

Abstract
A constant provision of ATP is of necessity for cardiac contraction. As the heart progresses toward failure following a myocardial infarction (MI), it undergoes metabolic alterations that have the potential to compromise the ability to meet energetic demands. This study evaluated the efficacy of mesenchymal stem cell (MSC) transplantation into the infarcted heart to minimize impairments in the metabolic processes that contribute to energy provision. Seven and twenty-eight days following the MI and MSC transplantation, MSC administration minimized cardiac systolic dysfunction. Hyperinsulinemic-euglycemic clamps, coupled with 2-[14C]deoxyglucose administration, were employed to assess systemic insulin sensitivity and tissue-specific, insulin-mediated glucose uptake 36 days following the MI in the conscious, unrestrained, C57BL/6 mouse. The improved systolic performance in MSC-treated mice was associated with a preservation of in vivo insulin-stimulated cardiac glucose uptake. Conserved glucose uptake in the heart was linked to the ability of the MSC treatment to diminish the decline in insulin signaling as assessed by Akt phosphorylation. The MSC treatment also sustained mitochondrial content, ADP-stimulated oxygen flux, and mitochondrial oxidative phosphorylation efficiency in the heart. Maintenance of mitochondrial function and density was accompanied by preserved peroxisome proliferator-activated receptor-γ coactivator-1α, a master regulator of mitochondrial biogenesis. These studies provide insight into mechanisms of action that lead to an enhanced energetic state in the infarcted heart following MSC transplantation that may assist in energy provision and dampen cardiac dysfunction.
Keywords: mitochondria, glucose uptake, myocardial infarction, stem cells
following a myocardial infarction (MI), the heart undergoes remodeling that often manifests as altered shape, size, and function (43). Changes in cardiac structure and performance post-MI are routinely accompanied by metabolic abnormalities that challenge the ability of the heart to meet the constant energetic requirements of contraction (27). The compromised matching of energy supply and demand in the failing myocardium is, in part, the result of potentially impaired insulin signaling, a decline in insulin-mediated glucose uptake, and the overall rate of glucose utilization being depressed (18, 23, 27). While the impediment in fuel delivery that accompanies insulin resistance may limit ATP synthesis, further detriment to energy provision is facilitated through alterations in mitochondria (32). Integrative mitochondrial oxidative phosphorylation (OXPHOS) is often repressed as indicated by a decline in ADP-stimulated oxygen consumption (35, 36). Impairments in function may be accompanied by defects in individual electron transport chain complex activities (14), and a lowering of mitochondrial density has been reported in the postinfarcted heart (8). These adverse mitochondrial characteristics are exacerbated by reduced peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) in the infarcted and failing heart (3). PGC-1α is a master regulator that influences mitochondrial respiration and coupling (21, 22). Also, this coactivator promotes the transcription and replication of nuclear and mitochondrial DNA via nuclear respiratory factors (NRFs) and mitochondrial transcription factor A (TFAM) (47).
Cell-based treatments have been investigated as a therapeutic agent for repairing cardiac tissue and attenuating dysfunction in the infarcted heart. However, the efficacy of cell transplantation to minimize disparities in ATP supply/demand has been largely unstudied. Bone marrow mononuclear cell transplantation into the heart has been reported to enhance the cardiac phospho- (p) Akt-to-Akt ratio following ischemia-reperfusion injury (24). This suggests potential utility in combating impaired insulin-mediated glucose transport. Additionally, following a MI, the surviving myocardium of mesenchymal stem cell (MSC)-treated hearts exhibited an elevation in the peri-infarct (∼30%) and whole left ventricle (LV) (∼16%) phosphocreatine (PCr)-to-ATP ratio (PCr/ATP) (6). The PCr/ATP reflects OXPHOS function, efficiency of myocardial energy provision, and correlates well with LV contractile function (6, 48).
The present report aimed to further these experimental studies, indicating stem cell administration provides the postinfarcted heart relief from “energy starvation” by examining the conservation of metabolic function post-MI with MSC treatment. To uncover contributors to energy preservation with MSC administration, cardiac mitochondrial function, content and regulators of mitochondrial biogenesis, such as PGC-1α, NRF-1, and TFAM, were assessed. Hyperinsulinemic-euglycemic clamps combined with a radioactive glucose tracer provided indexes of whole body and tissue-specific, insulin-stimulated glucose uptake in vivo. Together, these measures allowed for identification of potential means by which the transplanted cells improve cardiac ATP provision and act as a metabolic therapy for the infarcted heart.
MATERIALS AND METHODS
See Fig. 1 for a schematic of experimental outline.
Fig. 1.

Schematic outline of experimental procedures and timeline. Echocardiography on conscious mice was performed before as well as 7 and 28 days following ligation of the left anterior descending (LAD) coronary artery. Arterial and jugular chronic catheterization was performed 28 days following the myocardial infarction (MI) event for the arterial sampling and venous infusion protocols of the hyperinsulinemic-euglycemic (insulin) clamp. Insulin clamps were executed after 7 days of recovery from the catheterization surgeries (36 days postinfarct) to assess insulin sensitivity in the conscious, unrestrained mouse. Isotopic tracer {2-[14C]deoxyglucose (2-[14C]DG)} administration during insulin clamps allowed for tissue-specific glucose uptake to be assessed in vivo. Additional experiments included evaluation of mitochondrial respiration in permeabilized cardiac fibers and regulators of metabolism by immunoblotting. MSC, mesenchymal stem cell; PBS, phosphate-buffered saline.
Animals and MI.
Procedures were approved by the Vanderbilt University Animal Care and Use Committee and performed according to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Publication no. 85-23, revised 1996, A3227-01).
Thirteen-week-old, male C57BL/6J mice (Jackson Laboratories, Bar Harbour, ME) were randomly separated into three groups: Sham, MI + phosphate-buffered saline (MI+PBS), and MI + MSC (MI+MSC). A left anterior descending coronary artery chronic ligation model was employed to induce a MI, as previously described (17). Immediately following the ligation, 25 μl of PBS (pH 7.2) or 2.5 × 105 MSCs in 25 μl of PBS (pH 7.2) were administered via a single intramyocardial injection into the blanching peri-infarct area of the MI+PBS or MI+MSC mice, respectively. The mice were anesthetized with 50 mg/kg pentobarbital sodium via intraperitoneal injection before the surgeries. Adequate intraprocedural anesthesia was confirmed by the absence of withdrawal reflex and evaluating respiratory and/or heart rate.
MSCs.
MSCs reportedly exhibit immunoprivileged characteristics that make the use of this cell type appealing for transplantation purposes (10, 30, 45). These unique characteristics provide the prospect for utilization of MSCs in allogeneic and xenogeneic transplantation in addition to autologous administration. Given this, human bone marrow-derived MSCs were purchased from the Center for the Preparation and Distribution of Adult Stem Cells (Texas A&M Health Science Center College of Medicine Institute for Regenerative, Temple, TX) that provides standardized preparations of MSCs with NIH/Nationa Center for Research Resources (P40 RR 17447–06) support. The MSCs have been identified to consistently exhibit the capacity for osteoblast, adipocyte, and chondrocyte lineage differentiation, as well as be CD90+, CD105+, CD49c+, CD49f+, CD166+, CD59+, CD29+, CD44+, CD45−, CD34−, CD117−, and CD36− (29). MSCs were cultured in α-minimum essential medium with l-glutamine (Life Technologies, Burlington, ON, Canada), 16.5% defined FBS (HyClone, Logan, UT), streptomycin (100 μg/ml), and penicillin (100 U/ml). All MSCs for injection were obtained from a single donor, and passages 3–7 were utilized.
Cardiac function.
Cardiac systolic function was evaluated by M-mode echocardiography in conscious mice before the left anterior descending coronary artery ligation procedures, as well as 7 and 28 days post-MI, as previously described (5, 34).
Chronic catheterization procedures.
In preparation for the hyperinsulinemic-euglycemic clamps (insulin clamps), the mice underwent chronic venous and arterial catheterization surgery 28 days postligation, as previously described (17). The mice were housed individually for 7 days postcatheterization to ensure the mice were within 10% of presurgical weight for use in the insulin clamps.
Hyperinsulinemic-euglycemic clamps and isotopic tracer administration.
Insulin clamps were executed 36 days post-MI, as previously described (1). Mice were fasted at 7:00 AM for 5 h before the initiation of the experiments. One hour before the insulin clamps, the mice were given an opportunity to acclimate to their environment by connecting the externalized mouse catheters to all catheter leads and infusion syringes. Just before insulin clamp commencement, an arterial blood sample was obtained for fasted plasma glucose, insulin, nonesterified fatty acids (NEFAs) and hematocrit measures. To prevent a decline in hematocrit, a continuous, venous administration of saline-washed erythrocytes (5 μl/min) was performed. Each experiment maintained a constant, continuous infusion (4 mU·kg−1·min−1) of insulin for 133.17 ± 10.02 min. Euglycemia (∼7.0 mM) was maintained during the insulin clamps. Once a stable glucose infusion rate and euglycemia was a achieved for at least 30 min, arterial blood was sampled for glucose concentration (t = 0 min). Following this sample, a bolus containing 2-[14C]deoxyglucose (2-[14C]DG) (13 μCi) was administered via the jugular vein to provide an index of tissue-specific glucose uptake. At time t = 2, 5, 10, 15, and 20 min, arterial blood was sampled to determine glucose and 2-[14C]DG. At t = 30 min, arterial blood was taken for the measurement of glucose, experimental insulin, 2-[14C]DG, and hematocrit. Plasma samples were stored at −20°C until analysis. Once the insulin clamp was completed, mice were killed via cervical dislocation. Tissues (heart, soleus, gastrocnemius, superficial vastus lateralis, and white adipose tissue from epididymal deposits) were immediately excised for analysis or stored at −80°C. The LV peri-infarct was defined as the cardiac tissue directly adjacent to the infarcted region up to 2 mm. The remote LV was considered the LV tissue beyond this 2-mm region. Of note, the experimental ligation procedure creates an abrupt changeover from ischemic zone to perfused, viable tissue (13, 25). This poses restrictions in precisely modeling the clinical setting characterized by a progressive transition from infarct to viable tissue, however, it is also advantageous to the experiment's performance as it reduces the effect of cell viability in the peri-infarct region.
Plasma analyses.
Plasma insulin was evaluated by a double antibody method, as previously performed (26). Plasma NEFAs (NEFA C kit; Wako Chemicals, Richmond, VA) and glucose were determined spectrophotometrically (Molecular Devices, Sunnyvale, CA), as previously described (17). Plasma 2-[14C]DG was assessed as previously outlined (37).
Tissue-specific substrate kinetics.
Tissue 2-[14C]DG and phosphorylated 2-[14C]DG (2-[14C]DG-P) were evaluated as previously described (37). The metabolic index of glucose (Rg) uptake was calculated (38) and expressed (33) as previously outlined. Rg for tissues were expressed relative to the brain Rg, which represents constant reservoir of glucose uptake (33).
Mitochondrial oxygen consumption and enzymatic activity.
Saponin-permeabilized, cardiac fibers from the peri-infarct area were prepared as described (16). High-resolution respirometry (Oroboros Instruments, Innsbruck, Austria) was performed in duplicate at 37°C in MiR05 (0.5 mM EGTA, 3 mM MgCl2·6H2O, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 1 g/l BSA, 60 mM potassium-lactobionate, 110 mM sucrose, pH 7.1, adjusted at 30°C). Substrates included 10 mM glutamate plus 2 mM malate and 5mM pyruvate, 5 mM ADP, and 10 mM succinate. 10 mM cytochrome c were added to ensure the outer mitochondrial membrane was intact after processing. Following the respirometry studies, the fibers were stored at −80°C until use for citrate synthase activity determination as previously outlined (17).
Immunoblotting.
Cardiac tissue was homogenized in a lysis buffer containing 20 mM NaCl, 20 mM Tris·HCl, 0.1 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% β-mercaptoethanol (pH 7.4) in the presence of a protease inhibitor cocktail (Sigma-Aldrich, Oakville, ON, Canada) and phosphatase inhibitor cocktail (Thermo Fisher Scientific, Mississauga, ON, Canada). Tissue homogenate was centrifuged (10 min at 1,000 g and 4°C), and supernatant protein determination was assessed using the Bradford method. Cardiac (15–50 μg) proteins were separated on NuPAGE 4–12% Bis-Tris gels (Life Technologies) and transferred to a polyvinylidene fluoride membrane. Membranes were probed with PGC-1α (Santa Cruz Biotechnology, Santa Cruz, CA), glucose transporter-4 (Abcam, Cambridge, MA), hexokinase II (Chemicon, Temecula, CA), uncoupling protein 3 (Abcam), phospho-insulin receptor substrate 1 (Tyr608) (p-IRS-1; EMD Millipore Chemicals, Billerica, MA), IRS-1 (EMD Millipore Chemicals), phospho-Akt (Ser473) (p-Akt; Cell Signaling Technology, Whitby, ON, Canada), Akt (Cell Signaling Technology), NRF-1 (Santa Cruz Biotechnology), TFAM (Santa Cruz Biotechnology), and OXPHOS complexes I-V (OXPHOS CI–CV; Abcam) antibodies. Glyceraldehyde-3-phosphate dehydrogenase (Abcam) protein was utilized as a loading control.
Statistical analyses.
One-way ANOVA and two-way repeated-measures ANOVA were performed to detect statistical differences (P < 0.05) followed by Tukey's post hoc tests. All data are reported as means ± SE. Interpretations and conclusions as to the effect of the MSCs on all physiological parameters are based primarily on the ability of the MSCs to prevent statistically significant changes in relation to the Sham animals.
RESULTS
Stem cell administration minimizes cardiac contractile dysfunction.
Echocardiography was employed in the conscious mouse to evaluate cardiac contractile function (Table 1). Systolic dysfunction was identified in the MI+PBS group 7 and 28 days postinfarction, as indicated by the depression in ejection fraction (Fig. 2A) and fractional shortening (Fig. 2B). The MSC administration exhibited the ability to lessen the insult induced by the MI. The MSC-treated animals displayed a reduction in ejection fraction (Fig. 2A) and fractional shortening (Fig. 2B) at 7 and 28 days post-MI compared with the Sham group. However, the MSC-treated hearts exhibited a greater ejection fraction (Fig. 2A) and fractional shortening (Fig. 2B) than the MI+PBS mice at 7 and 28 days following cardiac insult. Cardiac hypertrophy was also assessed in the mice. The heart-to-body weight (Fig. 2C) was elevated in MI+PBS mice 36 days post-MI. The MSC administration minimized the increase in heart-to-body weight (Fig. 2C).
Table 1.
Cardiovascular parameters in conscious C57BL/6 mice
| Baseline |
7 days post-MI |
28 days post-MI |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Sham | MI+PBS | MI+MSC | Sham | MI+PBS | MI+MSC | Sham | MI+PBS | MI+MSC | |
| HR, beats/min | 691 ± 7 | 646 ± 34 | 666 ± 13 | 716 ± 8 | 692 ± 16 | 678 ± 7 | 777 ± 43 | 799 ± 69 | 689 ± 9*† |
| IVSd, mm | 0.79 ± 0.01 | 0.77 ± 0.01 | 0.76 ± 0.01 | 0.84 ± 0.01 | 0.81 ± 0.05 | 0.78 ± 0.02* | 0.77 ± 0.02 | 0.77 ± 0.03 | 0.81 ± 0.01 |
| LVIDd, mm | 2.93 ± 0.05 | 3.09 ± 0.10 | 3.33 ± 0.09 | 3.02 ± 0.04 | 4.44 ± 0.32* | 3.93 ± 0.28* | 2.98 ± 0.04 | 4.76 ± 0.33* | 4.35 ± 0.14* |
| LVPWd, mm | 0.78 ± 0.02 | 0.76 ± 0.03 | 0.74 ± 0.01 | 0.77 ± 0.02 | 0.77 ± 0.07 | 0.80 ± 0.02 | 0.74 ± 0.03 | 0.60 ± 0.06* | 0.83 ± 0.02*† |
| IVSs, mm | 0.93 ± 0.02 | 0.89 ± 0.02 | 0.93 ± 0.02 | 0.95 ± 0.03 | 0.91 ± 0.04* | 0.93 ± 0.02 | 0.88 ± 0.02 | 0.81 ± 0.03 | 0.93 ± 0.02† |
| LVIDs, mm | 1.45 ± 0.03 | 1.53 ± 0.06 | 1.73 ± 0.06 | 1.53 ± 0.03 | 3.36 ± 0.29* | 2.86 ± 0.14*† | 1.52 ± 0.03 | 3.69 ± 0.32* | 2.94 ± 0.16*† |
| LVPWs, mm | 0.97 ± 0.02 | 0.97 ± 0.02 | 1.02 ± 0.02 | 0.99 ± 0.03 | 0.86 ± 0.07* | 0.96 ± 0.02 | 0.92 ± 0.03 | 0.75 ± 0.07* | 1.01 ± 0.02*† |
Values are means ± SE; n = 6–14 mice/group. HR, heart rate; IVSd, interventricular septal thickness in diastole; LVIDd, left ventricle (LV) end-diastolic dimension; LVPWd, LV posterior wall thickness in diastole; IVSs, interventricular septal thickness in systole; LVIDs, LV end-systolic dimension; LVPWs, LV posterior wall thickness in systole; MI, myocardial infarction; PBS, phosphate-buffered saline; MSC, mesenchymal stem cell.
P < 0.05 vs. Sham at specified time point.
P < 0.05 vs. MI+PBS at specified time point.
Fig. 2.
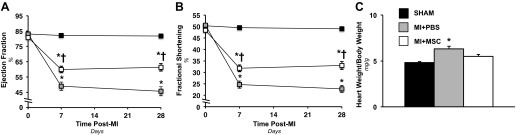
Cardiac functional and hypertropic indexes. A: cardiac ejection fraction (%) before a MI and 7 and 28 days following a MI. n = 6–14 mice/group. B: cardiac fractional shortening (%) before a MI and 7 and 28 days following a MI. n = 6–14 mice/group. C: heart weight-to-body weight ratio 36 days following a MI. n = 11–12 mice/group. Values are means ± SE. *P < 0.05 vs. Sham. †P < 0.05 vs. MI+PBS.
MSC therapy dampens impairment in glucose uptake in the infarcted heart.
Insulin clamps in the conscious, unrestrained mouse were employed to evaluate in vivo whole body glucose disposal in response to insulin and as a means of identifying a systemic effect of the MI and MSC treatment. The glucose infusion rate required to achieve euglycemia (∼7.0 mM) was similar between groups (Fig. 3A). Blood glucose levels were comparable between groups during the insulin clamp (Fig. 3B). Isotopic glucose (2-[14C]DG) was administered during the insulin clamp to assess insulin-stimulated, tissue-specific glucose uptake. Immediately following the insulin clamp, the LV of the heart was dissected based strictly on anatomical relation to the infarction into a remote LV region and peri-infarct area. Both the remote LV (MI+PBS LV) and the peri-infarct region (MI+PBS PI) in the MI+PBS animals exhibited a lower rate of glucose utilization compared the Sham group (Fig. 3, C and D). In contrast, a regional effect was observed in the MSC-treated mice. An intramyocardial injection of MSC preserved cardiac glucose uptake in response to insulin in the remote LV (MI+MSC LV), but was not able to rescue the peri-infarct region (MI+MSC PI) from impaired glucose uptake (Fig. 3, C and D).
Fig. 3.
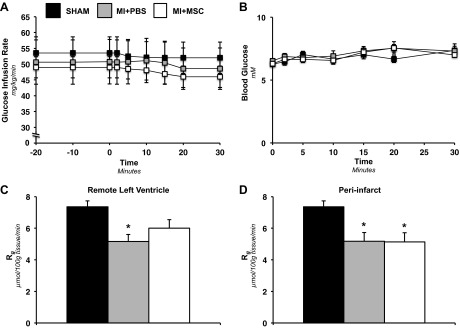
Insulin-stimulated whole body disposal and cardiac-specific glucose uptake. A: glucose infusion rate (GIR) during the insulin clamp. The GIR is equivalent to the whole body glucose disposal rate in response to venous infusion of insulin. The GIR is shown as a time course starting 20 min before administration of 2-[14C]DG (−20-min time point) to 30 min following 2-[14C]DG infusion (30-min time point). B: arterial blood glucose concentration following 2-[14C]DG for 30 min postadministration. Values are means ± SE for n = 6–10 mice/group. Metabolic index of glucose uptake (Rg) in the remote left ventricle (LV; C) and Rg in the peri-infarct (PI) region of the LV (D) are shown. Cardiac Rg values are relative to brain Rg. Values are means ± SE; n = 6–8 mice/group. *P < 0.05 vs. Sham.
Diminished impairments in regulators of glucose uptake by MSC transplantation.
Regulators of glucose metabolism were evaluated to identify the mechanisms by which MSC administration preserved cardiac glucose uptake in the remote LV. The insulin signaling pathway was probed by determining p-IRS-1 (Tyr608), total IRS-1, p-Akt (Ser473), and total Akt. Both infarct groups displayed a reduction in p-IRS-1 in the remote LV and peri-infarct region (Fig. 4, A–C). Total IRS-1 was similar between groups; however, total IRS-1 may have trended toward a significant decline, given that the p-IRS-1-to-IRS-1 ratio was comparable between groups (Fig. 4, A–C). The MI event resulted in a reduction of the p-Akt-to-Akt ratio in the remote LV of the MI+PBS mice (Fig. 4, D and F). The MSC administration minimized the p-Akt/Akt decline in the remote LV (Fig. 4, D and F). The remote LV region displayed similar PGC-1α between groups (Fig. 4, G and I). However, the MI-induced a depression in cardiac PGC-1α that was inhibited by the MSC treatment in the peri-infarct region (Fig. 4, H and I). Cardiac glucose transporter-4 and hexokinase II in the remote LV and peri-infarct regions were comparable between groups (Fig. 4, G–I).
Fig. 4.
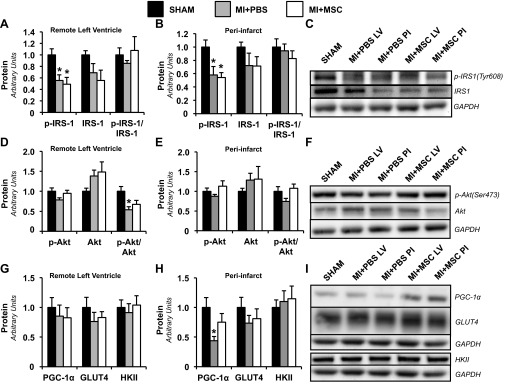
Mediators of cardiac glucose uptake. Remote LV (A) and PI (B) cardiac phospho-insulin receptor substrate-1 (p-IRS-1), IRS-1, and p-IRS-1-to-total IRS-1 ratio (p-IRS-1/IRS-1) as determined by immunoblotting are shown. C: representative immunoblotting performed to measure p-IRS-1 and IRS-1. Remote LV (D) and PI (E) cardiac phospho-Akt (p-Akt), Akt, and p-Akt-to-total Akt ratio (p-Akt/Akt), as determined by immunoblotting, are shown. F: representative immunoblotting performed to measure p-Akt and Akt. Remote LV (G) and PI (H) cardiac peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), glucose transporter-4 (GLUT4), and hexokinase II (HKII), as determined by immunoblotting, are shown. I: representative immunoblotting performed to measure PGC-1α, GLUT4, and HKII. Protein levels are normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) content and are relative to the Sham group. Values are means ± SE; n = 5–6 mice/group. *P < 0.05 vs. Sham.
Potential for increased peripheral tissue glucose uptake post-MSC therapy.
It has been proposed that cardiomyopathy is associated with generalized energetic failure and systemic insulin resistance (32, 42). Furthermore, previous studies have reported MSCs to persist, albeit low in number, in other tissues, such as the lungs, spleen, and liver, following an intramyocardial injection (2, 15). As such, insulin-stimulated glucose uptake in the soleus, white adipose tissue, superficial vastus lateralis, and gastrocnemius was assessed to identify potential for a systemic effect of the exogenous cells (Fig. 5, A–D). The MI+MSC soleus exhibited an elevated Rg compared with that of the Sham and MI+PBS mice (Fig. 5A). The rate of glucose utilization in the white adipose tissue, superficial vastus lateralis, and gastrocnemius was similar between groups (Fig. 5, B–D). Also, the 5-h fasted levels of arterial glucose and NEFAs were comparable between groups (Table 2).
Fig. 5.
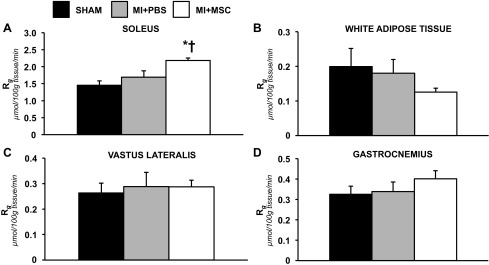
Insulin-stimulated peripheral tissue glucose uptake. A: Rg in the soleus. B: Rg in white adipose tissue. C: Rg in the superficial vastus lateralis. D: Rg in the gastrocnemius. Tissue Rg values are relative to brain Rg. Values are means ± SE; n = 6–9 mice/group. *P < 0.05 vs. Sham. †P < 0.05 vs. MI+PBS.
Table 2.
Biometric characteristics of C57BL/6 mice 36 days following a myocardial infarction
| Sham | MI+PBS | MI+MSC | |
|---|---|---|---|
| Fasting plasma glucose, mM | 9.05 ± 0.31 | 8.18 ± 0.37 | 8.43 ± 0.33 |
| Fasting plasma NEFA, mM | 0.9 ± 0.05 | 0.94 ± 0.05 | 1.0 ± 0.05 |
| Experimental plasma insulin, μU/ml | 103.87 ± 22.32 | 90.25 ± 13.58 | 91.67 ± 12.40 |
| Body weight, g | 26.61 ± 0.85 | 25.77 ± 0.98 | 27.9 ± 0.55 |
| Muscle, % | 90.88 ± 0.88 | 90.03 ± 0.41 | 89.15 ± 0.94 |
| Fat, % | 7.66 ± 0.68 | 7.93 ± 0.32 | 8.59 ± 0.97 |
| Free fluid, % | 1.46 ± 0.36 | 2.04 ± 0.20 | 2.21 ± 0.17 |
Values are means ± SE; n = 12–13 mice/group for plasma glucose; n = 11–12 for plasma NEFA, n = 6–9 for experimental plasma insulin, n = 7–9 for body weight, and n = 11–15 for body composition measurements. NEFA, nonesterified fatty acid.
MSC transplantation preserves cardiac mitochondrial content and lessens fall in ADP-stimulated oxygen flux.
Given both cardiac contraction and glucose utilization may be influenced by mitochondrial function, polarographic oxygen flux measurements were performed to evaluate mitochondrial OXPHOS. Basal oxygen consumption supported by complex I substrates malate, glutamate, and pyruvate was comparable between groups (Fig. 6A). The MI-only animals displayed a reduction in maximal ADP-stimulated oxygen flux (VMAX-CI; Fig. 6A). In contrast, ADP-stimulated oxygen consumption through complex I in MI + MSC animals was statistically comparable to the Sham group (Fig. 6A). The addition of succinate to assess ADP-stimulated oxygen flux via convergent electron flux through mitochondrial complexes I+II showed a significant reduction in the MI+PBS mice only (VMAX-CI+CII; Fig. 6A). Also, the succinate control ratio was heightened in the MI+PBS peri-infarct region (VMAX-CI+CII/VMAX-CI; Fig. 6B). Citrate synthase activity was assessed to identify a possible effect of mitochondrial heterogeneity on potential for energy provision. Citrate synthase activity was reduced in the MI+PBS group; however, MSC transplantation protected hearts from a decline in this mitochondrial content maker (Fig. 6C). The preservation of ADP-stimulated oxygen flux and cardiac citrate synthase activity in the MI+MSC mice prompted the evaluation of mitochondrial proteins involved in function and biogenesis. In the remote LV mitochondrial OXPHOS complexes I-V, NRF-1 and TFAM were similar between groups (Fig. 6, D and F). Peri-infarct complex I and TFAM were lowered in both infarct groups (Fig. 6, E and F). Uncoupling protein 3 in the peri-infarct region was lower in the MI+PBS mice (Fig. 6, E and F).
Fig. 6.
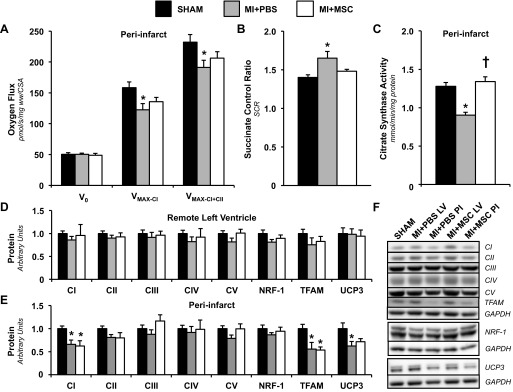
Cardiac mitochondrial function and characteristics. A: PI permeabilized cardiac fiber basal oxygen consumption supported by glutamate, malate, and pyruvate (V0), maximal oxygen consumption (ADP-stimulated) supported by glutamate, malate, and pyruvate through complex I (VMAX-CI), and maximal convergent oxygen flux supported by glutamate, malate, pyruvate, and succinate (VMAX-CI+CII). n = 8–10 mice/group. B: succinate control ratio (SCR; defined as VMAX-CI+CII/VMAX-CI). n = 9–10 mice per group. C: PI citrate synthase (CS) activity (mmol·min−1·mg protein−1). n = 8–10 mice/group. Remote LV (D) and PI (E) mitochondrial oxidative phosphorylation (OXPHOS) complexes I-V (CI-CV), nuclear respiratory factor-1 (NRF-1), mitochondrial transcription factor A (TFAM), and uncoupling protein-3 (UCP-3), as determined by immunoblotting, are shown. F: representative immunoblotting of the regional protein levels OXPHOS complexes I-V, NRF-1, TFAM, and UCP-3. Protein levels were normalized to GAPDH content and are expressed relative to the Sham group. n = 6 mice/group. Values are means ± SE. *P < 0.05 vs. Sham. †P < 0.05 vs. MI+PBS PI.
DISCUSSION
Owing to a limited, innate capacity for regeneration of lost cardiac tissue, the post-MI necrotic region of the heart undergoes a healing process to form a scar for structural integrity (4, 19). The severely depressed contractile properties of the scar places added mechanical responsibility on the uninjured myocardium to preserve function (41). Cell transplantation research has received significant attention as a potential method of repopulating the infarcted area of the heart with exogenous cells. However, cell-based therapies have been less than adequate at replacing the lost cells, in part, due to poor cell persistence following administration (12, 31). In light of the low cell survival and engraftment, cell-mediated paracrine effects have been proposed to be the major mediator for the improvement in cardiac function and lowering of maladaptive responses (11). Moreover, paracrine factors released by stem cells have been identified to hold a potential therapeutic role in several processes that impact cardiac function, including the modulation of apoptotic signaling, vascular density, inflammation, fibrosis, cardiac contractility, and metabolism (11, 46).
The present study aimed to evaluate the ability of MSC therapy to attenuate metabolic aberrations in cardiac glucose utilization and mitochondria. Such disturbances may hinder energy provision required to sustain contractile performance. Experimental studies have previously reported MSC-treated hearts to exhibit elevated high-energy phosphates compared with infarct-only hearts (6). Furthermore, this improvement in the energy state of the heart was accompanied by lower cardiac dysfunction (6). However, it remains to be completely determined which metabolic processes involved in energy provision are modulated by MSC therapy to achieve these therapeutic effects.
Our results indicated systolic performance was reduced 40–55%, as indicated by ejection fraction and fractional shortening following the MI. A single intramyocardial injection of MSC minimized the decline in contractile performance to ∼25–36%. Also, the exogenous cells mediated a mild inhibition of the hypertrophic response following a MI. The improvement in cardiac function and structure was associated with the ability of the MSC transplantation to lessen irregularities in in vivo cardiac glucose uptake. Thirty-six days after the MI, infarct-only hearts exhibited a decline in insulin-stimulated glucose uptake in the remote LV and peri-infarct regions. The MSC treatment impeded a significant reduction in glucose utilization in the remote LV. In contrast, the cell-based therapy could not prevent a decline in insulin-stimulated glucose uptake in the peri-infarct region. These results indicate that the intramyocardial injection of MSCs may exert regional influences and at the very least slow the onset of alterations in glucose utilization associated with cardiac remodeling post-MI.
Following evaluation of glucose uptake, immunoblotting was performed to identify potential mediators contributing to the maintenance of insulin-mediated glucose uptake in the MSC-treated remote LVs. The MSC administration did not rescue the infarcted heart from a fall in p-IRS-1. However, the stem cell transplantation did appear to augment components involved in insulin signaling through the attenuation of a significant decline in the remote LV p-Akt-to-Akt ratio. Constitutively active Akt has been reported to almost match the effect of insulin in vitro (20, 28). However, considering the p-Akt-to-Akt ratio was comparable between groups in the peri-infarct region, the ability of the MSCs to lessen the decline in Akt phosphorylation is, at best, of modest physiological significance to the beneficial effects the exogenous cells had on insulin-stimulated glucose uptake in the remote LV. Of note, the potential for MSC therapy to provide some protection to Akt phosphorylation is in agreement with previous studies that indicate the cell-based treatment enhances PI3K-Akt pathway signaling (40).
In addition to substrate flux, modulation mitochondrial OXPHOS may be a means by which the stem cell therapy may improve cardiac energetic abnormalities following a MI. The present study found MSC transplantation to minimize impairments in ADP-stimulated oxygen consumption supported by pyruvate, malate, and glutamate from decline. This mitochondrial ADP-responsiveness may contribute to the elevated high-energy phosphates previously identified following MSC transplantation in the infarcted heart. Although the enzymatic activities of the individual electron transport chain complexes were not evaluated, the combination of a depressed VMAX-CI and complex I protein and elevated succinate control ratio hints that complex I could be mediating this dysfunction, as there may be availability for increased flux downstream of complex I.
The preservation of cardiac ADP-stimulated oxygen flux in saponin-permeabilized fibers from the peri-infarct region led us to explore PGC-1α. PGC-1α is a transcriptional coactivator that augments mitochondrial function (44). Previous studies overexpressing PGC-1α in rodent ventricular myocytes have identified that this transcription factor coactivator promotes an increase in ADP-stimulated oxygen flux supported by malate and glutamate (21). Cardiac PGC-1α is of interest in our MI model because this master regulator of energy metabolism declines as the heart progresses toward overt failure (32). Similarly, the present study identified peri-infarct PGC-1α to be reduced in the MI-only mice.
Concurrent with its role in enhancing mitochondrial oxidative capability, PGC-1α promotes mitochondrial biogenesis (21). A loss of mitochondrial content has been observed in rodent models of heart failure (8). The present study identified a depression in citrate synthase activity, a marker of mitochondrial density, in the MI+PBS mice. However, the MSC-treated hearts exhibited no alterations in the activity of this tricarboxylic acid cycle enzyme. The preservation of cardiac mitochondria content following MSC transplantation may play a role in the improved PCr/ATP and cardiac energy homeostasis. A downstream effector of PGC-1α-mediated mitochondrial nuclear gene transcription is NRF-1 (9). Moreover, PGC-1α acts in concert with the NRFs and is proposed to be the limiting factor in NRF target gene expression (9). NRF-1 was comparable between groups, suggesting the ability of the MSC treatment to diminish a decline in PGC-1α may play an important role in the conservation of mitochondrial content. Of note, our results indicate that the lowering of TFAM following a MI was not blunted by the MSCs. TFAM is a nuclear-encoded transcription factor that regulates the replication and transcription of mitochondrial DNA (7, 39). As such, caution must be exerted when it comes to conclusions concerning the ability of the cell-based treatment to prevent long-term impairments in mitochondrial density.
In summary, the present study found MSC transplantation for the infarcted heart to lessen declines in in vivo, insulin-stimulated cardiac glucose uptake. Also, the stem cell administration exhibited a protective effect on integrative OXPHOS function and mitochondrial density following a MI. These mitochondrial improvements may have been conferred by the ability of MSC therapy to prevent alterations in PGC-1α and its downstream signaling. From a therapeutic perspective, the reduction in metabolic insults induced by the MSC transplantation could assist in allowing the heart to minimize disparities in energy supply and demand.
GRANTS
This work was supported by Canadian Institutes of Health Research (C. C. Hughey, J. Shearer), the Killam Trusts (C. C. Hughey), MitoCanada (J. Shearer), and National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-054902 (D. H. Wasserman) and DK-59637 (Vanderbilt University Mouse Metabolic Phenotyping Center).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: C.C.H., D.H.W., J.N.R., and J.S. conception and design of research; C.C.H., F.D.J., L.M., D.P.B., and Z.W. performed experiments; C.C.H. and J.S. analyzed data; C.C.H. interpreted results of experiments; C.C.H. prepared figures; C.C.H. drafted manuscript; C.C.H., F.D.J., L.M., D.P.B., Z.W., D.H.W., J.N.R., and J.S. edited and revised manuscript; C.C.H., F.D.J., L.M., D.P.B., Z.W., D.H.W., J.N.R., and J.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank the Vanderbilt University Analytical Resources Core for measuring plasma insulin concentrations.
REFERENCES
- 1.Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 55: 390–397, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Campbell NG, Suzuki K. Cell delivery routes for stem cell therapy to the heart: current and future approaches. J Cardiovasc Transl Res 5: 713–726, 2012 [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Wang Y, Chen J, Chen X, Cao W, Chen S, Xu S, Huang H, Liu P. Roles of transcriptional corepressor RIP140 and coactivator PGC-1alpha in energy state of chronically infarcted rat hearts and mitochondrial function of cardiomyocytes. Mol Cell Endocrinol 362: 11–18, 2012 [DOI] [PubMed] [Google Scholar]
- 4.Cleutjens JP, Blankesteijn WM, Daemen MJ, Smits JF. The infarcted myocardium: simply dead tissue, or a lively target for therapeutic interventions. Cardiovasc Res 44: 232–241, 1999 [DOI] [PubMed] [Google Scholar]
- 5.Exil VJ, Roberts RL, Sims H, McLaughlin JE, Malkin RA, Gardner CD, Ni G, Rottman JN, Strauss AW. Very-long-chain acyl-coenzyme a dehydrogenase deficiency in mice. Circ Res 93: 448–455, 2003 [DOI] [PubMed] [Google Scholar]
- 6.Feygin J, Mansoor A, Eckman P, Swingen C, Zhang J. Functional and bioenergetic modulations in the infarct border zone following autologous mesenchymal stem cell transplantation. Am J Physiol Heart Circ Physiol 293: H1772–H1780, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Fisher RP, Lisowsky T, Parisi MA, Clayton DA. DNA wrapping and bending by a mitochondrial high mobility group-like transcriptional activator protein. J Biol Chem 267: 3358–3367, 1992 [PubMed] [Google Scholar]
- 8.Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura-Clapier R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol 551: 491–501, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gleyzer N, Vercauteren K, Scarpulla RC. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol Cell Biol 25: 1354–1366, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gnecchi M, Danieli P, Cervio E. Mesenchymal stem cell therapy for heart disease. Vascul Pharmacol 57: 48–55, 2012 [DOI] [PubMed] [Google Scholar]
- 11.Gnecchi M, Zhang Z, Ni A, Dzau VJ. Paracrine mechanisms in adult stem cell signaling and therapy. Circ Res 103: 1204–1219, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haider H, Ashraf M. Strategies to promote donor cell survival: combining preconditioning approach with stem cell transplantation. J Mol Cell Cardiol 45: 554–566, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hearse DJ. Models and problems in the study of myocardial ischemia and tissue protection. Eur Heart J 4, Suppl C: 43–48, 1983 [DOI] [PubMed] [Google Scholar]
- 14.Heather LC, Carr CA, Stuckey DJ, Pope S, Morten KJ, Carter EE, Edwards LM, Clarke K. Critical role of complex III in the early metabolic changes following myocardial infarction. Cardiovasc Res 85: 127–136, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Hou D, Youssef EA, Brinton TJ, Zhang P, Rogers P, Price ET, Yeung AC, Johnstone BH, Yock PG, March KL. Radiolabeled cell distribution after intramyocardial, intracoronary, and interstitial retrograde coronary venous delivery: implications for current clinical trials. Circulation 112: I150–I156, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Hughey CC, Hittel DS, Johnsen VL, Shearer J. Respirometric oxidative phosphorylation assessment in saponin-permeabilized cardiac fibers. J Vis Exp 48: 2431, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughey CC, Johnsen VL, Ma L, James FD, Young PP, Wasserman DH, Rottman JN, Hittel DS, Shearer J. Mesenchymal stem cell transplantation for the infarcted heart: a role in minimizing abnormalities in cardiac-specific energy metabolism. Am J Physiol Endocrinol Metab 302: E163–E172, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD. Targeting fatty acid and carbohydrate oxidation–a novel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta 1813: 1333–1350, 2011 [DOI] [PubMed] [Google Scholar]
- 19.Kikuchi K, Poss KD. Cardiac regenerative capacity and mechanisms. Annu Rev Cell Dev Biol 28: 719–741, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3–L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem 271: 31372–31378, 1996 [DOI] [PubMed] [Google Scholar]
- 21.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest 106: 847–856, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lehman JJ, Boudina S, Banke NH, Sambandam N, Han X, Young DM, Leone TC, Gross RW, Lewandowski ED, Abel ED, Kelly DP. The transcriptional coactivator PGC-1α is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. Am J Physiol Heart Circ Physiol 295: H185–H196, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, Zeng H, Chen JX. Apelin-13 increases myocardial progenitor cells and improves repair postmyocardial infarction. Am J Physiol Heart Circ Physiol 303: H605–H618, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lovell MJ, Yasin M, Lee KL, Cheung KK, Shintani Y, Collino M, Sivarajah A, Leung KY, Takahashi K, Kapoor A, Yaqoob MM, Suzuki K, Lythgoe MF, Martin J, Munroe PB, Thiemermann C, Mathur A. Bone marrow mononuclear cells reduce myocardial reperfusion injury by activating the PI3K/Akt survival pathway. Atherosclerosis 213: 67–76, 2010 [DOI] [PubMed] [Google Scholar]
- 25.Miura T, Miki T. Limitation of myocardial infarct size in the clinical setting: current status and challenges in translating animal experiments into clinical therapy. Basic Res Cardiol 103: 501–513, 2008 [DOI] [PubMed] [Google Scholar]
- 26.Morgan CR, Lazarow A. Immunoassay of pancreatic and plasma insulin following alloxan injection of rats. Diabetes 14: 669–671, 1965 [DOI] [PubMed] [Google Scholar]
- 27.Neubauer S. The failing heart–an engine out of fuel. N Engl J Med 356: 1140–1151, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Ng Y, Ramm G, Lopez JA, James DE. Rapid activation of Akt2 is sufficient to stimulate GLUT4 translocation in 3T3–L1 adipocytes. Cell Metab 7: 348–356, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Oh JY, Lee RH, Yu JM, Ko JH, Lee HJ, Ko AY, Roddy GW, Prockop DJ. Intravenous mesenchymal stem cells prevented rejection of allogeneic corneal transplants by aborting the early inflammatory response. Mol Ther 20: 2143–2152, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pittenger MF, Martin BJ. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ Res 95: 9–20, 2004 [DOI] [PubMed] [Google Scholar]
- 31.Robey TE, Saiget MK, Reinecke H, Murry CE. Systems approaches to preventing transplanted cell death in cardiac repair. J Mol Cell Cardiol 45: 567–581, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosca MG, Hoppel CL. Mitochondrial dysfunction in heart failure. Heart Fail Rev 18: 607–622, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rottman JN, Bracy D, Malabanan C, Yue Z, Clanton J, Wasserman DH. Contrasting effects of exercise and NOS inhibition on tissue-specific fatty acid and glucose uptake in mice. Am J Physiol Endocrinol Metab 283: E116–E123, 2002 [DOI] [PubMed] [Google Scholar]
- 34.Rottman JN, Ni G, Khoo M, Wang Z, Zhang W, Anderson ME, Madu EC. Temporal changes in ventricular function assessed echocardiographically in conscious and anesthetized mice. J Am Soc Echocardiogr 16: 1150–1157, 2003 [DOI] [PubMed] [Google Scholar]
- 35.Sanbe A, Tanonaka K, Hanaoka Y, Katoh T, Takeo S. Regional energy metabolism of failing hearts following myocardial infarction. J Mol Cell Cardiol 25: 995–1013, 1993 [DOI] [PubMed] [Google Scholar]
- 36.Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN. Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol 32: 2361–2367, 2000 [DOI] [PubMed] [Google Scholar]
- 37.Shearer J, Fueger PT, Bracy DP, Wasserman DH, Rottman JN. Partial gene deletion of heart-type fatty acid-binding protein limits the severity of dietary-induced insulin resistance. Diabetes 54: 3133–3139, 2005 [DOI] [PubMed] [Google Scholar]
- 38.Shearer J, Fueger PT, Wang Z, Bracy DP, Wasserman DH, Rottman JN. Metabolic implications of reduced heart-type fatty acid binding protein in insulin resistant cardiac muscle. Biochim Biophys Acta 1782: 586–592, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi Y, Dierckx A, Wanrooij PH, Wanrooij S, Larsson NG, Wilhelmsson LM, Falkenberg M, Gustafsson CM. Mammalian transcription factor A is a core component of the mitochondrial transcription machinery. Proc Natl Acad Sci U S A 109: 16510–16515, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Si Y, Zhao Y, Hao H, Liu J, Guo Y, Mu Y, Shen J, Cheng Y, Fu X, Han W. Infusion of mesenchymal stem cells ameliorates hyperglycemia in type 2 diabetic rats: identification of a novel role in improving insulin sensitivity. Diabetes 61: 1616–1625, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun Y, Weber KT. Infarct scar: a dynamic tissue. Cardiovasc Res 46: 250–256, 2000 [DOI] [PubMed] [Google Scholar]
- 42.Swan JW, Anker SD, Walton C, Godsland IF, Clark AL, Leyva F, Stevenson JC, Coats AJ. Insulin resistance in chronic heart failure: relation to severity and etiology of heart failure. J Am Coll Cardiol 30: 527–532, 1997 [DOI] [PubMed] [Google Scholar]
- 43.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev 79: 215–262, 1999 [DOI] [PubMed] [Google Scholar]
- 44.Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res 79: 208–217, 2008 [DOI] [PubMed] [Google Scholar]
- 45.Williams AR, Hare JM. Mesenchymal stem cells: biology, pathophysiology, translational findings, and therapeutic implications for cardiac disease. Circ Res 109: 923–940, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wollert KC, Drexler H. Cell therapy for the treatment of coronary heart disease: a critical appraisal. Nat Rev Cardiol 7: 204–215, 2010 [DOI] [PubMed] [Google Scholar]
- 47.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115–124, 1999 [DOI] [PubMed] [Google Scholar]
- 48.Zeng L, Hu Q, Wang X, Mansoor A, Lee J, Feygin J, Zhang G, Suntharalingam P, Boozer S, Mhashilkar A, Panetta CJ, Swingen C, Deans R, From AH, Bache RJ, Verfaillie CM, Zhang J. Bioenergetic and functional consequences of bone marrow-derived multipotent progenitor cell transplantation in hearts with postinfarction left ventricular remodeling. Circulation 115: 1866–1875, 2007 [DOI] [PubMed] [Google Scholar]