

Abstract
Glucose transporters and the glycolysis enzyme lactate dehydrogenase A (LDH-A) are both overexpressed in cancer cells, two proliferation tactics that underlie the phenomenon known as the Warburg effect. Herein we report the development and activity of a glucose-conjugated LDH-A inhibitor designed to target both of these tumor-promoting facets. In addition to the promise of this conjugate, dual targeting of the Warburg effect using glycoconjugation as an anticancer strategy could be applied to inhibitors of many of the enzymes involved in glycolysis or tumor metabolism.
Keywords: Warburg effect, glycoconjugation, targeted anticancer agents, drug delivery, cancer
Nearly a century ago, German scientist Otto Warburg observed that solid tumors deviate from most normal tissues in their ravenous consumption of glucose and high rates of aerobic glycolysis.[1] This dysfunctional metabolism has been proposed to convey a survival advantage to tumor cells, allowing them to proliferate in normoxic or hypoxic environments and to evade killing by the immune system.[2] The molecular mechanisms underlying the Warburg effect have been elucidated, most notably tumor cells’ overexpression of the glucose transporter GLUT-1[3] and the enzymes of glycolysis, including lactate dehydrogenase isoform A (LDH-A).[4] Targeting dysregulated tumor cell metabolism is emerging as a tantalizing anticancer strategy.[2a] Herein we report the first demonstration of dually targeting the Warburg effect using a glucose-conjugated LDH-A inhibitor, thus exploiting both the preferential glucose uptake and increased glycolysis of cancer cells (Figure 1A).
Figure 1.

A) Dual-targeting of the Warburg effect by a glucose-conjugated LDH-A inhibitor. B) Structures and in vitro Ki values vs. NADH in LDH-A of unconjugated and glucose-conjugated N-hydroxyindole (NHI) class compounds. Values are reported as the mean ± SD of three or more independent experiments.
A common clinical application of the selective uptake of glucose into cancerous versus normal tissues is the use of the radiolabeled glucose analog 2-deoxy-2-(18F)fluoro-D-glucose (18F-FDG). 18F-FDG is a ubiquitous imaging tool for diagnosing and staging many types of cancers, including lung, breast, endometrial and colorectal carcinomas, several types of sarcomas, and both Hodgkin’s and non-Hodgkin’s lymphomas.[5] In addition, the conjugation of glucose[6] or similar sugars potentially recognized by GLUT-1 receptors[7] to anticancer agents offers potential selective targeting of cytotoxic drugs,[8] with the most clinically advanced glycoconjugate, glufosfamide, reaching phase II and III clinical trials in Europe and the United States.[9]
LDH-A is a key enzyme in glycolysis, catalyzing the reduction of pyruvate to lactate (Figure 1Aa), generating NAD+ and thus enabling continued glycolysis and ATP production even in the absence of aerobic oxidation of NADH.[10] Much of the lactate produced in this reaction is excreted into the tumor microenvironment, acidifying it to limit immune access to tumor tissue.[11] Overexpression of LDH-A has been noted in numerous solid tumors and has been found to correlate with poor clinical outcome in patients;[12] these data have been corroborated by a number of studies demonstrating that cancer cells in which LDH-A activity has been attenuated (through RNA interference) are less viable and less tumorigenic.[13] Importantly, LDH-A inhibition is unlikely to harm normal tissues: LDH-A deficiency is present in the human population at a frequency of 0.0012,[14] and those individuals heterozygous for LDH-A deficiency have no clinical presentation, while homozygotes present with myoglobinuria only upon extreme exertion.[15]
We recently reported the discovery of N-hydroxyindole (NHI)-based LDH-A inhibitors (exemplified by compound NHI-1, Figure 1B) as anticancer agents.[16] While other classes of in vitro LDH-A inhibitors exist,[17] including the natural product gossypol, [18] its derivative FX-11,[19] the pyruvate mimetic oxamate,[20] the gallic acid derivative galloflavin,[21] compounds developed in a fragment-based approach by AstraZeneca[22] and by ARIAD Pharmaceuticals,[23] and in screening by Genentech,[24] the NHI inhibitors are attractive candidates due to their facile syntheses, selective toxicity toward cancerous cells, and in vitro and cell culture efficacy.[16a] Thus the NHIs are an outstanding compound class to demonstrate the concept of dually targeting the Warburg effect by linking glucose to a glycolytic enzyme inhibitor.
We previously reported compound NHI-1 (Figure 1B) as a competitive inhibitor of LDH-A in vitro, with the ability to inhibit the conversion of 13C glucose to 13C lactate in HeLa human cervical carcinoma cells when used at a high concentration (500 µM).[16a] Later, methyl ester NHI-2 was found to inhibit LDH-A in vitro and kill cancer cells in culture.[16b] Further, NHI-2 proved to be stable after uptake by cancer cells, suggesting its improved anti-proliferative activity is due improved cell uptake compared to NHI-1.[16b] In efforts to enhance the tumor cell selectivity and efficacy of NHI-1 and NHI-2, their glucose conjugates NHI-Glc-1 and NHI-Glc-2 (Figure 1B) were synthesized and evaluated (see supporting information for synthetic routes).
Evaluation versus LDH-A in vitro revealed that non-conjugated (NHI-1 and NHI-2)[16b] and glucose-conjugated derivatives (NHI-Glc-1 and NHI-Glc-2) are competitive inhibitors of the NADH binding pocket of LDH-A, with conjugation to the sugar moiety of the NHI derivatives lowering the inhibitory potency of the resulting conjugates by 2- (NHI-Glc-1) and 7-fold (NHI-Glc-2) (Figure 1B). To rule out inhibition by aggregation, additional assays were conducted in the presence of Triton X detergent and bovine serum albumin (BSA) using conditions described previously.[25] The NHI series, as exemplified by NHI-1, NHI-2, and NHI-Glc-2, retained its inhibitory potency against LDH-A in the presence of both Triton X and BSA (Figure S1).
Docking studies followed by molecular dynamic (MD) simulations were carried out to examine the interaction of the glucose conjugates with LDH-A. Starting from the average structure of the minimized LDH-A/NHI-1 complex that we recently reported,[16a] compound NHI-Glc-2 was docked in the protein by using GOLD 5.1,[21] and the minimized complex was then subjected to 10 ns of MD simulation using Amber 11.[22] As shown in Figure 2, the ester of NHI-Glc-2 forms a H-bond with R169, similar to that found by MD simulation of ester-aglycone NHI-2.[16b] The indole portion is located in a pocket defined by N138, H193, G194, A238, and Y239, with its 6-phenyl substituent protruding toward the enzyme cavity entrance channel. The glucose moiety establishes strong interactions in the NADH-binding pocket; in particular, H-bonds with Asn138 and with the backbone of Val136 and Ser137 (Figure 2B and Table S1). These additional interactions largely compensate for the loss of those involving the N-OH group of non-conjugated derivatives Interestingly, both Val136 and Asn138 were previously found to be similarly involved in crucial interactions with the enzyme cofactor in the X-ray structure of the complex of LDH-A with NADH and oxamate (1I10 PDB code).[23]
Figure 2.
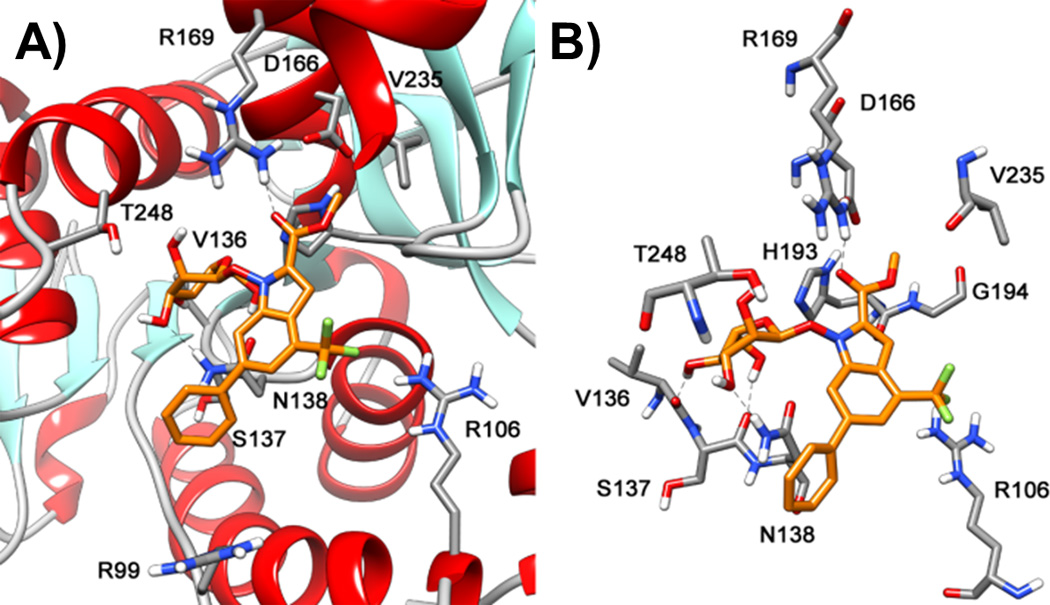
Binding pose resulting from MD simulation of the LDH-A complex with compound NHI-Glc-2. A) disposition of the ligand into the enzyme active site displaying the protein backbone; B) skewed view of the complex showing the protein residues that are most relevant for interaction with the inhibitor.
Evaluation of these compounds versus a panel of cancer cell lines representing the types of cancers in which LDH-A is commonly shown to be overexpressed[12b, 26] and which highly express GLUT-1[27] demonstrate that, while NHI-Glc-1 is inactive (IC50 > 200 µM in HeLa cells), NHI-Glc-2 has 3–5-fold and 6–9-fold enhanced potencies, compared to NHI-2 and NHI-1, respectively (Table 1). All compounds are significantly less potent against non-cancerous mouse embryonic fibroblasts (WT-MEF, Table 1).
Table 1.
Cancer cell toxicity of NHI class LDH-A inhibitors [a]
| Cancer cell line |
Tissue of origin |
IC50 values
(µM) |
||
|---|---|---|---|---|
| NHI-1 | NHI-2 | NHI-Glc-2 | ||
| HeLa | Cervix | 43.8 ± 4.6 [16b] | 33.4 ± 1.0[16b] | 7.2 ± 0.2 |
| A549 | Lung | 131.0 ± 17.6 | 44.1 ± 6.2 | 17.2 ± 3.0 |
| H1299 | Lung | 141.0 ± 11.1 | 61.1 ± 11.8 | 18.0 ± 1.5 |
| H226 | Lung | 120.7 ± 7.8 | 43.4 ± 5.3 | 16.8 ± 2.8 |
| MCF-7 | Breast | 124.3 ± 7.1 | 64.9 ± 13.1 | 16.7 ± 1.1 |
| BT549 | Breast | 110.1 ± 9.5 | 34.5 ± 10.0 | 12.7 ± 0.4 |
| IGROV-1 | Ovary | 123.3 ± 6.8 | 57.4 ± 7.3 | 15.5 ± 3.0 |
| WT-MEF | Normal fibroblast | 245.0 ± 13.0 | 80.5 ± 8.2 | 32.2 ± 0.2 |
All cells were seeded at 5000 cells/well in plates in which vehicle or compound in DMSO was pre-dispensed (1% final concentration DMSO in all wells). Following a 72 hour incubation, biomass was quantified using the Sulforhodamine B (SRB) assay, and IC50 values (in µM) were calculated from logistical dose response curves. Averages were obtained from three independent experiments, and error is standard error (n=3).
To assess efficacy in inhibiting lactate production in cells, HeLa cells were treated with varying concentrations of NHIs and other reported LDH-A inhibitors including FX-11,[19a] galloflavin,[21] and the AstraZeneca compound AZ 33[22] (structures are depicted in Figure S2). After 8 hours of treatment, the lactate present in cell culture media was quantified by GC-MS. This GC-MS based assay for lactate detection has several-fold increased sensitivity over the 13C NMR-based assay previously employed,[16a] allowing for precise detection of low micromolar lactate (versus the low millimolar detection limit afforded by 13C NMR). As shown in Figure 3A, consistent with the proliferation assay results, NHI-Glc-1 has only modest effects at 200 µM, similar to its aglycone NHI-1 (which required 500 µM for substantial efficacy, as previously reported[16a]). On the contrary, treatment with NHI-Glc-2 leads to significant, dose-dependent reduction in cellular lactate production, and is more potent than its aglycone NHI-2. The hexokinase inhibitor 2-deoxyglucose also has a modest effect at very high concentrations (10 mM), whereas negligible effects are observed for LDH-A inhibitors FX-11, galloflavin, and AZ 33 (each tested at 100 µM). Cytotoxic compounds that do not impact glucose metabolism, such as the topoisomerase II inhibitor etoposide, have no effect on lactate production. Furthermore, the reduction in lactate production observed with the NHIs precedes the onset of cell death (Figure S3).
Figure 3.
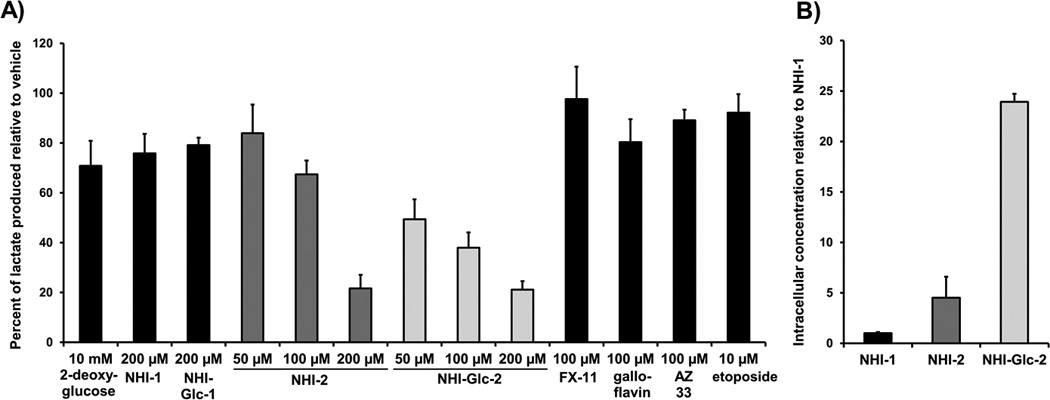
A)Treatment of HeLa cells with various compounds indicates that NHI-Glc-2 dose-dependently reduces lactate production, comparing favorably in this regard to NHI-2. The effect of NHI-Glc-2 is significantly more potent than that of NHI-1, NHI-Glc-1, and other reported LDH-A inhibitors, FX-11, galloflavin, and AZ 33. HeLa cells were treated for 8 hours with compound or DMSO vehicle (1% final concentration DMSO) in DMEM media. To quantify the lactate produced by the cells, derivatized cell culture media was analyzed by GC-MS. Lactate peaks were normalized using an internal standard present in each sample, and are presented as percent of vehicle lactate production. Averages are shown, with error bars denoting standard error of three or more independent experiments. B) Compound NHI-Glc-2 is more readily taken up by cancer cells than NHI-1 or NHI-2. A549 cells were treated with compound (100 µM) or vehicle (0.2% final concentration DMSO). Cells were collected after 4 hours, washed twice in PBS, sonicated in methanol, and analyzed via LC-MS. UV trace integration areas, standardized by sample fresh weights, were converted to relative concentrations using calibration of known concentrations (Figure S4). Relative concentrations are presented as ratios of the concentration of NHI-1. Error is standard error of three or more independent experiments.
To test if the enhanced cancer cell toxicity and lactate production inhibition by NHI-2 and NHI-Glc-2 was due to enhanced cell uptake, the ability of these compounds to penetrate A549 cells was evaluated. To compare the relative intracellular concentrations, A549 cells were treated with equimolar compounds or vehicle for 4 hours, and cell lysates were subjected to LC-MS analysis using calibrations of known concentrations generated using the same LC-MS protocol (Figure S4). In A549 cells, NHI-2 was present in approximately 4.5-fold higher concentrations in the lysate of samples compared to NHI-1, and NHI-Glc-2 was present at approximately 24-fold higher concentrations in the lysate samples (Figure 3B). NHI-Glc-2 does not appear to be appreciably cleaved to NHI-2 or NHI-Glc-1 inside the cell (representative UV traces are shown in Figure S5).
To examine whether NHI-Glc-2 is entering cells via GLUT transporters, a competition assay was performed between NHI-Glc-2 and GB2-Cy3, a fluorescent Cy3-linked glucose bioprobe recently developed by Park and coworkers.[28] While a number of glucose bioprobes are known,[29] GB2-Cy3 has been shown to possess an enhanced fluorescent signal in live cells and an enhanced ability to compete with glucose for uptake in cultured cells compared to the known 2-deoxyglucose analog 2-[N-(7-nitrobenx-2-oxa-1,3-diazol-4-yl)-amino]-2-deoxy-D-glucose (2-NBDG), with 5 µM GB2-Cy3 more potently inhibiting glucose uptake than 50 µM 2-NBDG.[30] For our purposes, cellular fluorescence was used as a readout for whether the cellular uptake of GB2-Cy3 was hindered by co-incubation with compound. A549 cells, which highly express GLUT-1[27b], were treated for 30 minutes with 2.5 µM GB2-Cy3 in the presence of either vehicle or 10 µM NHI-2, NHI-Glc-2, or glucose. Cells were then imaged by confocal laser scanning microscopy, and fluorescence was quantified and averaged over 40–60 cells per treatment. Treatment with NHI-Glc-2 and glucose caused a statistically significant decrease in fluorescence compared to vehicle, whereas treatment with NHI-2 did not (Figure 4), thus suggesting that NHI-Glc-2 and glucose are competing with GB2-Cy3 for cellular entry through GLUT transporters. To confirm that loss of cell viability was not skewing this result, 30 minute toxicity assays were performed for both NHI-2 and NHI-Glc-2; the results show that no appreciable loss of viability is observed at compound concentrations up to 200 µM at 30 minute treatment times.
Figure 4.
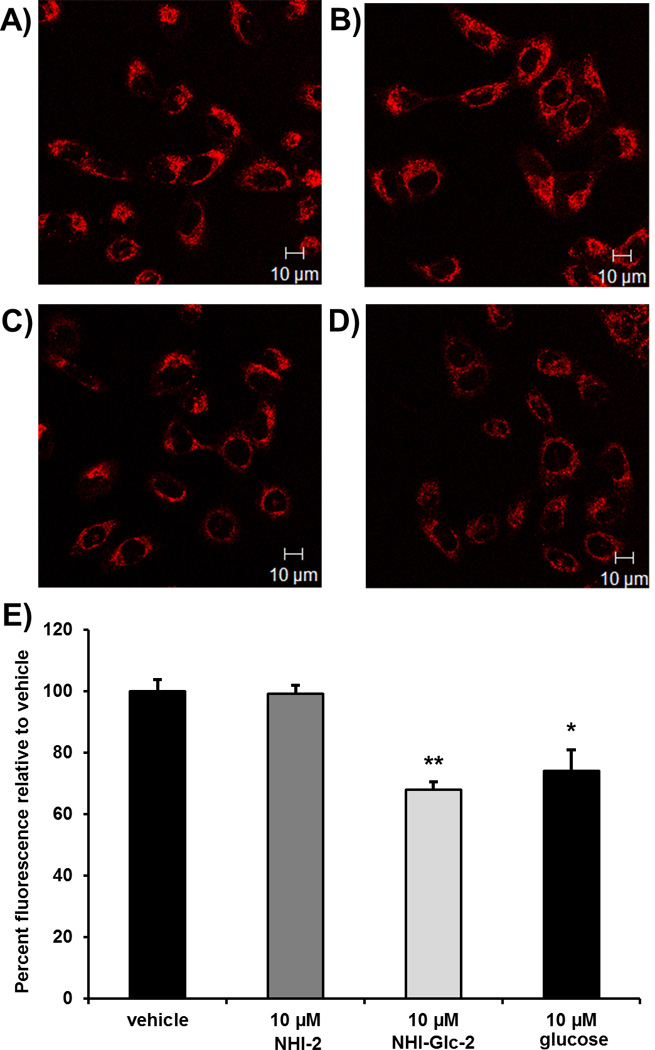
A549 cells were grown in RPMI 1640 growth media on cover glass bottom dishes (60,000 cells/dish). When cells reached 70% confluence, they were treated with GB2-Cy3 (2.5 µM) A) in the absence or B) the presence of NHI-2 (10 µM), C) NHI-Glc-2 (10 µM), or D) glucose (10 µM) for 30 minutes at 37 °C. Cellular fluorescence was observed using a Zeiss LSM700 confocal microscope, using a photomultiplier gain of 844 and a laser power of 555 nm (representative images are shown). E) The intracellular fluorescence of NHI-Glc-2- and glucose-treated cells is statistically significantly less than that of vehicle-treated cells, indicating that the uptake of the fluorescent GB2-Cy3 probe in these cells was inhibited by treatment with NHI-Glc-2 and glucose. The mean fluorescence intensities of each sample were calculated by averaging the fluorescence intensities of 40–60 cells per treatment over three independent experiments. Error bars denote standard error (n=3); statistical analysis was performed using an unpaired, two-tailed Student’s t test. * denotes p<0.05; ** denotes p<0.01.
In summary, NHI-Glc-2 has been designed as the first compound aimed at dual targeting of the Warburg effect, created to exploit a) the enhancement in glucose uptake, and b) the increased glycolysis that characterizes many aggressive tumors. NHI-Glc-2 has improved potency against cancer cells and increased cell permeability compared to its aglycone, showing a modest reduction in its inhibition potency on isolated enzyme that is highly compensated by its improved cell uptake via GLUT transporters. This compound will be an outstanding tool to fully probe the tractability of LDH-A inhibition in advanced mammalian tumor models. In addition, these results suggest application of this dual targeting strategy to inhibitors of the various other enzymes involved in glycolysis.
Acknowledgments
Alexander Ulanov and Lucas Li at the UIUC Roy J. Carver Metabolomics Center are acknowledged for their assistance in designing GC-MS experiment parameters, and Furong Sun at the UIUC Mass Spectrometry Laboratory is acknowledged for assistance with LC-MS experiments. F.M. thanks Drs. Mario Varasi, Daniele Fancelli, and Simon Plyte of the European Institute of Oncology (I.E.O.), Milano – Italy for helpful discussions. We are grateful to the NIH (R01GM098453), the University of Pisa, and the University of Illinois for funding. E.C.C. is an NIH Ruth L. Kirschstein National Research Service Award predoctoral fellow (1F30CA168323-01) and a UIUC Department of Biochemistry Herbert Carter fellow. R.W.H. III is an American Cancer Society postdoctoral fellow. H.Y.L. is a National Research Foundation of Korea postdoctoral fellow. The Q-Tof Ultima mass spectrometer was purchased in part with a grant from the National Science Foundation, Division of Biological Infrastructure (DBI-0100085). Dr. Giorgio Placanica of the University of Pisa is gratefully acknowledged for technical assistance in the analysis of chemical products. GB2-Cy3 was a kind gift from Prof. Seung Bum Park at Seoul National University.
Contributor Information
Filippo Minutolo, Email: filippo.minutolo@farm.unipi.it.
Paul J. Hergenrother, Email: hergenro@illinois.edu.
References
- 1.Warburg O. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 2.a) Vander Heiden MG. Nat Rev Drug Discov. 2011;10:671–684. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]; b) Jones NP, Schulze A. Drug Discov Today. 2012;17:232–241. doi: 10.1016/j.drudis.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 3.a) Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, Reynolds GE, Chi JT, Wu J, Solow-Cordero DE, Bonnet M, Flanagan JU, Bouley DM, Graves EE, Denny WA, Hay MP, Giaccia AJ. Sci Transl Med. 2011;3:94ra70. doi: 10.1126/scitranslmed.3002394. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Luo F, Liu X, Yan N, Li S, Cao G, Cheng Q, Xia Q, Wang H. BMC Cancer. 2006;6:26. doi: 10.1186/1471-2407-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altenberg B, Greulich KO. Genomics. 2004;84:1014–1020. doi: 10.1016/j.ygeno.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Bensinger SJ, Christofk HR. Semin Cell Dev Biol. 2012;23:352–361. doi: 10.1016/j.semcdb.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 6.a) Pohl J, Bertram B, Hilgard P, Nowrousian MR, Stuben J, Wiessler M. Cancer Chemother Pharmacol. 1995;35:364–370. doi: 10.1007/s002800050248. [DOI] [PubMed] [Google Scholar]; b) Lin YS, Tungpradit R, Sinchaikul S, An FM, Liu DZ, Phutrakul S, Chen ST. J Med Chem. 2008;51:7428–7441. doi: 10.1021/jm8006257. [DOI] [PubMed] [Google Scholar]; c) Kumar P, Shustov G, Liang H, Khlebnikov V, Zheng W, Yang XH, Cheeseman C, Wiebe LI. J Med Chem. 2012;55:6033–6046. doi: 10.1021/jm2017336. [DOI] [PubMed] [Google Scholar]; d) Oliveri V, Giuffrida ML, Vecchio G, Aiello C, Viale M. Dalton Trans. 2012;41:4530–4535. doi: 10.1039/c2dt12371a. [DOI] [PubMed] [Google Scholar]; e) Lee HY, Kwon J-T, Koh M, Cho M-H, Park SB. Bioorg Med Chem Lett. 2007;17:6335–6339. doi: 10.1016/j.bmcl.2007.08.071. [DOI] [PubMed] [Google Scholar]
- 7.a) Goff RD, Thorson JS. J Med Chem. 2010;53:8129–8139. doi: 10.1021/jm101024j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Peltier-Pain P, Timmons SC, Grandemange A, Benoit E, Thorson JS. ChemMedChem. 2011;6:1347–1350. doi: 10.1002/cmdc.201100178. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Goff RD, Thorson JS. Org Lett. 2012;14:2454–2457. doi: 10.1021/ol300703z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calvaresi E, Hergenrother PJ. Chem Sci. 2013;4:2319–2333. doi: 10.1039/C3SC22205E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Chiorean EG, Dragovich T, Hamm J, Barrios CH, Gorini CF, Langmuir VK, Kroll S, Jung DT, Tidmarsh GT, Loehrer PJ. Am J Clin Oncol. 2010;33:111–116. doi: 10.1097/COC.0b013e3181979204. [DOI] [PubMed] [Google Scholar]; b) Ciuleanu TE, Pavlovsky AV, Bodoky G, Garin AM, Langmuir VK, Kroll S, Tidmarsh GT. Eur J Cancer. 2009;45:1589–1596. doi: 10.1016/j.ejca.2008.12.022. [DOI] [PubMed] [Google Scholar]; c) Giaccone G, Smit EF, de Jonge M, Dansin E, Briasoulis E, Ardizzoni A, Douillard JY, Spaeth D, Lacombe D, Baron B, Bachmann P, Fumoleau P. Eur J Cancer. 2004;40:667–672. doi: 10.1016/j.ejca.2003.10.027. [DOI] [PubMed] [Google Scholar]; d) Briasoulis E, Pavlidis N, Terret C, Bauer J, Fiedler W, Schoffski P, Raoul JL, Hess D, Selvais R, Lacombe D, Bachmann P, Fumoleau P. Eur J Cancer. 2003;39:2334–2340. doi: 10.1016/s0959-8049(03)00629-4. [DOI] [PubMed] [Google Scholar]; e) van den Bent MJ, Grisold W, Frappaz D, Stupp R, Desir JP, Lesimple T, Dittrich C, de Jonge MJ, Brandes A, Frenay M, Carpentier AF, Chollet P, Oliveira J, Baron B, Lacombe D, Schuessler M, Fumoleau P. Ann Oncol. 2003;14:1732–1734. doi: 10.1093/annonc/mdg491. [DOI] [PubMed] [Google Scholar]
- 10.Fiume L, Manerba M, Vettraino M, Di Stefano G. Pharmacology. 2010;86:157–162. doi: 10.1159/000317519. [DOI] [PubMed] [Google Scholar]
- 11.a) Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. Cancer Res. 2011;71:2550–2560. doi: 10.1158/0008-5472.CAN-10-2828. [DOI] [PubMed] [Google Scholar]; b) Husain Z, Huang Y, Seth P, Sukhatme VP. J Immunol. doi: 10.4049/jimmunol.1202702. Epub ahead of print: July 1, 2013. [DOI] [PubMed] [Google Scholar]
- 12.a) Kolev Y, Uetake H, Takagi Y, Sugihara K. Ann Surg Oncol. 2008;15:2336–2344. doi: 10.1245/s10434-008-9955-5. [DOI] [PubMed] [Google Scholar]; b) Koukourakis MI, Giatromanolaki A, Sivridis E, Bougioukas G, Didilis V, Gatter KC, Harris AL. Br J Cancer. 2003;89:877–885. doi: 10.1038/sj.bjc.6601205. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Giatromanolaki A, Sivridis E, Kouskoukis C, Gatter KC, Harris AL, Koukourakis MI. Melanoma Res. 2003;13:493–501. doi: 10.1097/00008390-200310000-00008. [DOI] [PubMed] [Google Scholar]
- 13.a) Seth P, Grant A, Tang J, Vinogradov E, Wang X, Lenkinski R, Sukhatme VP. Neoplasia. 2011;13:60–71. doi: 10.1593/neo.101020. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fantin VR, St-Pierre J, Leder P. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 14.Maekawa M, Kanda S, Sudo K, Kanno T. Am J Hum Genet. 1984;36:1204–1214. [PMC free article] [PubMed] [Google Scholar]
- 15.Maekawa M, Kanno T. Clin Chim Acta. 1989;185:299–308. doi: 10.1016/0009-8981(89)90220-9. [DOI] [PubMed] [Google Scholar]
- 16.a) Granchi C, Roy S, Giacomelli C, Macchia M, Tuccinardi T, Martinelli A, Lanza M, Betti L, Giannaccini G, Lucacchini A, Funel N, Leon LG, Giovannetti E, Peters GJ, Palchaudhuri R, Calvaresi EC, Hergenrother PJ, Minutolo F. J Med Chem. 2011;54:1599–1612. doi: 10.1021/jm101007q. [DOI] [PubMed] [Google Scholar]; b) Granchi C, Calvaresi EC, Tuccinardi T, Paterni I, Macchia M, Martinelli A, Hergenrother PJ, Minutolo F. Org Biomol Chem. 2013 doi: 10.1039/c3ob40870a. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Granchi C, Bertini S, Macchia M, Minutolo F. Curr Med Chem. 2010;17:672–697. doi: 10.2174/092986710790416263. [DOI] [PubMed] [Google Scholar]
- 18.Gomez MS, Piper RC, Hunsaker LA, Royer RE, Deck LM, Makler MT, Vander Jagt DL. Mol Biochem Parasitol. 1997;90:235–246. doi: 10.1016/s0166-6851(97)00140-0. [DOI] [PubMed] [Google Scholar]
- 19.a) Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Proc Natl Acad Sci U S A. 2010;107:2037–2042. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dutta P, Le A, Vander Jagt DL, Tsukamoto T, Martinez GV, Dang CV, Gillies RJ. Cancer Res. 2013;73:4190–4195. doi: 10.1158/0008-5472.CAN-13-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thornburg JM, Nelson KK, Clem BF, Lane AN, Arumugam S, Simmons A, Eaton JW, Telang S, Chesney J. Breast Cancer Res. 2008;10:R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manerba M, Vettraino M, Fiume L, Di Stefano G, Sartini A, Giacomini E, Buonfiglio R, Roberti M, Recanatini M. ChemMedChem. 2012;7:311–317. doi: 10.1002/cmdc.201100471. [DOI] [PubMed] [Google Scholar]
- 22.Ward RA, Brassington C, Breeze AL, Caputo A, Critchlow S, Davies G, Goodwin L, Hassall G, Greenwood R, Holdgate GA, Mrosek M, Norman RA, Pearson S, Tart J, Tucker JA, Vogtherr M, Whittaker D, Wingfield J, Winter J, Hudson K. J Med Chem. 2012;55:3285–3306. doi: 10.1021/jm201734r. [DOI] [PubMed] [Google Scholar]
- 23.Kohlmann A, Zech SG, Li F, Zhou T, Squillace RM, Commodore L, Greenfield MT, Lu X, Miller DP, Huang W-S, Qi J, Thomas RM, Wang Y, Zhang S, Dodd R, Liu S, Xu R, Xu Y, Miret JJ, Rivera V, Clackson T, Shakespeare WC, Zhu X, Dalgarno DC. J Med Chem. 2013;56:1023–1040. doi: 10.1021/jm3014844. [DOI] [PubMed] [Google Scholar]
- 24.Dragovich PS, Fauber BP, Corson LB, Ding CZ, Eigenbrot C, Ge H, Giannetti AM, Hunsaker T, Labadie S, Liu Y, Malek S, Pan B, Peterson D, Pitts K, Purkey HE, Sideris S, Ultsch M, VanderPorten E, Wei B, Xu Q, Yen I, Yue Q, Zhang H, Zhang X. Bioorg Med Chem Lett. 2013;23:3186–3194. doi: 10.1016/j.bmcl.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 25.Coan KED, Shoichet BK. Mol BioSyst. 2007;3:208–213. doi: 10.1039/b616314a. [DOI] [PubMed] [Google Scholar]
- 26.a) Chow SN, Lin JK, Li SS, Chien CH. Gynecol Oncol. 1997;64:114–120. doi: 10.1006/gyno.1996.4531. [DOI] [PubMed] [Google Scholar]; b) Huang L, Zheng M, Zhou QM, Zhang MY, Jia WH, Yun JP, Wang HY. Cancer. 2011;117:3363–3373. doi: 10.1002/cncr.25870. [DOI] [PubMed] [Google Scholar]; c) Simpson NE, Tryndyak VP, Beland FA, Pogribny IP. Breast Cancer Res Treat. 2011 doi: 10.1007/s10549-011-1871-x. s10549-10011-11871. [DOI] [PubMed] [Google Scholar]
- 27.a) Young CD, Lewis AS, Rudolph MC, Ruehle MD, Jackman MR, Yun UJ, Ilkun O, Pereira R, Abel ED, Anderson SM. PLoS One. 2011;6:e23205. doi: 10.1371/journal.pone.0023205. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ito S, Fukusato T, Nemoto T, Sekihara H, Seyama Y, Kubota S. J Natl Cancer Inst. 2002;94:1080–1091. doi: 10.1093/jnci/94.14.1080. [DOI] [PubMed] [Google Scholar]
- 28.Lee HY, Lee JJ, Park J, Park SB. Chem. Eur. J. 2011;17:143–150. doi: 10.1002/chem.201002560. [DOI] [PubMed] [Google Scholar]
- 29.Kim WH, Lee J, Jung DW, Williams DR. Sensors. 2012;12:5005–5027. doi: 10.3390/s120405005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a) Yamada K, Nakata M, Horimoto N, Saito M, Matsuoka H, Inagaki N. J Biol Chem. 2000;275:22278–22283. doi: 10.1074/jbc.M908048199. [DOI] [PubMed] [Google Scholar]; b) Yoshioka K, Takahashi H, Homma T, Saito M, Oh KB, Nemoto Y, Matsuoka H. Biochim Biophys Acta. 1996;1289:5–9. doi: 10.1016/0304-4165(95)00153-0. [DOI] [PubMed] [Google Scholar]