Then, the mixture was diluted with 1,4-dioxane to a concentration of 1.1×10−3 M. Then, the stability of each compound was monitored by UV-vis spectroscopy. UV-vis spectra were measured at 25°C on a UV-vis spectrophotometer (JASCO V-650, Tokyo, Japan). For comparison, UV-vis spectra of 1.1×10−3 M solution of C18:1-acyl plumbagin (2f), and C18:1-acyl juglone (5f) in 1,4-dioxane and MeOH (19/1, v/v) were measured.
Statistical analysis
All data are expressed as the mean value ± the standard deviation (SD) of at least three independent determinations for each experiment. Statistical significance between each experimental group was analyzed using Student's t-test, and a probability level of 0.01 and 0.05 was used as the criterion of significance.
Results and Discussion
Synthesis of 5-O-acyl plumbagins (2a–j)
Synthesis of 5-O-acyl plumbagins (2a–j) is summarized in Fig. 2. C2:0-Acyl plumbagin (2a) was prepared by treatment of plumbagin (1) with acetic anhydride in pyridine in 90% yield (Fig. 2A). 5-O-Acyl plumbagins 2b–f were prepared by condensation of plumbagin (1) with the corresponding carboxylic acids, using MNBA with triethylamine and DMAP in CH2Cl2 (Fig. 2B). 5-O-Acyl plumbagins 2g–j were prepared by acylation of plumbagin (1) with acyl chlorides in the presence of DMAP in pyridine (Fig. 2C).
Figure 2. Synthesis of 5-O-acyl plumbagins (2a–j).

(A) Synthesis of 5-O-acyl plumbagin (2a). (B) Synthesis of 5-O-acyl plumbagins (2b–f). (C) Synthesis of 5-O-acyl plumbagins (2g–j).
Stability of C18:1-acyl plumbagin (2f) and C18:1-acyl juglone (5f) under basic conditions
We have found that C18:1-acyl plumbagin (2f) is more stable than C18:1-acyl juglone (5f) [23] under basic conditions. C18:1-acyl plumbagin (2f) and C18:1-acyl juglone (5f) were treated with Triton B (benzyltrimethylammonium hydroxide) in 1,4-dioxane and MeOH. As shown in Fig. 3, UV spectra of the mixture were recorded at different reaction times. Almost no changes in UV spectra of C18:1-acyl plumbagin (2f) were observed before and after treatment of Triton B (Fig. 3A). UV spectra of the mixture of C18:1-acyl plumbagin (2f) and Triton B rarely changed during the reaction. In contrast, the changes in UV absorption of C18:1-acyl juglone (5f) were clearly observed (Fig. 3B). UV absorption at 675 nm and 400 nm was observed soon after the addition of Triton B. The absorption at 675 nm suggests the formation of an extended aromatic compound by the reaction of C18:1-acyl juglone with Triton B in 1,4-dioxane and MeOH. The absorption at 675 nm became smaller during the reaction, suggesting that the extended aromatic compound had decomposed. The absorption at 400 nm increased with reaction time. We observed weak and broad absorption in the wavelength range 300–700 nm. These results suggest that C18:1-acyl juglone (5f) readily decomposes under basic conditions.
Figure 3. Stability of C18:1-acyl plumbagin (2f) and C18:1-acyl juglone (5f) under basic conditions.

C18:1-acyl plumbagin (2f) (A) and C18:1-acyl juglone (5f) (B) were treated with 1 equivalent of Triton B in 1,4-dioxane and MeOH. The mixtures were monitored by UV-vis spectroscopy over time. Conditions: 1.1×10−3 M, 25°C, light path length = 1 mm.
Effect of synthesized 5-O-acyl plumbagins (2a–j) on the activities of mammalian pols
Initially, the inhibitory activity of each 5-O-acyl plumbagin (2a–j) toward mammalian pols was investigated using calf pol α, and human pols γ, κ, and λ. In mammalian pols, pols α, γ, κ, and λ were used as the representative pols for families B, A, Y, and X, respectively [8], [9]. Assessment of the relative activity of each pol at a set concentration (10 µM) of plumbagin (1) and the ten chemically synthesized compounds showed that some 5-O-acyl plumbagins were stronger inhibitors of these four mammalian pols than plumbagin (1) (Fig. 4). The plumbagins conjugated with C18 and longer chain unsaturated fatty acids (i.e., C18:1 to C22:6-acyl plumbagins 2f–j) strongly inhibited the activities of pols α and γ, suggesting that the group of unsaturated longer acyl side chains might be an important structural characteristic of 5-O-acyl plumbagin for pol inhibition. C22:6-acyl plumbagin (2j) was the strongest inhibitor of human pols κ and λ among the compounds tested. In contrast, C2:0-acyl plumbagin (2a) hardly inhibited the activities of human pols κ and λ, and C18:0-acyl plumbagin (2e) did not inhibit pol α activity. When activated DNA (bovine deoxyribonuclease I-treated DNA) was used as the DNA template-primer substrate instead of synthesized DNA [poly(dA)/oligo(dT)18 (A/T = 2/1)] and dNTP was used as the nucleotide substrate instead of dTTP, the inhibitory effects of these compounds did not differ (data not shown).
Figure 4. Inhibitory effects of plumbagin (1) and 5-O-acyl plumbagins (2a–j) on the activity of mammalian pols.

Each compound (10 µM) was incubated with calf pol α (B-family pol), human pol γ (A-family pol), human pol κ (Y-family pol), and human pol λ (X-family pol) (0.05 units each). Pol activity in the absence of the compound (control) was taken as 100%, and the relative activity is shown. Data are shown as the mean ± SD (n = 3). ** P<0.01 and * P<0.05 vs. controls.
Effect of synthesized 5-O-acyl plumbagins (2a–j) on cultured human cancer cells
Pols have emerged as important cellular targets for chemical intervention in the development of anti-cancer agents [2]. Therefore, the synthesized 5-O-acyl plumbagins (2a–j) could be useful in chemotherapy. Hence, we investigated the cytotoxic effect of these compounds against HCT116 human colon carcinoma cells. As shown in Fig. 5, 100 µM plumbagin (1) approximately 50% suppressed cell proliferation, but 10 µM of this compound did not. Of the ten synthesized compounds, the nine 5-O-acyl plumbagins except for C18:0-acyl plumbagin (2e) were stronger cell proliferation inhibitors than plumbagin (1). C18:1-Acyl plumbagin (2f) and C18:2-acyl plumbagin (2g) had the first and second strongest proliferation inhibitory effect on HCT116 cells among the compounds tested, and at 10 µM compound these cells showed less than 40 and 60% of the cell proliferation rate, respectively.
Figure 5. Effect of plumbagin (1) and 5-O-acyl plumbagins (2a–j) on the proliferation of HCT116 human colon carcinoma cells.

Each compound (10 and 100 µM) was added to cultured HCT116 cells. The cells were incubated for 48 h, and the rate of proliferation inhibition was determined by MTT assay. Cell proliferation inhibition of the cancer cells in the absence of the compound (control) was taken as 100%. Data are shown as the mean ± SD (n = 5). ** P<0.01 and * P<0.05 vs. controls.
Relationship of mammalian pol inhibition and human cancer cell proliferation suppression by 5-O-acyl plumbagins (2a–j)
The possible relationship between the observed inhibition of four mammalian pol families and HCT116 human colon cancer cell proliferation inhibitory activity was confirmed by comparing the effects of plumbagin (1) and the ten synthesized 5-O-acyl plumbagins (2a–j) on these biological activities (Fig. 6). The effect of 10 µM of these compounds on the relative activity of pol α, which is a DNA replicative pol of the B family, showed the highest correlation with the effect of 100 µM of these compounds on the cancer cell proliferation among those mammalian pol families tested, with a correlation coefficient of 0.844. The relative activity of pol γ, a mitochondrial DNA replicative pol of the A family, showed a moderate correlation (R2 = 0.545) with the cancer cell proliferation rate. Conversely, neither the activities of pols κ and λ, which are DNA repair-related pols of the Y and X families respectively, were related to cytotoxicity, with a correlation coefficient between these activities and cytotoxicity of <0.25. These results led to the speculation that the inhibition of the activities of DNA replicative pols, such as pols α and γ; in particular, inhibition of both the A and B families of pols by compounds 1 and 2a–j might cause the suppression of human cancer cell proliferation.
Figure 6. Relationship between mammalian pol inhibitory activities versus human cancer cell proliferation inhibition by plumbagin (1) and 5-O-acyl plumbagins (2a–j).

X-axis indicates mammalian pol relative activity at 10 µM compound. (A) Calf pol α, (B) human pol γ, (C) human pol κ, and (D) human pol λ. Y-axis indicates rate of HCT116 human colon carcinoma cell proliferation at 100 µM compound. These data are based on Figs. 4 and 5; correlation coefficient values are shown in each panel.
In particular, C18:1-acyl plumbagin (2f) showed the strongest inhibition of pol γ of the ten synthesized 5-O-acyl plumbagins (2a–j) tested. This compound also exhibited the strongest effects on cancer cell proliferation suppression (Fig. 5). Therefore, C18:1-acyl plumbagin (2f) was used in the latter part of this study.
Effects of C18:1-acyl plumbagin (2f) on the activities of various pols and other DNA metabolic enzymes
As described briefly in the Introduction, we succeeded in obtaining eleven mammalian pol species, including pols α, β, γ, δ, ε, η, ι, κ, λ, and μ, and TdT; however, pols ζ, θ and ν, and REV1 were not yet available at the time of the study. Currently, eukaryotes are thought to express at least 15 species of pols [6], [7], and we are still in an era when most pols are very difficult to obtain in their purified form in a laboratory. Table 1 shows the inhibitory effect (50% inhibitory concentration; IC50 value) of C18:1-acyl plumbagin (2f) against various pol species including the 11 mammalian pols that could be obtained. This compound inhibited the activity of all of the mammalian pols with IC50 values of 4.5–10.6 µM, and 50% inhibition of the A, B, X, and Y families of pols was observed at doses of 4.5, 4.8–5.3, 9.2–10.6, and 7.6–8.2 µM, respectively; therefore, the inhibitory effect of this compound on the A and B families of pols was 1.5- to 2-fold stronger than that on the X and Y families of pols. Because the IC50 values of aphidicolin, a known eukaryotic DNA replicative pol α, δ, and ε inhibitor, were 20, 13, and 16 µM, respectively [45], the pol inhibitory activity of C18:1-acyl plumbagin (2f) was >2-fold more potent than that of aphidicolin.
Table 1. IC50 of C18:1-acyl plumbagin (2f) on the activities of mammalian pols, other species pols, and various DNA metabolic enzymes.
| Enzyme | IC50 values (µM) |
| – Mammalian pols – | |
| [A family of pol] | |
| Human pol γ | 4.5±0.40 ** |
| [B family of pols] | |
| Calf pol α | 4.8±0.38 ** |
| Human pol δ | 5.3±0.41 ** |
| Human pol ε | 4.9±0.40 ** |
| [X family of pols] | |
| Rat pol β | 10.3±0.46 ** |
| Human pol λ | 9.2±0.43 ** |
| Human pol μ | 9.5±0.48 ** |
| Calf TdT | 10.6±0.50 ** |
| [Y family of pols] | |
| Human pol η | 7.9±0.42 ** |
| Mouse pol ι | 8.2±0.43 ** |
| Human pol κ | 7.6±0.41 ** |
| – Plant pol – | |
| Cauliflower pol α | >100 |
| Rice pol λ | >100 |
| – Prokaryotic pols – | |
| E. coli pol I | >100 |
| Taq pol | >100 |
| T4 pol | >100 |
| – Other DNA metabolic enzymes – | |
| Calf primase of pol α | >100 |
| T7 RNA polymerase | >100 |
| Mouse IMP dehydrogenase (type II) | >100 |
| T4 polynucleotide kinase | >100 |
| Bovine deoxyribonuclease I | >100 |
Compounds were incubated with each enzyme (0.05 units). Enzyme activity in the absence of an inhibitor (control) was taken as 100%; data, mean ± SD (n = 3).
P<0.01 vs. controls.
In contrast, C18:1-acyl plumbagin (2f) had no effect on plant pols such as cauliflower pol α or rice pol λ, or prokaryotic pols, such as E. coli pol I, Taq pol, or T4 pol (Table 1). The three-dimensional structures of eukaryotic pols are likely to differ greatly from those of prokaryotic pols. This compound did not inhibit the activity of other DNA metabolic enzymes, such as calf primase pol α, 7 RNA polymerase, mouse IMP dehydrogenase (type II), T4 polynucleotide kinase, or bovine deoxyribonuclease I. These results suggest that 5-O-acyl plumbagins (2) may be selective inhibitors of mammalian pols; in particular, plumbagin (1) conjugated with unsaturated fatty acids, such as C18:1-acyl plumbagin (2f), potently inhibited the activities of the A and B families of pols.
Influence of C18:1-acyl plumbagin (2f) on the hyperchromicity of dsDNA
Specific assays were performed to determine whether C18:1-acyl plumbagin (2f)-induced inhibition resulted from the ability of the compound to bind to DNA or the enzyme. The interaction of C18:1-acyl plumbagin (2f) with dsDNA was investigated by studying its thermal transition. For this, the melting temperature (Tm) of dsDNA in the presence of an excess of C18:1-acyl plumbagin (2f) (100 µM) was observed using a spectrophotometer equipped with a thermoelectric cell holder. As shown in Fig. 7, a thermal transition (i.e., Tm) from 75 to 90°C was not observed within the concentration range used in the assay, whereas when a typical intercalating compound, such as ethidium bromide (EtBr, 15 µM), was used as a positive control, an obvious thermal transition was observed.
Figure 7. Effect of C18:1-acyl plumbagin (2f) on the thermal transition of dsDNA.

Control (open-square), C18:1-acyl plumbagin (2f) (100 µM, closed-circle), and EtBr (15 µM, open-diamond) were incubated with 6 µg/mL of calf thymus dsDNA in 0.1 M Na-phosphate buffer (pH 7.0). Data are shown as the mean ± SD (n = 3). ** P<0.01 and * P<0.05 vs. controls.
The question of whether the inhibitory effect of C18:1-acyl plumbagin (2f) resulted from nonspecific adhesion to mammalian pols or from its binding to these enzymes was investigated by determining if an excessive amount of nucleic acid [poly(rC)] or protein (BSA; bovine serum albumin) prevented the inhibitory effect of C18:1-acyl plumbagin (2f). Poly(rC) and BSA had little or no influence on pol inhibition by C18:1-acyl plumbagin (2f) (data not shown), suggesting that this compound selectively bound to the pol molecule. These observations indicated that C18:1-acyl plumbagin (2f) did not act as a DNA intercalating agent or as a template-primer substrate.
Collectively, these results suggested that C18:1-acyl plumbagin (2f) might be a potent and specific inhibitor of mammalian pols. Subsequently, we investigated whether pol inhibition by C18:1-acyl plumbagin (2f) resulted in reduced human cancer cell proliferation.
Effect of C18:1-acyl plumbagin (2f) on cultured human cancer cell lines
C18:1-Acyl plumbagin (2f) treatment for 48 h suppressed the proliferation of various human cancer cells in a dose-dependent manner. As shown in Table 2, C18:1-acyl plumbagin (2f) prevented the proliferation of nine human cancer cell lines, such as A549, DU145, HCT116, HeLa, HepG2, HT-29, MCF-7, PANC-1, and PC3 cells, with LD50 values of 2.9–21.0 µM. These results suggested that this compound could have suppressive activity against the different type of cancer cell lines. In particular, this compound showed the strongest cell proliferation suppression in the colon cancer cell lines, HCT116 and HT-29, with LD50 values of 6.5 and 2.9 µM, respectively. These dose-response curves by MTT detection were the same as that obtained by trypan blue staining (data not shown), suggesting that C18:1-acyl plumbagin (2f) might cause a direct toxic effect on the cells. Because these LD50s were similar to the IC50s for pols (Table 1), this inhibition must be mostly led by the function of pols, such as DNA replicative pol α. C18:1-Acyl plumbagin (2f) more strongly suppressed the proliferation of these human cancer cell lines than aphidicolin, which is an inhibitor of eukaryotic DNA replicative pols (data not shown).
Table 2. LD50 values of C18:1-acyl plumbagin (2f) on the proliferation of human cancer cells.
| Human cancer cell line | LD50 values (µM) |
| A549 (lung cancer) | 15.4±1.7 ** |
| DU145 (prostate cancer) | 8.8±0.9 ** |
| HCT116 (colon cancer) | 6.5±0.6 ** |
| HeLa (cervical cancer) | 21.0±2.5 ** |
| HepG2 (hepatocellular liver cancer) | 7.4±0.8 ** |
| HT-29 cells (colon cancer) | 2.9±0.4 ** |
| MCF-7 (breast cancer) | 7.2±0.7 ** |
| PANC-1 (pancreatic cancer) | 16.1±1.8 ** |
| PC3 (prostate cancer) | 10.0±1.1 ** |
The nine human cancer cell lines were incubated with C18:1-acyl plumbagin (2f) for 48 h. Cell viability was determined by MTT assay, and this viability in the absence of an inhibitor (control) was taken as 100%; data, mean ± SD (n = 5).
P<0.01 vs. controls.
Effect of C18:1-acyl plumbagin (2f) on in vivo anti-tumor activity
Because the cell proliferation inhibitory effect of C18:1-acyl plumbagin (2f) was the strongest on HT-29 cells among the nine human cancer cell lines tested (Table 2), this cell line was used in vivo anti-tumor assays. In this assay, we investigated whether C18:1-acyl plumbagin (2f) was more stable and/or had stronger bioactivity than C18:1-acyl juglone (5f) in vivo; thus, C18:1-acyl plumbagin (2f) was compared with vitamin K3 (3), juglone (4), and C18:1-acyl juglone (5f).
HT-29 cells were subcutaneously injected into 40 nude mice. At 12 days after the implantation, these nude mice were sorted five groups (one control group and four treatment groups) and each mouse group contained 6 mice bearing solid tumor volume of 96–102 mm3. The sorted nude mice were injected with each test compound dissolved in PBS (5 mg/kg) at 1-day intervals until 40 days. As shown in Fig. 8A, these compounds suppressed tumor growth as compared with the control (PBS) group, and the decreased rates of tumor volume at 40 days following injection with vitamin K3 (3), juglone (4), C18:1-acyl juglone (5f), and C18:1-acyl plumbagin (2f) were 7.0, 9.2, 10.3, and 30.5%, respectively. C18:1-acyl plumbagin (2f) showed more than a 3-fold stronger anti-tumor effect than the other compounds tested, suggesting that C18:1-acyl plumbagin (2f) must be stable in vivo, but C18:1-acyl juglone (5f) did not. A significant correlation was found between 5-O-acyl plumbagins (2) and the inhibition of mammalian pols, especially DNA replicative pol α (Fig. 6A). C18:1-Acyl plumbagin (2f) may be able to penetrate cancer cells of tumor in mouse and reach the nucleus, inhibiting the activities of pols and then the inhibition of pol activity by this compound may lead to cell proliferative suppression and prevent tumor growth.
Figure 8. In vivo anti-tumor effects of C18:1-acyl plumbagin (2f).

Nude mice bearing HT-29 solid tumors were administered with PBS (control), vitamin K3 (3), juglone (4), C18:1-acyl juglone (5f), and C18:1-acyl plumbagin (2f) at a dose of 5 mg/kg. (A) Inhibitory effect on tumor volume in nude mice. (B) Body weight changes of nude mice; data, means ± SE (n = 6). * P<0.05 vs. controls.
None of the nude mice showed any significant loss of body weight throughout the experimental period (Fig. 8B). It was also noted that the main visceral organs, such as the liver, lung, kidney, spleen, heart, stomach, small intestine, large intestine, pancreas, and testis of all the groups showed no toxic or degenerative histological appearance (data not shown); therefore, C18:1-acyl plumbagin (2f) is of interest as a candidate material for anti-cancer treatment.
Conclusions
We previously found that vitamin K3 (2-methyl-1,4-naphthoquinone), juglone (5-hydroxy-1,4-naphthoquinone) conjugated with fatty acids inhibited the activity of mammalian pols. In this study, 5-O-acyl plumbagins (2), which are plumbagins (5-hydroxy-2-methyl-1,4-naphthoquinone) conjugated with fatty acid, were produced to establish an efficient chemical synthesis method. In the synthesized ten 5-O-acyl plumbagins (2a–j), 2c–j are novel compounds. These synthesized compounds were stronger inhibitors of the mammalian pols α, γ, κ, and λ representing the pol families B, A, Y, and X, respectively, than plumbagin (1). Of the synthesized compounds, C18:1-acyl plumbagin (2f) showed the strongest suppression of human cancer cell proliferation. The human cancer cytotoxicity of this compound was realized through the inhibition of pols, which are essential for DNA replication as well as cell division. Because C18:1-acyl plumbagin (2f) potently inhibited the activities of replicative pols, such as pols α and γ and suppressed human cancer cell proliferation, they might show in vivo anti-tumor activity without any side effects. The in vivo anti-tumor effect of C18:1-acyl plumbagin (2f) was stronger than that of C18:1-acyl juglone (5f), because C18:1-acyl plumbagin (2f) is more stable than C18:1-acyl juglone (5f) under basic conditions. These results suggested that 5-O-acyl plumbagins (2), such as C18:1-acyl plumbagin (2f), could be used as anti-cancer chemotherapy agents based on their mammalian pol inhibition.
Acknowledgments
We are grateful for the following donations: calf pol α by Dr. M. Takemura of Tokyo University of Science (Tokyo, Japan); rat pol β, and human pols δ and ε by Dr. K. Sakaguchi of Tokyo University of Science (Chiba, Japan); human pol γ by Dr. M. Suzuki of Nagoya University School of Medicine (Nagoya, Japan); mouse pol η and human pol ι by Dr. F. Hanaoka of Gakushuin University (Tokyo, Japan) and Dr. C. Masutani of Nagoya University (Nagoya, Japan); human pol κ by Dr. H. Ohmori of Kyoto University (Kyoto, Japan); and human pols λ and μ by Dr. O. Koiwai of Tokyo University of Science (Chiba, Japan). This study was carried out on mass spectrometers at the Joint Usage/Research Center (JURC) at Institute for Chemical Research, Kyoto University.
Funding Statement
This study was supported in part by the MEXT (Ministry of Education, Culture, Sports, Science and Technology, Japan)-Supported Program for the Strategic Research Foundation at Private Universities, 2012–2016. Y.M. acknowledges Grant-in-Aids for Scientific Research (C) (No. 24580205) from MEXT, and the Hyogo Science and Technology Association (Japan). I.K. acknowledges a Grant-in-Aid for Young Scientists (B) (No. 23710262) from MEXT. K.K and Y.M. acknowledge a Grant-in-Aid for A-STEP (Adaptable and Seamless Technology Transfer Program through target-driven R&D) (Exploratory Research) from JST (Japan Science and Technology Agency). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Berdis AJ (2008) DNA polymerases as therapeutic targets. Biochemistry (Moscow) 47: 8253–8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sakaguchi K, Sugawara F, Mizushina Y (2002) Inhibitors of eukaryotic DNA polymerases. Seikagaku 74: 244–251. [PubMed] [Google Scholar]
- 3. Surh YJ (2003) Cancer chemoprevention with dietary phytochemicals. Nature Rev Cancer 3: 768–780. [DOI] [PubMed] [Google Scholar]
- 4. Liu RH (2004) Potential synergy of phytochemicals in cancer prevention: mechanism of action. J Nutr 134: 3479S–3485S. [DOI] [PubMed] [Google Scholar]
- 5.Kornberg K, Baker TA (1992) DNA replication. FreemanW.D. and Co. New York, University Science Books. 197–225p. [Google Scholar]
- 6. Bebenek K, Kunkel TA (2004) Functions of DNA polymerases. Adv Protein Chem 69: 137–165. [DOI] [PubMed] [Google Scholar]
- 7. Hubscher U, Maga G, Spadari S (2002) Eukaryotic DNA polymerases. Ann Rev Biochem 71: 133–163. [DOI] [PubMed] [Google Scholar]
- 8. Loeb LA, Monnat RJ Jr (2008) DNA polymerases and human disease. Nature Rev Genet 9: 594–604. [DOI] [PubMed] [Google Scholar]
- 9. Lange SS, Takata K, Wood RD (2011) DNA polymerases and cancer. Nature Rev Cancer 11: 96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mizushina Y (2009) Specific inhibitors of mammalian DNA polymerase species. Biosci Biotechnol Biochem 73: 1239–1251. [DOI] [PubMed] [Google Scholar]
- 11. Mizushina Y (2011) Screening of novel bioactive compounds from food components and nutrients. J Jpn Soc Nutr Food Sci 64: 377–384. [Google Scholar]
- 12. Mizushina Y, Yonezawa Y, Yoshida Y (2007) Selective inhibition of animal DNA polymerases by fat-soluble vitamins A, D, E and K, and their related compounds. Curr Enzyme Inhibition 3: 61–75. [Google Scholar]
- 13. Sasaki R, Suzuki Y, Yonezawa Y, Ota Y, Okamoto Y, et al. (2008) DNA polymerase γ inhibition by vitamin K3 induces mitochondria-mediated cytotoxicity in human cancer cells. Cancer Sci 99: 1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Monks TJ, Hanzlik RP, Cohen GM, Ross D, Graham DG (1992) Quinone chemistry and toxicity. Toxicol Appl Pharmacol 112: 2–16. [DOI] [PubMed] [Google Scholar]
- 15. O'Brien PJ (1991) Molecular mechanisms of quinone cytotoxicity. Chem Biol Interact 80: 1–41. [DOI] [PubMed] [Google Scholar]
- 16. Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ (2000) Role of quinones in toxicology. Chem Res Toxicol 13: 135–160. [DOI] [PubMed] [Google Scholar]
- 17. Cho AK, Stefano ED, You Y, Rodriguez CE, Debra A, et al. (2004) Determination of Four Quinones in Diesel Exhaust Particles, SRM 1649a, and Atmospheric PM2.5. Aerosol Sci Technol 38: 68–81. [Google Scholar]
- 18. Mossa JS, El-Feraly FS, Muhammad I (2004) Antimycobacterial constituents from Juniperus procera, Ferula communis and Plumbago zeylanica and their in vitro synergistic activity with isonicotinic acid hydrazide. Phytother Res 18: 934–937. [DOI] [PubMed] [Google Scholar]
- 19. Srinivas P, Gopinath G, Banerji A, Dinakar A, Srinivas G (2003) Plumbagin induces reactive oxygen species, which mediate apoptosis in human cervical cancer cells. Mol Carcinog 40: 201–211. [DOI] [PubMed] [Google Scholar]
- 20. Tilak JC, Adhikari S, Devasagayam TP (2004) Antioxidant properties of Plumbagozeylanica, an Indian medicinal plant and its active ingredient, plumbagin. Redox Rep 9: 219–227. [DOI] [PubMed] [Google Scholar]
- 21. Ding Y, Chen ZJ, Liu S, Che D, Vetter M, et al. (2005) Inhibition of Nox-4 activity by plumbagin, a plant-derived bioactive naphthoquinone. J Pharm Pharmacol 57: 111–116. [DOI] [PubMed] [Google Scholar]
- 22. Lee JH, Yeon JH, Kim H, Roh W, Chae J, et al. (2012) The natural anticancer agent plumbagin induces potent cytotoxicity in MCF-7 human breast cancer cells by inhibiting a PI-5 kinase for ROS generation. PLoS One 7: e45023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maruo S, Kuriyama I, Kuramochi K, Tsubaki K, Yoshida H, et al. (2011) Inhibitory effect of novel 5-O-acyl juglones on mammalian DNA polymerase activity, cancer cell growth and inflammatory response. Bioorg Med Chem 19: 5803–5812. [DOI] [PubMed] [Google Scholar]
- 24. Tamai K, Kojima K, Hanaichi T, Masaki S, Suzuki M, et al. (1988) Structural study of immunoaffinity-purified DNA polymerase α-DNA primase complex from calf thymus. Biochim Biophys Acta 950: 263–273. [DOI] [PubMed] [Google Scholar]
- 25. Date T, Yamaguchi M, Hirose F, Nishimoto Y, Tanihara K, et al. (1998) Expression of active rat DNA polymerase β in Escherichia coli . Biochemistry 27: 2983–2990. [DOI] [PubMed] [Google Scholar]
- 26. Umeda S, Muta T, Ohsato T, Takamatsu C, Hamasaki N, et al. (2000) The d-loop structure of human mtDNA is destabilized directly by 1-methyl-4-phenylpyridinium ion (MPP+), a parkinsonism-causing toxin. Eur J Biochem 267: 200–206. [DOI] [PubMed] [Google Scholar]
- 27. Oshige M, Takeuchi R, Ruike R, Kuroda K, Sakaguchi K (2004) Subunit protein-affinity isolation of Drosophila DNA polymerase catalytic subunit. Protein Expr Purif 35: 248–256. [DOI] [PubMed] [Google Scholar]
- 28. Kusumoto R, Masutani C, Shimmyo S, Iwai S, Hanaoka F (2004) DNA binding properties of human DNA polymerase η: implications for fidelity and polymerase switching of translesion synthesis. Genes Cells 9: 1139–1150. [DOI] [PubMed] [Google Scholar]
- 29. Biertümpfel C, Zhao Y, Kondo Y, Ramón-Maiques S, Gregory M, et al. (2010) Structure and mechanism of human DNA polymerase η. Nature 465: 1044–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ohashi E, Murakumo Y, Kanjo N, Akagi J, Masutani C, et al. (2004) Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells 9: 523–531. [DOI] [PubMed] [Google Scholar]
- 31. Shimazaki N, Yoshida K, Kobayashi T, Toji S, Tamai T, et al. (2002) Over-expression of human DNA polymerase λ in E. coli and characterization of the recombinant enzyme. Genes Cells 7: 639–651. [DOI] [PubMed] [Google Scholar]
- 32. Sakaguchi K, Hotta Y, Stern H (1980) Chromatin-associated DNA polymerase activity in meiotic cells of lily and mouse. Cell Struct Funct 5: 323–334. [Google Scholar]
- 33. Uchiyama Y, Kimura S, Yamamoto T, Ishibashi T, Sakaguchi K (2004) Plant DNA polymerase λ, a DNA repair enzyme that functions in plant meristematic and meiotic tissues. Eur J Biochem 271: 2799–2807. [DOI] [PubMed] [Google Scholar]
- 34. Mizushina Y, Tanaka N, Yagi H, Kurosawa T, Onoue M, et al. (1996) Fatty acids selectively inhibit eukaryotic DNA polymerase activities in vitro. Biochim Biophys Acta 1308: 256–262. [DOI] [PubMed] [Google Scholar]
- 35. Mizushina Y, Yoshida S, Matsukage A, Sakaguchi K (1997) The inhibitory action of fatty acids on DNA polymerase β. Biochim Biophys Acta 1336: 509–521. [DOI] [PubMed] [Google Scholar]
- 36. Ogawa A, Murate T, Suzuki M, Nimura Y, Yoshida S (1998) Lithocholic acid, a putative tumor promoter, inhibits mammalian DNA polymerase β. Jpn J Cancer Res 89: 1154–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tamiya-Koizumi K, Murate T, Suzuki M, Simbulan CM, Nakagawa M, et al. (1997) Inhibition of DNA primase by sphingosine and its analogues parallels with their growth suppression of cultured human leukemic cells. Biochem Mol Biol Int 41: 1179–1189. [DOI] [PubMed] [Google Scholar]
- 38. Nakayama C, Saneyoshi M (1985) Inhibitory effects of 9-β-d-xylofuranosyladenine 5′-triphosphate on DNA-dependent RNA polymerase I and II from cherry salmon (Oncorhynchus masou). J Biochem (Tokyo) 97: 1385–1389. [DOI] [PubMed] [Google Scholar]
- 39. Mizushina Y, Dairaku I, Yanaka N, Takeuchi T, Ishimaru C, et al. (2007) Inhibitory action of polyunsaturated fatty acids on IMP dehydrogenase. Biochimie 89: 581–590. [DOI] [PubMed] [Google Scholar]
- 40. Soltis DA, Uhlenbeck OC (1982) Isolation and characterization of two mutant forms of T4 polynucleotide kinase. J Biol Chem 257: 11332–11339. [PubMed] [Google Scholar]
- 41. Lu BC, Sakaguchi K (1991) An endo-exonuclease from meiotic tissues of the basidiomycete Coprinus cinereus: Its purification and characterization. J Biol Chem 266: 21060–21066. [PubMed] [Google Scholar]
- 42. Mizushina Y, Murakami C, Ohta K, Takikawa H, Mori K, et al. (2002) Selective inhibition of the activities of both eukaryotic DNA polymerases and DNA topoisomerases by elenic acid. Biochem Pharmacol 63: 421–427. [DOI] [PubMed] [Google Scholar]
- 43. Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65: 55–63. [DOI] [PubMed] [Google Scholar]
- 44. Mathew R, Kruthiventi AK, Prasad JV, Kumar SP, Srinu G, et al. (2010) Inhibition of mycobacterial growth by plumbagin derivatives. Chem Biol Drug Des 76: 34–42. [DOI] [PubMed] [Google Scholar]
- 45. Mizushina Y, Kamisuki S, Mizuno T, Takemura M, Asahara H, et al. (2000) Dehydroaltenusin, a mammalian DNA polymerase α inhibitor. J Biol Chem 275: 33957–33961. [DOI] [PubMed] [Google Scholar]